

Untargeted Metabolomics Identifies Potential Hypertrophic Cardiomyopathy Biomarkers in Carriers of MYBPC3 Founder Variants

 , , , , ,
, , , , ,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
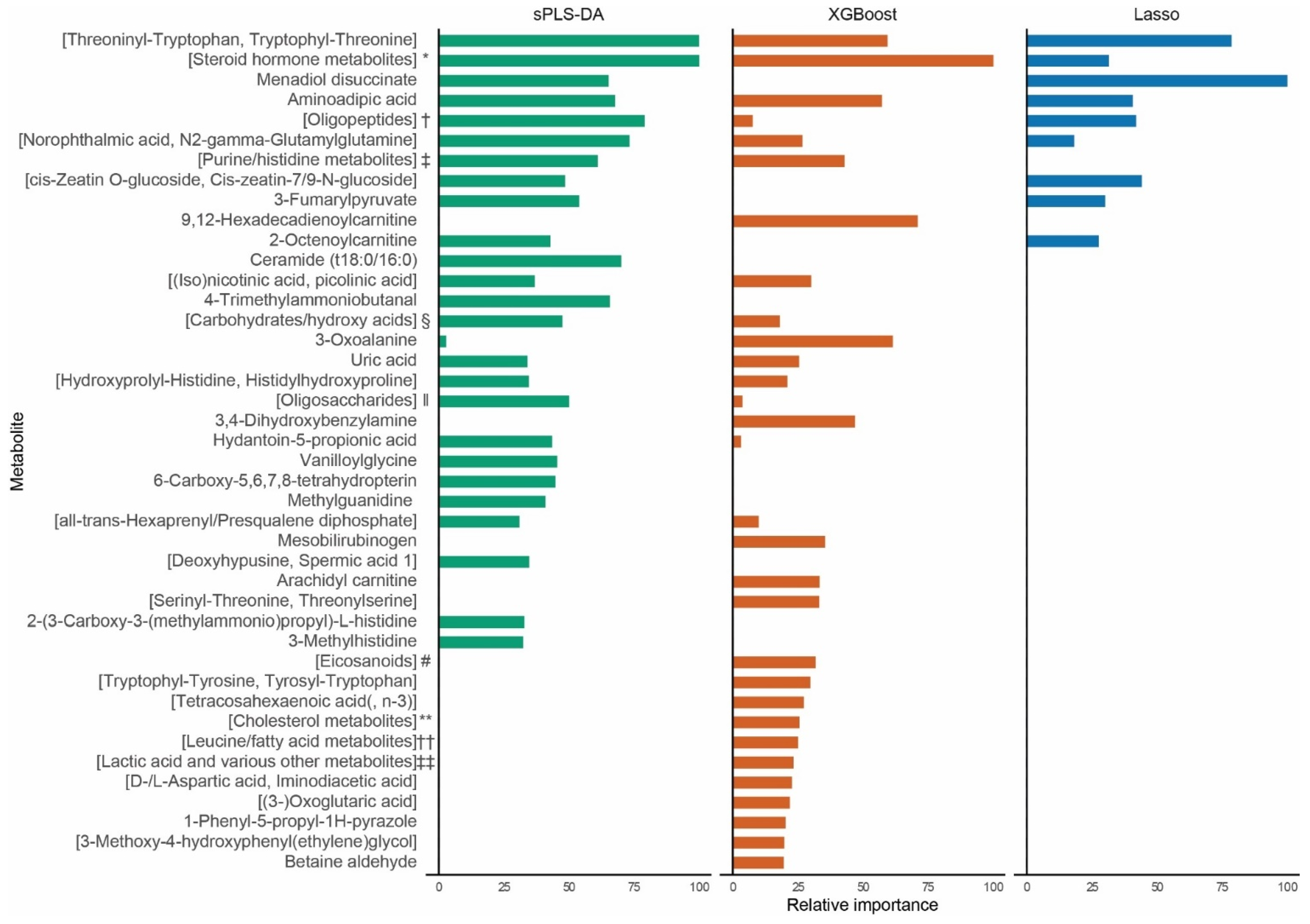
2.1. Biomarker Identification
2.2. Sensitivity Analyses and External Validation
3. Discussion
3.1. Energy Metabolism
3.2. Other Pathways
3.3. Previous Metabolomics Studies
3.4. Limitations and Future Directions
4. Materials and Methods
4.1. Subject Inclusion
4.2. Metabolomics
4.3. Biomarker Identification
4.4. Statistical Analysis
4.5. Sensitivity Analyses and External Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Charron, P.; Carrier, L.; Ledeuil, C.; Cheav, T.; Pichereau, C.; Benaiche, A.; Isnard, R.; Dubourg, O.; Burban, M.; et al. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003, 107, 2227–2232. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.W.; Li, A.; Lal, S.; Bos, J.M.; Harris, S.P.; van der Velden, J.; Ackerman, M.J.; Cooke, R.; Dos Remedios, C.G. MYBPC3 mutations are associated with a reduced super-relaxed state in patients with hypertrophic cardiomyopathy. PLoS ONE 2017, 12, e0180064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christiaans, I.; Nannenberg, E.A.; Dooijes, D.; Jongbloed, R.J.; Michels, M.; Postema, P.G.; Majoor-Krakauer, D.; van den Wijngaard, A.; Mannens, M.M.; van Tintelen, J.P.; et al. Founder mutations in hypertrophic cardiomyopathy patients in the Netherlands. Neth. Heart J. 2010, 18, 248–254. [Google Scholar] [CrossRef]
- van Dijk, S.J.; Dooijes, D.; dos Remedios, C.; Michels, M.; Lamers, J.M.; Winegrad, S.; Schlossarek, S.; Carrier, L.; ten Cate, F.J.; Stienen, G.J.; et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: Haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 2009, 119, 1473–1483. [Google Scholar] [CrossRef] [Green Version]
- Nannenberg, E.A.; Michels, M.; Christiaans, I.; Majoor-Krakauer, D.; Hoedemaekers, Y.M.; van Tintelen, J.P.; Lombardi, M.P.; ten Cate, F.J.; Schinkel, A.F.; Tijssen, J.G.; et al. Mortality risk of untreated myosin-binding protein C-related hypertrophic cardiomyopathy: Insight into the natural history. J. Am. Coll. Cardiol. 2011, 58, 2406–2414. [Google Scholar] [CrossRef] [Green Version]
- Christiaans, I.; Birnie, E.; van Langen, I.M.; van Spaendonck-Zwarts, K.Y.; van Tintelen, J.P.; van den Berg, M.P.; Atsma, D.E.; Helderman-van den Enden, A.T.; Pinto, Y.M.; Hermans-van Ast, J.F.; et al. The yield of risk stratification for sudden cardiac death in hypertrophic cardiomyopathy myosin-binding protein C gene mutation carriers: Focus on predictive screening. Eur. Heart J. 2010, 31, 842–848. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.J.; Rowin, E.J.; Casey, S.A.; Link, M.S.; Lesser, J.R.; Chan, R.H.; Garberich, R.F.; Udelson, J.E.; Maron, M.S. Hypertrophic Cardiomyopathy in Adulthood Associated with Low Cardiovascular Mortality With Contemporary Management Strategies. J. Am. Coll. Cardiol. 2015, 65, 1915–1928. [Google Scholar] [CrossRef] [Green Version]
