
The Role of the NLRP3 Inflammasome and Programmed Cell Death in Acute Liver Injury


Abstract
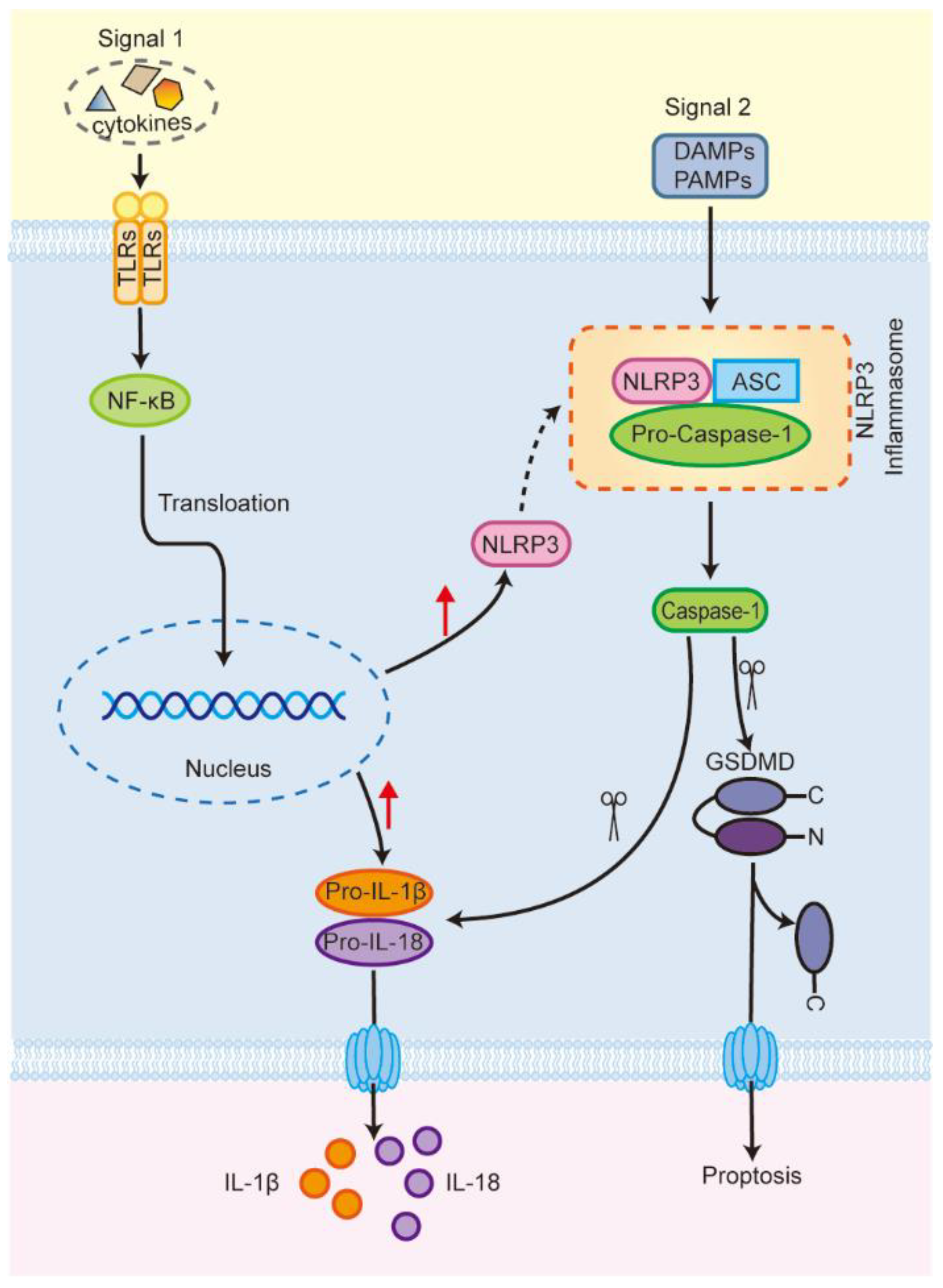
:1. Introduction
2. NLRP3 Inflammasome and Programmed Cell Death
2.1. Apoptosis
2.2. Necroptosis
2.3. Pyroptosis
2.4. Autophagy
3. NLRP3 Inflammasome and Programmed Cell Death in ALI
3.1. NLRP3 Inflammasome and Programmed Cell Death in Acetaminophen (APAP)-Induced Acute Liver Injury
3.2. NLRP3 Inflammasome and Programmed Cell Death in Liver Ischemia-Reperfusion Injury
3.3. NLRP3 Inflammasome and Programmed Cell Death in CCl4-Induced Acute Liver Injury
3.4. NLRP3 Inflammasome and Programmed Cell Death in Alcohol-Induced Acute Liver Injury
3.5. NLRP3 Inflammasome and Programmed Cell Death in Con A-Induced Autoimmune Hepatitis
3.6. NLRP3 Inflammasome and Programmed Cell Death in LPS/D-GalN-Induced Acute Liver Injury
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviation
| ACD | accidental cell death |
| AIH | autoimmune hepatitis |
| ALI | acute liver injury |
| APAP | acetaminophen |
| ASC | apoptosis-associated spotted protein |
| Caspase-1 | cysteine aspartate protease 1 |
| Con A | concanavalin A |
| CYP | cytochrome P450 enzymes |
| CYP2E1 | cytochrome P450 2E1 |
| DAMPs | damage-associated molecular patterns |
| D-GalN | D-galactosamine |
| DILI | drug-induced liver injury |
| FOXO1 | forkhead box protein O1 |
| GSDMD | gasdermin D |
| GSH | glutathione |
| HO-1 | heme oxygenase 1 |
| HMGB1 | high mobility group protein B1 |
| IL-1β | interleukin-1β |
| IL-1R1 | interleukin-1 receptor type 1 |
| IL-18 | interleukin-18 |
| KA | kaempferol |
| LPS | lipopolysaccharide |
| MCP-1 | monocyte chemoattractant protein-1 |
| MLKL | mixed lineage kinase domain-like |
| NAC | N-acetylcysteine |
| NAPQI | n-acetyl-p-benzoquinoneimine |
| Nec-1 | Necrostatin-1 |
| NLRP3 | nod-like receptor protein 3 |
| NF-ĸB | nuclear factor kappa-B |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| OGD/R | oxygen-glucose deprivation/reoxygenation |
| ORY | γ- Oryzanol |
| PAMPs | pathogen-associated molecular patterns |
| PCD | programmed cell death |
| PEITC | phenethyl isothiocyanate |
| PKA | protein kinase A |
| RCD | regulated cell death |
| ROS | reactive oxygen species |
| RIPK1 | receptor-interacting serine-threonine kinase 1 |
| RIPK3 | receptor-interacting serine-threonine kinase 3 |
| Sal | salidroside |
| SULT | sulfotransferase |
| S100A9 | S100 calcium-binding protein A9 |
| TNF-α | tumor necrosis factor-α |
| TLR4 | toll-like receptor 4 |
| UGT | UDP-glucuronosyltransferase |
| ZBP1 | Z-DNA binding protein 1 |
| 25HC | 25 hydroxycholesterol |
References
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Matsubara, Y.; Kiyohara, H.; Teratani, T.; Mikami, Y.; Kanai, T. Organ and brain crosstalk: The liver-brain axis in gastrointestinal, liver, and pancreatic diseases. Neuropharmacology 2022, 205, 108915. [Google Scholar] [CrossRef]
- Harrell, C.R.; Pavlovic, D.; Djonov, V.; Volarevic, V. Therapeutic potential of mesenchymal stem cells in the treatment of acute liver failure. World J. Gastroenterol. 2022, 28, 3627–3636. [Google Scholar] [CrossRef] [PubMed]
- Tujios, S.; Stravitz, R.T.; Lee, W.M. Management of Acute Liver Failure: Update 2022. Semin. Liver Dis. 2022, 42, 362–378. [Google Scholar] [CrossRef]
- Stravitz, R.T.; Lee, W.M. Acute liver failure. Lancet 2019, 394, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, R.; Chen, P. Gut Microbiota and Chemical-Induced Acute Liver Injury. Front. Physiol. 2021, 12, 688780. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.M.; Chauhan, A. The role of platelet mediated thromboinflammation in acute liver injury. Front. Immunol. 2022, 13, 1037645. [Google Scholar] [CrossRef]
- Liu, J.; Yuan, Z.; Wang, Q. Pluripotent Stem Cell-derived Strategies to Treat Acute Liver Failure: Current Status and Future Directions. J. Clin. Transl. Hepatol. 2022, 10, 692–699. [Google Scholar] [CrossRef]
- Bernal, W.; Auzinger, G.; Dhawan, A.; Wendon, J. Acute liver failure. Lancet 2010, 376, 190–201. [Google Scholar] [CrossRef]
- Hirao, H.; Nakamura, K.; Kupiec-Weglinski, J.W. Liver ischaemia-reperfusion injury: A new understanding of the role of innate immunity. Nat. Reviews. Gastroenterol. Hepatol. 2022, 19, 239–256. [Google Scholar] [CrossRef]
- Malik, A.; Kanneganti, T.D. Inflammasome activation and assembly at a glance. J. Cell Sci. 2017, 130, 3955–3963. [Google Scholar] [CrossRef] [PubMed]
- Akbal, A.; Dernst, A.; Lovotti, M.; Mangan, M.S.J.; McManus, R.M.; Latz, E. How location and cellular signaling combine to activate the NLRP3 inflammasome. Cell. Mol. Immunol. 2022, 19, 1201–1214. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.R.; Kanneganti, T.D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- Wang, L.; Hauenstein, A.V. The NLRP3 inflammasome: Mechanism of action, role in disease and therapies. Mol. Asp. Med. 2020, 76, 100889. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Ding, X.; Zhang, H.; Li, S.; Yang, P.; Tan, Q. Relevance of NLRP3 Inflammasome-Related Pathways in the Pathology of Diabetic Wound Healing and Possible Therapeutic Targets. Oxidative Med. Cell. Longev. 2022, 2022, 9687925. [Google Scholar] [CrossRef] [PubMed]
- Christgen, S.; Place, D.E.; Kanneganti, T.D. Toward targeting inflammasomes: Insights into their regulation and activation. Cell Res. 2020, 30, 315–327. [Google Scholar] [CrossRef]