- Güçlü, A.; Knaapen, P.; Harms, H.J.; Parbhudayal, R.Y.; Michels, M.; Lammertsma, A.A.; van Rossum, A.C.; Germans, T.; van der Velden, J. Disease Stage-Dependent Changes in Cardiac Contractile Performance and Oxygen Utilization Underlie Reduced Myocardial Efficiency in Human Inherited Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Imaging 2017, 10, e005604. [Google Scholar] [CrossRef] [Green Version]
- Coats, C.J.; Heywood, W.E.; Virasami, A.; Ashrafi, N.; Syrris, P.; Dos Remedios, C.; Treibel, T.A.; Moon, J.C.; Lopes, L.R.; McGregor, C.G.A.; et al. Proteomic Analysis of the Myocardium in Hypertrophic Obstructive Cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e001974. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.; Schuldt, M.; Nagyova, E.; Gu, Z.; El Bouhaddani, S.; Yiangou, L.; Jansen, M.; Calis, J.J.A.; Dorsch, L.M.; Blok, C.S.; et al. Multi-omics integration identifies key upstream regulators of pathomechanisms in hypertrophic cardiomyopathy due to truncating MYBPC3 mutations. Clin. Epigenet. 2021, 13, 61. [Google Scholar] [CrossRef] [PubMed]
- Ranjbarvaziri, S.; Kooiker, K.B.; Ellenberger, M.; Fajardo, G.; Zhao, M.; Roest, A.S.V.; Woldeyes, R.A.; Koyano, T.T.; Fong, R.; Ma, N.; et al. Altered Cardiac Energetics and Mitochondrial Dysfunction in Hypertrophic Cardiomyopathy. Circulation 2021, 144, 1714–1731. [Google Scholar] [CrossRef] [PubMed]
- Ussher, J.R.; Elmariah, S.; Gerszten, R.E.; Dyck, J.R. The Emerging Role of Metabolomics in the Diagnosis and Prognosis of Cardiovascular Disease. J. Am. Coll. Cardiol. 2016, 68, 2850–2870. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, C.; Jichi, F.; Pavlou, M.; Monserrat, L.; Anastasakis, A.; Rapezzi, C.; Biagini, E.; Gimeno, J.R.; Limongelli, G.; McKenna, W.J.; et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur. Heart J. 2014, 35, 2010–2020. [Google Scholar] [CrossRef]
- van der Velden, J.; Tocchetti, C.G.; Varricchi, G.; Bianco, A.; Sequeira, V.; Hilfiker-Kleiner, D.; Hamdani, N.; Leite-Moreira, A.F.; Mayr, M.; Falcao-Pires, I.; et al. Metabolic changes in hypertrophic cardiomyopathies: Scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 1273–1280. [Google Scholar] [CrossRef]
- Ruiz, M.; Labarthe, F.; Fortier, A.; Bouchard, B.; Legault, J.T.; Bolduc, V.; Rigal, O.; Chen, J.; Ducharme, A.; Crawford, P.A.; et al. Circulating acylcarnitine profile in human heart failure: A surrogate of fatty acid metabolic dysregulation in mitochondria and beyond. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H768–H781. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Sugihara, H.; Kinoshita, N.; Ito, K.; Adachi, Y.; Hirasaki, S.; Matsuo, A.; Azuma, A.; Kodo, N.; Nakagawa, M. Serum carnitine concentrations in patients with idiopathic hypertrophic cardiomyopathy: Relationship with impaired myocardial fatty acid metabolism. Clin. Sci. 1999, 97, 493–501. [Google Scholar] [CrossRef]
- Verdonschot, J.A.J.; Wang, P.; Van Bilsen, M.; Hazebroek, M.R.; Merken, J.J.; Vanhoutte, E.K.; Henkens, M.; Van Den Wijngaard, A.; Glatz, J.F.C.; Krapels, I.P.C.; et al. Metabolic Profiling Associates with Disease Severity in Nonischemic Dilated Cardiomyopathy. J. Card. Fail. 2020, 26, 212–222. [Google Scholar] [CrossRef] [Green Version]
- Leslie, N.D.; Valencia, C.A.; Strauss, A.W.; Zhang, K. Very Long-Chain Acyl-Coenzyme a Dehydrogenase Deficiency. In GeneReviews(®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Andersson, C.; Liu, C.; Cheng, S.; Wang, T.J.; Gerszten, R.E.; Larson, M.G.; Vasan, R.S. Metabolomic signatures of cardiac remodelling and heart failure risk in the community. ESC Heart Fail. 2020, 7, 3707–3715. [Google Scholar] [CrossRef]
- Wang, T.J.; Ngo, D.; Psychogios, N.; Dejam, A.; Larson, M.G.; Vasan, R.S.; Ghorbani, A.; O’Sullivan, J.; Cheng, S.; Rhee, E.P.; et al. 2-Aminoadipic acid is a biomarker for diabetes risk. J. Clin. Investig. 2013, 123, 4309–4317. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Wang, J.; Wang, Y.; Jia, L.; Sun, K.; Wang, H.; Zou, Y.; Tian, T.; Liu, Y.; Zou, J.; et al. Plasma Uric Acid as a Prognostic Marker in Patients with Hypertrophic Cardiomyopathy. Can. J. Cardiol. 2015, 31, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xu, Y.; Liao, H.; Chen, X.; He, S. U-shaped association between serum uric acid concentration and mortality in hypertrophic cardiomyopathy patients. Upsala J. Med. Sci. 2020, 125, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Cheng, J.D. Uric Acid and Cardiovascular Disease: An Update from Molecular Mechanism to Clinical Perspective. Front. Pharmacol. 2020, 11, 582680. [Google Scholar] [CrossRef] [PubMed]
- Kouzu, H.; Katano, S.; Yano, T.; Ohori, K.; Nagaoka, R.; Inoue, T.; Takamura, Y.; Ishigo, T.; Watanabe, A.; Koyama, M.; et al. Plasma amino acid profiling improves predictive accuracy of adverse events in patients with heart failure. ESC Heart Fail. 2021, 8, 5045–5056. [Google Scholar] [CrossRef]
- Hwang, J.J.; Allen, P.D.; Tseng, G.C.; Lam, C.W.; Fananapazir, L.; Dzau, V.J.; Liew, C.C. Microarray gene expression profiles in dilated and hypertrophic cardiomyopathic end-stage heart failure. Physiol. Genom. 2002, 10, 31–44. [Google Scholar] [CrossRef]