- Banerjee, S.K.; Chatterjee, A.; Gupta, S.; Nagar, A. Activation and Regulation of NLRP3 by Sterile and Infectious Insults. Front. Immunol. 2022, 13, 896353. [Google Scholar] [CrossRef]
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 Inflammasome Pathway: A Review of Mechanisms and Inhibitors for the Treatment of Inflammatory Diseases. Front. Aging Neurosci. 2022, 14, 879021. [Google Scholar] [CrossRef]
- Chao, Y.Y.; Puhach, A.; Frieser, D.; Arunkumar, M.; Lehner, L.; Seeholzer, T.; Garcia-Lopez, A.; van der Wal, M.; Fibi-Smetana, S.; Dietschmann, A.; et al. Human T(H)17 cells engage gasdermin E pores to release IL-1α on NLRP3 inflammasome activation. Nat. Immunol. 2023, 24, 295–308. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ren, W.; Wu, Q.; Liu, T.; Wei, Y.; Ding, J.; Zhou, C.; Xu, H.; Yang, S. NLRP3 Inflammasome Activation: A Therapeutic Target for Cerebral Ischemia-Reperfusion Injury. Front. Mol. Neurosci. 2022, 15, 847440. [Google Scholar] [CrossRef]
- Xu, J.; Núñez, G. The NLRP3 inflammasome: Activation and regulation. Trends Biochem. Sci. 2022; in press. [Google Scholar]
- Chen, C.; Xu, P. Activation and Pharmacological Regulation of Inflammasomes. Biomolecules 2022, 12, 1005. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho Ribeiro, M.; Szabo, G. Role of the Inflammasome in Liver Disease. Annu. Rev. Pathol. 2022, 17, 345–365. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Gu, Y.; Li, H.; Liang, B.; Han, C.; Zhang, Y.; Liu, Q.; Wei, W.; Ma, Y. NLRP3 inflammasome activation mechanism and its role in autoimmune liver disease. Acta Biochim. Biophys. Sin. 2022, 54, 1577–1586. [Google Scholar] [CrossRef]
- Torres, S.; Segalés, P.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondria and the NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Cells 2022, 11, 1475. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Hong, W.; Lu, S.; Li, Y.; Guan, Y.; Weng, X.; Feng, Z. The NLRP3 Inflammasome in Non-Alcoholic Fatty Liver Disease and Steatohepatitis: Therapeutic Targets and Treatment. Front. Pharmacol. 2022, 13, 780496. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Huang, H.; Fu, X.; Chen, C.; Liu, H.; Wang, H.; Wu, D. The Role of Endoplasmic Reticulum Stress and NLRP3 Inflammasome in Liver Disorders. Int. J. Mol. Sci. 2022, 23, 3528. [Google Scholar] [CrossRef] [PubMed]
- Wallace, H.L.; Wang, L.; Gardner, C.L.; Corkum, C.P.; Grant, M.D.; Hirasawa, K.; Russell, R.S. Crosstalk Between Pyroptosis and Apoptosis in Hepatitis C Virus-induced Cell Death. Front. Immunol. 2022, 13, 788138. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, R.; Fusco, R.; Cordaro, M.; Siracusa, R.; Peritore, A.F.; Gugliandolo, E.; Crupi, R.; Scuto, M.; Cuzzocrea, S.; Di Paola, R.; et al. Modulation of NLRP3 Inflammasome through Formyl Peptide Receptor 1 (Fpr-1) Pathway as a New Therapeutic Target in Bronchiolitis Obliterans Syndrome. Int. J. Mol. Sci. 2020, 21, 2144. [Google Scholar] [CrossRef] [Green Version]
- Zuo, R.M.; Jiao, J.Y.; Chen, N.; Jiang, X.L.; Wu, Y.L.; Nan, J.X.; Lian, L.H. Carnosic acid suppressed the formation of NETs in alcoholic hepatosteatosis based on P2X7R-NLRP3 axis. Phytomed. Int. J. Phytother. Phytopharm. 2023, 110, 154599. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R. Cell Death in Development. Cold Spring Harb. Perspect. Biol. 2022, 14, a041095. [Google Scholar] [CrossRef]
- Peng, F.; Liao, M.; Qin, R.; Zhu, S.; Peng, C.; Fu, L.; Chen, Y.; Han, B. Regulated cell death (RCD) in cancer: Key pathways and targeted therapies. Signal Transduct. Target. Ther. 2022, 7, 286. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Bedoui, S.; Herold, M.J.; Strasser, A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat. Rev. Mol. Cell Biol. 2020, 21, 678–695. [Google Scholar] [CrossRef] [PubMed]
- Christgen, S.; Tweedell, R.E.; Kanneganti, T.D. Programming inflammatory cell death for therapy. Pharmacol. Ther. 2022, 232, 108010. [Google Scholar] [CrossRef]
- Liu, J.; Hong, M.; Li, Y.; Chen, D.; Wu, Y.; Hu, Y. Programmed Cell Death Tunes Tumor Immunity. Front. Immunol. 2022, 13, 847345. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 inflammasome activation and cell death. Cell. Mol. Immunol. 2021, 18, 2114–2127. [Google Scholar] [CrossRef]
- Zheng, M.; Williams, E.P.; Malireddi, R.K.S.; Karki, R.; Banoth, B.; Burton, A.; Webby, R.; Channappanavar, R.; Jonsson, C.B.; Kanneganti, T.D. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J. Biol. Chem. 2020, 295, 14040–14052. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Hornung, V. The NLRP3 Inflammasome Renders Cell Death Pro-inflammatory. J. Mol. Biol. 2018, 430, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Govaere, O.; Cockell, S.; Tiniakos, D.; Queen, R.; Younes, R.; Vacca, M.; Alexander, L.; Ravaioli, F.; Palmer, J.; Petta, S.; et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci. Transl. Med. 2020, 12, eaba4448. [Google Scholar] [CrossRef] [PubMed]
- Chapin, C.A.; Taylor, S.A.; Malladi, P.; Neighbors, K.; Melin-Aldana, H.; Kreiger, P.A.; Bowsher, N.; Schipma, M.J.; Loomes, K.M.; Behrens, E.M.; et al. Transcriptional Analysis of Liver Tissue Identifies Distinct Phenotypes of Indeterminate Pediatric Acute Liver Failure. Hepatol. Commun. 2021, 5, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.L.; Nakib, D.; Perciani, C.T.; MacParland, S.A. The immune niche of the liver. Clin. Sci. 2021, 135, 2445–2466. [Google Scholar] [CrossRef]
- Steensels, S.; Qiao, J.; Ersoy, B.A. Transcriptional Regulation in Non-Alcoholic Fatty Liver Disease. Metabolites 2020, 10, 283. [Google Scholar] [CrossRef] [PubMed]
- Schattenberg, J.M.; Galle, P.R.; Schuchmann, M. Apoptosis in liver disease. Liver Int. Off. J. Int. Assoc. Study Liver 2006, 26, 904–911. [Google Scholar] [CrossRef]
- Fleisher, T.A. Apoptosis. Ann. Allergy Asthma Immunol. 1997, 78, 245–249. [Google Scholar] [CrossRef]
- Hockenbery, D. Defining apoptosis. Am. J. Pathol. 1995, 146, 16–19. [Google Scholar]
- O’Reilly, L.A.; Strasser, A. Apoptosis and autoimmune disease. Inflamm. Res. 1999, 48, 5–21. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotter, T.G. Apoptosis and cancer: The genesis of a research field. Nat. Rev. Cancer 2009, 9, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Carter-Kent, C.; Feldstein, A.E. Apoptosis in nonalcoholic fatty liver disease: Diagnostic and therapeutic implications. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Delhalle, S.; Duvoix, A.; Schnekenburger, M.; Morceau, F.; Dicato, M.; Diederich, M. An introduction to the molecular mechanisms of apoptosis. Ann. NY Acad. Sci. 2003, 1010, 1–8. [Google Scholar] [CrossRef]
- Roberts, J.Z.; Crawford, N.; Longley, D.B. The role of Ubiquitination in Apoptosis and Necroptosis. Cell Death Differ. 2022, 29, 272–284. [Google Scholar] [CrossRef]
- Hague, A.; Paraskeva, C. Apoptosis and disease: A matter of cell fate. Cell Death Differ. 2004, 11, 1366–1372. [Google Scholar] [CrossRef]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K⁺ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Joza, N.; Susin, S.A.; Daugas, E.; Stanford, W.L.; Cho, S.K.; Li, C.Y.; Sasaki, T.; Elia, A.J.; Cheng, H.Y.; Ravagnan, L.; et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 2001, 410, 549–554. [Google Scholar] [CrossRef]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef]
- Khoury, M.K.; Gupta, K.; Franco, S.R.; Liu, B. Necroptosis in the Pathophysiology of Disease. Am. J. Pathol. 2020, 190, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhong, C.Q.; Zhang, D.W. Programmed necrosis: Backup to and competitor with apoptosis in the immune system. Nat. Immunol. 2011, 12, 1143–1149. [Google Scholar] [CrossRef]
- Vercammen, D.; Beyaert, R.; Denecker, G.; Goossens, V.; Van Loo, G.; Declercq, W.; Grooten, J.; Fiers, W.; Vandenabeele, P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 1998, 187, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Wang, X.; Zhou, Y.; Wang, X.; Yu, Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct. Target. Ther. 2022, 7, 196. [Google Scholar] [CrossRef]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflammation 2018, 15, 199. [Google Scholar] [CrossRef]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef]
- Speir, M.; Lawlor, K.E. RIP-roaring inflammation: RIPK1 and RIPK3 driven NLRP3 inflammasome activation and autoinflammatory disease. Semin. Cell Dev. Biol. 2021, 109, 114–124. [Google Scholar] [CrossRef]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Núñez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Fu, R.; Zhou, M.; Wang, S.; Huang, Y.; Hu, H.; Zhao, J.; Gaskin, F.; Yang, N.; Fu, S.M. Pathogenesis of lupus nephritis: RIP3 dependent necroptosis and NLRP3 inflammasome activation. J. Autoimmun. 2019, 103, 102286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wang, Y.; Inuzuka, H.; Wei, W. Necroptosis pathways in tumorigenesis. Semin. Cancer Biol. 2022, 86, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Kanneganti, T.D. The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis). Immunol. Rev. 2020, 297, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Malireddi, R.K.S.; Kesavardhana, S.; Kanneganti, T.D. ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis). Front. Cell. Infect. Microbiol. 2019, 9, 406. [Google Scholar] [CrossRef]
- Zychlinsky, A.; Prevost, M.C.; Sansonetti, P.J. Shigella flexneri induces apoptosis in infected macrophages. Nature 1992, 358, 167–169. [Google Scholar] [CrossRef]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Liu, T.; Zhou, Y.T.; Wang, L.Q.; Li, L.Y.; Bao, Q.; Tian, S.; Chen, M.X.; Chen, H.X.; Cui, J.; Li, C.W. NOD-like receptor family, pyrin domain containing 3 (NLRP3) contributes to inflammation, pyroptosis, and mucin production in human airway epithelium on rhinovirus infection. J. Allergy Clin. Immunol. 2019, 144, 777–787.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downs, K.P.; Nguyen, H.; Dorfleutner, A.; Stehlik, C. An overview of the non-canonical inflammasome. Mol. Asp. Med. 2020, 76, 100924. [Google Scholar] [CrossRef] [PubMed]
- Schnappauf, O.; Chae, J.J.; Kastner, D.L.; Aksentijevich, I. The Pyrin Inflammasome in Health and Disease. Front. Immunol. 2019, 10, 1745. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Liu, X.; Tang, Y.; Cui, Y.; Zhang, H.; Zhang, D. Autophagy is associated with cell fate in the process of macrophage-derived foam cells formation and progress. J. Biomed. Sci. 2016, 23, 57. [Google Scholar] [CrossRef]
- Kanayama, M.; Shinohara, M.L. Roles of Autophagy and Autophagy-Related Proteins in Antifungal Immunity. Front. Immunol. 2016, 7, 47. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef]
- Kovács, A.L.; Pálfia, Z.; Réz, G.; Vellai, T.; Kovács, J. Sequestration revisited: Integrating traditional electron microscopy, de novo assembly and new results. Autophagy 2007, 3, 655–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Liu, H.; Yang, Y.; Wang, H. The Role of Autophagy and Pyroptosis in Liver Disorders. Int. J. Mol. Sci. 2022, 23, 6208. [Google Scholar] [CrossRef] [PubMed]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay Between NLRP3 Inflammasome and Autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Bae, S.H.; Ryu, J.C.; Kwon, Y.; Oh, J.H.; Kwon, J.; Moon, J.S.; Kim, K.; Miyawaki, A.; Lee, M.G.; et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy 2016, 12, 1272–1291. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Jain, A.; Choi, S.W.; Mandell, M.A.; Johansen, T.; Deretic, V. TRIM-directed selective autophagy regulates immune activation. Autophagy 2017, 13, 989–990. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Han, X.; Sun, S.; Sun, Y.; Song, Q.; Zhu, J.; Song, N.; Chen, M.; Sun, T.; Xia, M.; Ding, J.; et al. Small molecule-driven NLRP3 inflammation inhibition via interplay between ubiquitination and autophagy: Implications for Parkinson disease. Autophagy 2019, 15, 1860–1881. [Google Scholar] [CrossRef]
- Spalinger, M.R.; Lang, S.; Gottier, C.; Dai, X.; Rawlings, D.J.; Chan, A.C.; Rogler, G.; Scharl, M. PTPN22 regulates NLRP3-mediated IL1B secretion in an autophagy-dependent manner. Autophagy 2017, 13, 1590–1601. [Google Scholar] [CrossRef]
- Allaeys, I.; Marceau, F.; Poubelle, P.E. NLRP3 promotes autophagy of urate crystals phagocytized by human osteoblasts. Arthritis Res. Ther. 2013, 15, R176. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.; Yao, H.; Shah, S.Z.A.; Wu, W.; Wang, D.; Zhao, Y.; Wang, L.; Zhou, X.; Zhao, D.; Yang, L. The NLRP3-Caspase 1 Inflammasome Negatively Regulates Autophagy via TLR4-TRIF in Prion Peptide-Infected Microglia. Front. Aging Neurosci. 2018, 10, 116. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519. [Google Scholar] [CrossRef] [PubMed]
- Bernal, W.; Wendon, J. Acute liver failure. N. Engl. J. Med. 2013, 369, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- McAtee, C. Drug-Induced Liver Injury. Crit. Care Nurs. Clin. N. Am. 2022, 34, 267–275. [Google Scholar] [CrossRef]
- Jaeschke, H.; Adelusi, O.B.; Akakpo, J.Y.; Nguyen, N.T.; Sanchez-Guerrero, G.; Umbaugh, D.S.; Ding, W.X.; Ramachandran, A. Recommendations for the use of the acetaminophen hepatotoxicity model for mechanistic studies and how to avoid common pitfalls. Acta Pharm. Sin. B 2021, 11, 3740–3755. [Google Scholar] [CrossRef]
- Luo, G.; Huang, L.; Zhang, Z. The molecular mechanisms of acetaminophen-induced hepatotoxicity and its potential therapeutic targets. Exp. Biol. Med. 2023, 15353702221147563. [Google Scholar] [CrossRef]
- Jaeschke, H. Acetaminophen: Dose-Dependent Drug Hepatotoxicity and Acute Liver Failure in Patients. Dig. Dis. 2015, 33, 464–471. [Google Scholar] [CrossRef]
- Bernal, W.; Williams, R. Acute Liver Failure. Clin. Liver Dis. 2020, 16, 45–55. [Google Scholar] [CrossRef]
- González-Recio, I.; Simón, J.; Goikoetxea-Usandizaga, N.; Serrano-Maciá, M.; Mercado-Gómez, M.; Rodríguez-Agudo, R.; Lachiondo-Ortega, S.; Gil-Pitarch, C.; Fernández-Rodríguez, C.; Castellana, D.; et al. Restoring cellular magnesium balance through Cyclin M4 protects against acetaminophen-induced liver damage. Nat. Commun. 2022, 13, 6816. [Google Scholar] [CrossRef]
- Sun, X.; Cui, Q.; Ni, J.; Liu, X.; Zhu, J.; Zhou, T.; Huang, H.; OuYang, K.; Wu, Y.; Yang, Z. Gut Microbiota Mediates the Therapeutic Effect of Monoclonal Anti-TLR4 Antibody on Acetaminophen-Induced Acute Liver Injury in Mice. Microbiol. Spectr. 2022, 10, e0064722. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Ramachandran, A.; Jaeschke, H. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol. 2016, 10, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Hua, S.; Deng, J.; Du, Z.; Zhang, D.; Liu, Z.; Khan, N.U.; Zhou, M.; Chen, Z. Astaxanthin Activated the Nrf2/HO-1 Pathway to Enhance Autophagy and Inhibit Ferroptosis, Ameliorating Acetaminophen-Induced Liver Injury. ACS Appl. Mater. Interfaces 2022, 14, 42887–42903. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Cui, Y.; Wang, J.; Liu, D.; Tian, Y.; Liu, K.; Wang, X.; Liu, L.; He, Y.; Pei, Y.; et al. Mesenchymal stem cells protect against acetaminophen hepatotoxicity by secreting regenerative cytokine hepatocyte growth factor. Stem Cell Res. Ther. 2022, 13, 94. [Google Scholar] [CrossRef]
- Yan, M.; Huo, Y.; Yin, S.; Hu, H. Mechanisms of acetaminophen-induced liver injury and its implications for therapeutic interventions. Redox Biol. 2018, 17, 274–283. [Google Scholar] [CrossRef]
- Larson, A.M.; Polson, J.; Fontana, R.J.; Davern, T.J.; Lalani, E.; Hynan, L.S.; Reisch, J.S.; Schiødt, F.V.; Ostapowicz, G.; Shakil, A.O.; et al. Acetaminophen-induced acute liver failure: Results of a United States multicenter, prospective study. Hepatology 2005, 42, 1364–1372. [Google Scholar] [CrossRef]
- Laine, J.E.; Auriola, S.; Pasanen, M.; Juvonen, R.O. Acetaminophen bioactivation by human cytochrome P450 enzymes and animal microsomes. Xenobiotica Fate Foreign Compd. Biol. Syst. 2009, 39, 11–21. [Google Scholar] [CrossRef]
- Mitchell, J.R.; Thorgeirsson, S.S.; Potter, W.Z.; Jollow, D.J.; Keiser, H. Acetaminophen-induced hepatic injury: Protective role of glutathione in man and rationale for therapy. Clin. Pharmacol. Ther. 1974, 16, 676–684. [Google Scholar] [CrossRef]
- Lee, W.M. Acetaminophen (APAP) hepatotoxicity-Isn’t it time for APAP to go away? J. Hepatol. 2017, 67, 1324–1331. [Google Scholar] [CrossRef]
- Jaeschke, H.; McGill, M.R.; Ramachandran, A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev. 2012, 44, 88–106. [Google Scholar] [CrossRef]
- McGill, M.R.; Jaeschke, H. Animal models of drug-induced liver injury. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wang, Q.; Sun, L.; Wu, M.; Li, S.; Hua, H.; Sun, Y.; Ni, T.; Zhou, C.; Huang, S.; et al. Acetaminophen-induced reduction of NIMA-related kinase 7 expression exacerbates acute liver injury. JHEP Rep. Innov. Hepatol. 2022, 4, 100545. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Duan, L.; Akakpo, J.Y.; Farhood, A.; Ramachandran, A. The role of apoptosis in acetaminophen hepatotoxicity. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2018, 118, 709–718. [Google Scholar] [CrossRef]
- Du, Y.C.; Lai, L.; Zhang, H.; Zhong, F.R.; Cheng, H.L.; Qian, B.L.; Tan, P.; Xia, X.M.; Fu, W.G. Kaempferol from Penthorum chinense Pursh suppresses HMGB1/TLR4/NF-κB signaling and NLRP3 inflammasome activation in acetaminophen-induced hepatotoxicity. Food Funct. 2020, 11, 7925–7934. [Google Scholar] [CrossRef] [PubMed]
- Elshal, M.; Abdelmageed, M.E. Diacerein counteracts acetaminophen-induced hepatotoxicity in mice via targeting NLRP3/caspase-1/IL-1β and IL-4/MCP-1 signaling pathways. Arch. Pharmacal Res. 2022, 45, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Wang, Z.; Sun, R.; Zou, B.; Li, R.; Liu, D.; Lin, M.; Zhou, J.; Ning, S.; et al. Peroxiredoxin 3 Inhibits Acetaminophen-Induced Liver Pyroptosis Through the Regulation of Mitochondrial ROS. Front. Immunol. 2021, 12, 652782. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, M.; Graffeo, C.S.; Rokosh, R.; Pansari, M.; Ochi, A.; Levie, E.M.; Van Heerden, E.; Tippens, D.M.; Greco, S.; Barilla, R.; et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis. 2015, 6, e1759. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shan, S.; Kang, K.; Zhang, C.; Kou, R.; Song, F. The cross-talk of NLRP3 inflammasome activation and necroptotic hepatocyte death in acetaminophen-induced mice acute liver injury. Hum. Exp. Toxicol. 2021, 40, 673–684. [Google Scholar] [CrossRef]
- Huang, J.; Xian, S.; Liu, Y.; Chen, X.; Pu, K.; Wang, H. A Renally Clearable Activatable Polymeric Nanoprobe for Early Detection of Hepatic Ischemia-Reperfusion Injury. Adv. Mater. 2022, 34, e2201357. [Google Scholar] [CrossRef]
- Tang, S.P.; Mao, X.L.; Chen, Y.H.; Yan, L.L.; Ye, L.P.; Li, S.W. Reactive Oxygen Species Induce Fatty Liver and Ischemia-Reperfusion Injury by Promoting Inflammation and Cell Death. Front. Immunol. 2022, 13, 870239. [Google Scholar] [CrossRef]
- Peng, Y.; Yin, Q.; Yuan, M.; Chen, L.; Shen, X.; Xie, W.; Liu, J. Role of hepatic stellate cells in liver ischemia-reperfusion injury. Front. Immunol. 2022, 13, 891868. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Cai, H.; Han, B.; Xia, Y.; Kong, X.; Gu, J. Natural Killer Cells in Hepatic Ischemia-Reperfusion Injury. Front. Immunol. 2022, 13, 870038. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; He, S.; Mao, X.; Zhang, Y.; Cai, Y.; Li, S. Effect of Hepatic Macrophage Polarization and Apoptosis on Liver Ischemia and Reperfusion Injury During Liver Transplantation. Front. Immunol. 2020, 11, 1193. [Google Scholar] [CrossRef]
- Zhai, Y.; Petrowsky, H.; Hong, J.C.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Ischaemia-reperfusion injury in liver transplantation--from bench to bedside. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Mendes-Braz, M.; Elias-Miró, M.; Jiménez-Castro, M.B.; Casillas-Ramírez, A.; Ramalho, F.S.; Peralta, C. The current state of knowledge of hepatic ischemia-reperfusion injury based on its study in experimental models. J. Biomed. Biotechnol. 2012, 2012, 298657. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Castro, M.B.; Cornide-Petronio, M.E.; Gracia-Sancho, J.; Peralta, C. Inflammasome-Mediated Inflammation in Liver Ischemia-Reperfusion Injury. Cells 2019, 8, 1131. [Google Scholar] [CrossRef] [PubMed]
- Sagulenko, V.; Thygesen, S.J.; Sester, D.P.; Idris, A.; Cridland, J.A.; Vajjhala, P.R.; Roberts, T.L.; Schroder, K.; Vince, J.E.; Hill, J.M.; et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013, 20, 1149–1160. [Google Scholar] [CrossRef]
- Inoue, Y.; Shirasuna, K.; Kimura, H.; Usui, F.; Kawashima, A.; Karasawa, T.; Tago, K.; Dezaki, K.; Nishimura, S.; Sagara, J.; et al. NLRP3 regulates neutrophil functions and contributes to hepatic ischemia-reperfusion injury independently of inflammasomes. J. Immunol. 2014, 192, 4342–4351. [Google Scholar] [CrossRef]
- Shu, G.; Qiu, Y.; Hao, J.; Fu, Q.; Deng, X. γ-Oryzanol alleviates acetaminophen-induced liver injury: Roles of modulating AMPK/GSK3β/Nrf2 and NF-κB signaling pathways. Food Funct. 2019, 10, 6858–6872. [Google Scholar] [CrossRef]
- Guo, X.X.; Zeng, Z.; Qian, Y.Z.; Qiu, J.; Wang, K.; Wang, Y.; Ji, B.P.; Zhou, F. Wheat Flour, Enriched with γ-Oryzanol, Phytosterol, and Ferulic Acid, Alleviates Lipid and Glucose Metabolism in High-Fat-Fructose-Fed Rats. Nutrients 2019, 11, 1697. [Google Scholar] [CrossRef]
- Du, Y.; Zhong, F.; Cheng, H.; Li, T.; Chen, Y.; Tan, P.; Huang, M.; Liang, T.; Liu, Y.; Xia, X.; et al. The Dietary Supplement γ-Oryzanol Attenuates Hepatic Ischemia Reperfusion Injury via Inhibiting Endoplasmic Reticulum Stress and HMGB1/NLRP3 Inflammasome. Oxidative Med. Cell. Longev. 2021, 2021, 4628050. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.R.; Verweij, M.; Brand, K.; van de Ven, M.; Goemaere, N.; van den Engel, S.; Chu, T.; Forrer, F.; Müller, C.; de Jong, M.; et al. Short-term dietary restriction and fasting precondition against ischemia reperfusion injury in mice. Aging Cell 2010, 9, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Rickenbacher, A.; Jang, J.H.; Limani, P.; Ungethüm, U.; Lehmann, K.; Oberkofler, C.E.; Weber, A.; Graf, R.; Humar, B.; Clavien, P.A. Fasting protects liver from ischemic injury through Sirt1-mediated downregulation of circulating HMGB1 in mice. J. Hepatol. 2014, 61, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, T.; Uchida, Y.; Kadono, K.; Hirao, H.; Kawasoe, J.; Watanabe, T.; Ueda, S.; Okajima, H.; Terajima, H.; Uemoto, S. Up-regulation of FOXO1 and reduced inflammation by β-hydroxybutyric acid are essential diet restriction benefits against liver injury. Proc. Natl. Acad. Sci. USA 2019, 116, 13533–13542. [Google Scholar] [CrossRef]
- El-Sisi, A.E.E.; Sokar, S.S.; Shebl, A.M.; Mohamed, D.Z.; Abu-Risha, S.E. Octreotide and melatonin alleviate inflammasome-induced pyroptosis through inhibition of TLR4-NF-κB-NLRP3 pathway in hepatic ischemia/reperfusion injury. Toxicol. Appl. Pharmacol. 2021, 410, 115340. [Google Scholar] [CrossRef]
- Cao, Q.; Luo, J.; Xiong, Y.; Liu, Z.; Ye, Q. 25-Hydroxycholesterol mitigates hepatic ischemia reperfusion injury via mediating mitophagy. Int. Immunopharmacol. 2021, 96, 107643. [Google Scholar] [CrossRef]
- Xue, R.; Qiu, J.; Wei, S.; Liu, M.; Wang, Q.; Wang, P.; Sha, B.; Wang, H.; Shi, Y.; Zhou, J.; et al. Lycopene alleviates hepatic ischemia reperfusion injury via the Nrf2/HO-1 pathway mediated NLRP3 inflammasome inhibition in Kupffer cells. Ann. Transl. Med. 2021, 9, 631. [Google Scholar] [CrossRef]
- Wang, Z.; Han, S.; Chen, X.; Li, X.; Xia, N.; Pu, L. Eva1a inhibits NLRP3 activation to reduce liver ischemia-reperfusion injury via inducing autophagy in kupffer cells. Mol. Immunol. 2021, 132, 82–92. [Google Scholar] [CrossRef]
- Zhang, T.; Huang, W.; Ma, Y. Down-regulation of TRPM2 attenuates hepatic ischemia/reperfusion injury through activation of autophagy and inhibition of NLRP3 inflammasome pathway. Int. Immunopharmacol. 2022, 104, 108443. [Google Scholar] [CrossRef]
- Yang, J.; Li, Y.; Wang, F.; Wu, C. Hepatoprotective effects of apple polyphenols on CCl4-induced acute liver damage in mice. J. Agric. Food Chem. 2010, 58, 6525–6531. [Google Scholar] [CrossRef]
- Kamel, R.; El Morsy, E.M. Hepatoprotective effect of methylsulfonylmethane against carbon tetrachloride-induced acute liver injury in rats. Arch. Pharmacal Res. 2013, 36, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wen, X.; Liu, J.; Kan, J.; Qian, C.; Wu, C.; Jin, C. Protective effect of an arabinogalactan from black soybean against carbon tetrachloride-induced acute liver injury in mice. Int. J. Biol. Macromol. 2018, 117, 659–664. [Google Scholar] [CrossRef]
- Chen, M.; Huang, W.; Wang, C.; Nie, H.; Li, G.; Sun, T.; Yang, F.; Zhang, Y.; Shu, K.; Wang, C.; et al. High-mobility group box 1 exacerbates CCl₄-induced acute liver injury in mice. Clin. Immunol. 2014, 153, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhou, J.; Xu, S.; Li, J.; Liu, J.; Lu, Y.; Shi, J.; Zhou, S.; Wu, Q. Induction of Nrf2 pathway by Dendrobium nobile Lindl. alkaloids protects against carbon tetrachloride induced acute liver injury. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 117, 109073. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kuang, G.; Wan, J.; Jiang, R.; Ma, L.; Gong, X.; Liu, X. Salidroside protects mice against CCl4-induced acute liver injury via down-regulating CYP2E1 expression and inhibiting NLRP3 inflammasome activation. Int. Immunopharmacol. 2020, 85, 106662. [Google Scholar] [CrossRef]
- Zhao, J.; He, B.; Zhang, S.; Huang, W.; Li, X. Ginsenoside Rg1 alleviates acute liver injury through the induction of autophagy and suppressing NF-κB/NLRP3 inflammasome signaling pathway. Int. J. Med. Sci. 2021, 18, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Prim. 2018, 4, 16. [Google Scholar] [CrossRef]
- Ge, Y.; Sun, H.; Xu, L.; Zhang, W.; Lv, J.; Chen, Y. The amelioration of alcohol-induced liver and intestinal barrier injury by Lactobacillus rhamnosus Gorbach-Goldin (LGG) is dependent on Interleukin 22 (IL-22) expression. Bioengineered 2022, 13, 12650–12660. [Google Scholar] [CrossRef]
- Hughes, E.; Hopkins, L.J.; Parker, R. Survival from alcoholic hepatitis has not improved over time. PLoS ONE 2018, 13, e0192393. [Google Scholar]
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol Res. Health J. Natl. Inst. Alcohol Abus. Alcohol. 2006, 29, 245–254. [Google Scholar]
- Yang, P.; Wang, Z.; Zhan, Y.; Wang, T.; Zhou, M.; Xia, L.; Yang, X.; Zhang, J. Endogenous A1 adenosine receptor protects mice from acute ethanol-induced hepatotoxicity. Toxicology 2013, 309, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Tian, L.; Chai, G.; Wen, B.; Wang, B. Targeting heme oxygenase-1 by quercetin ameliorates alcohol-induced acute liver injury via inhibiting NLRP3 inflammasome activation. Food Funct. 2018, 9, 4184–4193. [Google Scholar] [CrossRef] [PubMed]
- Dalekos, G.N.; Samakidou, A.; Lyberopoulou, A.; Banakou, E.; Gatselis, N.K. Recent advances in the diagnosis and management of autoimmune hepatitis. Pol. Arch. Intern. Med. 2022, 132, 16334. [Google Scholar] [CrossRef]
- Liberal, R.; de Boer, Y.S.; Heneghan, M.A. Established and novel therapeutic options for autoimmune hepatitis. Lancet Gastroenterol. Hepatol. 2021, 6, 315–326. [Google Scholar] [CrossRef]
- Tiegs, G.; Hentschel, J.; Wendel, A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J. Clin. Investig. 1992, 90, 196–203. [Google Scholar] [CrossRef]
- Lee, W.Y.; Salmi, M.; Kelly, M.M.; Jalkanen, S.; Kubes, P. Therapeutic advantage of anti-VAP-1 over anti-α4 integrin antibody in concanavalin a-induced hepatitis. Hepatology 2013, 58, 1413–1423. [Google Scholar] [CrossRef]
- Luan, J.; Zhang, X.; Wang, S.; Li, Y.; Fan, J.; Chen, W.; Zai, W.; Wang, S.; Wang, Y.; Chen, M.; et al. NOD-Like Receptor Protein 3 Inflammasome-Dependent IL-1β Accelerated ConA-Induced Hepatitis. Front. Immunol. 2018, 9, 758. [Google Scholar] [CrossRef]
- Aggarwal, M.; Saxena, R.; Sinclair, E.; Fu, Y.; Jacobs, A.; Dyba, M.; Wang, X.; Cruz, I.; Berry, D.; Kallakury, B.; et al. Reactivation of mutant p53 by a dietary-related compound phenethyl isothiocyanate inhibits tumor growth. Cell Death Differ. 2016, 23, 1615–1627. [Google Scholar] [CrossRef]
- Ioannides, C.; Konsue, N. A principal mechanism for the cancer chemopreventive activity of phenethyl isothiocyanate is modulation of carcinogen metabolism. Drug Metab. Rev. 2015, 47, 356–373. [Google Scholar] [CrossRef]
- Wang, J.; Shi, K.; An, N.; Li, S.; Bai, M.; Wu, X.; Shen, Y.; Du, R.; Cheng, J.; Wu, X.; et al. Direct Inhibition of GSDMD by PEITC Reduces Hepatocyte Pyroptosis and Alleviates Acute Liver Injury in Mice. Front. Immunol. 2022, 13, 825428. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.L.; Ni, S.T.; Luo, S.Q.; Hu, B.; Xu, R.; Liu, S.Y.; Huang, X.D.; Zeng, B.; Liang, Q.Q.; Chen, S.Y.; et al. Dimethyl fumarate ameliorates autoimmune hepatitis in mice by blocking NLRP3 inflammasome activation. Int. Immunopharmacol. 2022, 108, 108867. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Cho, H.I.; Kim, S.J.; Park, J.H.; Kim, J.S.; Kim, Y.H.; Lee, S.K.; Kwak, J.H.; Lee, S.M. Protective effect of linarin against D-galactosamine and lipopolysaccharide-induced fulminant hepatic failure. Eur. J. Pharmacol. 2014, 738, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.; Pervin, M.; Kuramochi, M.; Karim, M.R.; Izawa, T.; Kuwamura, M.; Yamate, J. M1/M2-macrophage Polarization-based Hepatotoxicity in d-galactosamine-induced Acute Liver Injury in Rats. Toxicol. Pathol. 2018, 46, 764–776. [Google Scholar] [CrossRef]
- Gehrke, N.; Wörns, M.A.; Mann, A.; Hövelmeyer, N.; Waisman, A.; Straub, B.K.; Galle, P.R.; Schattenberg, J.M. Hepatocyte Bcl-3 protects from death-receptor mediated apoptosis and subsequent acute liver failure. Cell Death Dis. 2022, 13, 510. [Google Scholar] [CrossRef]
- Tao, Y.C.; Wang, Y.H.; Wang, M.L.; Jiang, W.; Wu, D.B.; Chen, E.Q.; Tang, H. Upregulation of microRNA-125b-5p alleviates acute liver failure by regulating the Keap1/Nrf2/HO-1 pathway. Front. Immunol. 2022, 13, 988668. [Google Scholar] [CrossRef]
- Gong, X.; Yang, Y.; Huang, L.; Zhang, Q.; Wan, R.Z.; Zhang, P.; Zhang, B. Antioxidation, anti-inflammation and anti-apoptosis by paeonol in LPS/d-GalN-induced acute liver failure in mice. Int. Immunopharmacol. 2017, 46, 124–132. [Google Scholar] [CrossRef]
- Huang, S.; Wang, Y.; Xie, S.; Lai, Y.; Mo, C.; Zeng, T.; Kuang, S.; Deng, G.; Zhou, C.; Chen, Y.; et al. Hepatic TGFβr1 Deficiency Attenuates Lipopolysaccharide/D-Galactosamine-Induced Acute Liver Failure Through Inhibiting GSK3β-Nrf2-Mediated Hepatocyte Apoptosis and Ferroptosis. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 1649–1672. [Google Scholar] [CrossRef]
- Lv, H.; Yang, H.; Wang, Z.; Feng, H.; Deng, X.; Cheng, G.; Ci, X. Nrf2 signaling and autophagy are complementary in protecting lipopolysaccharide/d-galactosamine-induced acute liver injury by licochalcone A. Cell Death Dis. 2019, 10, 313. [Google Scholar] [CrossRef]
- Lv, H.; Fan, X.; Wang, L.; Feng, H.; Ci, X. Daphnetin alleviates lipopolysaccharide/d-galactosamine-induced acute liver failure via the inhibition of NLRP3, MAPK and NF-κB, and the induction of autophagy. Int. J. Biol. Macromol. 2018, 119, 240–248. [Google Scholar] [CrossRef]
- Liu, X.; Wang, T.; Liu, X.; Cai, L.; Qi, J.; Zhang, P.; Li, Y. Biochanin A protects lipopolysaccharide/D-galactosamine-induced acute liver injury in mice by activating the Nrf2 pathway and inhibiting NLRP3 inflammasome activation. Int. Immunopharmacol. 2016, 38, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.W.; Pan, Z.Z.; Hu, J.J.; Chen, W.L.; Zhou, G.Y.; Lin, W.; Jin, L.X.; Xu, C.L. Mangiferin alleviates lipopolysaccharide and D-galactosamine-induced acute liver injury by activating the Nrf2 pathway and inhibiting NLRP3 inflammasome activation. Eur. J. Pharmacol. 2016, 770, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Huang, J.; Wang, K.; Yu, Q.; Zhu, C.; Ren, H. Pterostilbene Protects Against Lipopolysaccharide/D-Galactosamine-Induced Acute Liver Failure by Upregulating the Nrf2 Pathway and Inhibiting NF-κB, MAPK, and NLRP3 Inflammasome Activation. J. Med. Food 2020, 23, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Tao, K.; Zhang, P.; Chen, X.; Sun, X.; Li, R. Maresin 1 protects against lipopolysaccharide/d-galactosamine-induced acute liver injury by inhibiting macrophage pyroptosis and inflammatory response. Biochem. Pharmacol. 2022, 195, 114863. [Google Scholar] [CrossRef]
- Gehrke, N.; Hövelmeyer, N.; Waisman, A.; Straub, B.K.; Weinmann-Menke, J.; Wörns, M.A.; Galle, P.R.; Schattenberg, J.M. Hepatocyte-specific deletion of IL1-RI attenuates liver injury by blocking IL-1 driven autoinflammation. J. Hepatol. 2018, 68, 986–995. [Google Scholar] [CrossRef]
- Bai, L.; Kong, M.; Duan, Z.; Liu, S.; Zheng, S.; Chen, Y. M2-like macrophages exert hepatoprotection in acute-on-chronic liver failure through inhibiting necroptosis-S100A9-necroinflammation axis. Cell Death Dis. 2021, 12, 93. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| The Type of Acute Liver Injury | Animal | Experimental Model | The Role of NLRP3 Inflammasome and PCD | Reference |
|---|---|---|---|---|
| Acetaminophen-induced acute liver injury | C57BL/6 mice | Intraperitoneal injection of APAP (300 mg/kg) | Kaempferol inhibits HMGB1/TLR4/NF-κB signaling pathway and activation of the NLRP3 inflammasome, thereby alleviating apoptosis | [125] |
| BALB/c mice | Intraperitoneal injection of APAP (400 mg/kg) | Diacetoreine downregulates NLRP3/caspase-1/IL-1β, IL-4/MCP-1, and TNF-α/ NF-κB proinflammatory signal pathway mediated oxidative stress, mitochondrial dysfunction, and apoptosis. | [126] | |
| C57BL/6 mice | Oral APAP (300 mg/kg) | Peroxiredoxin 3 inhibits NLRP3 inflammasome activation and prevents APAP-induced pyroptosis, | [127] | |
| C57BL/6 mice | Intraperitoneal injection of APAP (300 mg/kg) | Neurostatin-1, an inhibitor of RIPK1, alleviates APAP-induced liver injury by inhibiting the interaction between necroptosis and the NLRP3 inflammasome | [129] | |
| Liver ischemia-reperfusion injury | C57BL/6 mice | 70% liver ischemia-reperfusion model (1 h ischemia, 6 h reperfusion) | γ-Oryzanol protects the liver from I/R-induced inflammasome activation and apoptosis by inhibiting HMGB1/NLRP3. | [142] |
| SD rats | 70% liver ischemia-reperfusion model (30 min ischemia, 24 h reperfusion) | Octreotide plays a protective role in LIRI by disrupting TLR4-mediated NLRP3 inflammasome activation and pyroptosis | [146] | |
| SD rats | 70% liver ischemia-reperfusion model (1 h ischemia, 3/24 h reperfusion) | 25-hydroxycholesterol exerts protective effects by upregulating mitophagy and inhibiting NLRP3 inflammasome activation | [147] | |
| CCl4-induced acute liver injury | C57BL/6 mice | Intraperitoneal injection of 10% CCl4 (500 μL/kg, diluted with olive oil) | Salidroside exerts protective effects against CCl4-induced ALI by reducing hepatocyte apoptosis and inhibiting the activation of NLRP3 inflammasome | [156] |
| C57BL/6 mice | Intraperitoneal injection of 50% CCl4 (2 mL/kg, diluted with olive oil) | Ginsenoside Rg1 ameliorated acute liver injury via autophagy and may be associated with NF- κB/NLRP3 inflammasome signaling pathway | [157] | |
| Alcohol-induced acute liver injury | Wistar rats | 50% (v/v) ethanol by gavage (5 g/kg, three times) | Quercetin can induce the expression of HO-1 and then downregulate the activation of NLRP3 inflammasome | [164] |
| Con A-induced autoimmune hepatitis | ICR mice | Con A (10 mg/kg) was injected via tail vein | Phenethyl isothiocyanate can reduce NLRP3 production in the liver and directly interact with the cysteine at position 191 of GSDMD to inhibit hepatocyte pyroptosis | [172] |
| LPS/D-GalN-induced acute liver injury | C57BL/6 mice with Nrf2−/− and wild-type | Intraperitoneal injection of LPS/GalN (30 μg/kg and 600 mg/kg) | Licochalcone A attenuates liver injury by inhibiting the activation of NLRP3 inflammasome via inducing autophagy | [180] |
| C57BL/6 mice | Intraperitoneal injection of LPS/GalN (30 μg/kg and 600 mg/kg) | Maresin 1 inhibits mitogen-activated protein kinase/NF-κB signaling pathway and NLRP3 inflammasome-induced apoptosis | [185] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, C.; Chen, P.; Miao, L.; Di, G. The Role of the NLRP3 Inflammasome and Programmed Cell Death in Acute Liver Injury. Int. J. Mol. Sci. 2023, 24, 3067. https://doi.org/10.3390/ijms24043067
Yu C, Chen P, Miao L, Di G. The Role of the NLRP3 Inflammasome and Programmed Cell Death in Acute Liver Injury. International Journal of Molecular Sciences. 2023; 24(4):3067. https://doi.org/10.3390/ijms24043067
Chicago/Turabian StyleYu, Chaoqun, Peng Chen, Longyu Miao, and Guohu Di. 2023. "The Role of the NLRP3 Inflammasome and Programmed Cell Death in Acute Liver Injury" International Journal of Molecular Sciences 24, no. 4: 3067. https://doi.org/10.3390/ijms24043067
APA StyleYu, C., Chen, P., Miao, L., & Di, G. (2023). The Role of the NLRP3 Inflammasome and Programmed Cell Death in Acute Liver Injury. International Journal of Molecular Sciences, 24(4), 3067. https://doi.org/10.3390/ijms24043067