- Leary, P.J.; Tedford, R.J.; Bluemke, D.A.; Bristow, M.R.; Heckbert, S.R.; Kawut, S.M.; Krieger, E.V.; Lima, J.A.; Masri, C.S.; Ralph, D.D.; et al. Histamine H2 Receptor Antagonists, Left Ventricular Morphology, and Heart Failure Risk: The MESA Study. J. Am. Coll. Cardiol. 2016, 67, 1544–1552. [Google Scholar] [CrossRef]
- Kim, J.; Ogai, A.; Nakatani, S.; Hashimura, K.; Kanzaki, H.; Komamura, K.; Asakura, M.; Asanuma, H.; Kitamura, S.; Tomoike, H.; et al. Impact of blockade of histamine H2 receptors on chronic heart failure revealed by retrospective and prospective randomized studies. J. Am. Coll. Cardiol. 2006, 48, 1378–1384. [Google Scholar] [CrossRef] [Green Version]
- Koh, A.; Molinaro, A.; Ståhlman, M.; Khan, M.T.; Schmidt, C.; Mannerås-Holm, L.; Wu, H.; Carreras, A.; Jeong, H.; Olofsson, L.E.; et al. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through mTORC1. Cell 2018, 175, 947–961.e917. [Google Scholar] [CrossRef] [Green Version]
- van Driel, B.; Nijenkamp, L.; Huurman, R.; Michels, M.; van der Velden, J. Sex differences in hypertrophic cardiomyopathy: New insights. Curr. Opin. Cardiol. 2019, 34, 254–259. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Z.; Hu, F.; Yang, W.; Yuan, J.; Cui, J.; Hao, S.; Hu, J.; Zhou, Y.; Qiao, S. 17β-estradiol prevents cardiac diastolic dysfunction by stimulating mitochondrial function: A preclinical study in a mouse model of a human hypertrophic cardiomyopathy mutation. J. Steroid Biochem. Mol. Biol. 2015, 147, 92–102. [Google Scholar] [CrossRef]
- Dorsch, L.M.; Schuldt, M.; dos Remedios, C.G.; Schinkel, A.F.L.; de Jong, P.L.; Michels, M.; Kuster, D.W.D.; Brundel, B.; van der Velden, J. Protein Quality Control Activation and Microtubule Remodeling in Hypertrophic Cardiomyopathy. Cells 2019, 8, 741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlossarek, S.; Englmann, D.R.; Sultan, K.R.; Sauer, M.; Eschenhagen, T.; Carrier, L. Defective proteolytic systems in Mybpc3-targeted mice with cardiac hypertrophy. Basic Res. Cardiol. 2012, 107, 235. [Google Scholar] [CrossRef] [PubMed]
- Jorgenrud, B.; Jalanko, M.; Helio, T.; Jaaskelainen, P.; Laine, M.; Hilvo, M.; Nieminen, M.S.; Laakso, M.; Hyotylainen, T.; Oresic, M.; et al. The Metabolome in Finnish Carriers of the MYBPC3-Q1061X Mutation for Hypertrophic Cardiomyopathy. PLoS ONE 2015, 10, e0134184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, Y.J.; Batra, J.; Kochav, S.M.; Patel, P.; Jung, J.; Maurer, M.S.; Hasegawa, K.; Reilly, M.P.; Fifer, M.A. Difference in Metabolomic Response to Exercise between Patients with and without Hypertrophic Cardiomyopathy. J. Cardiovasc. Transl. Res. 2020, 14, 246–255. [Google Scholar] [CrossRef]
- Ravi, R.; Silva, L.F.; Vangipurapu, J.; Maria, M.; Raivo, J.; Helisalmi, S.; Laakso, M. Metabolite Signature in the Carriers of Pathogenic Genetic Variants for Cardiomyopathy: A Population-Based METSIM Study. Metabolites 2022, 12, 437. [Google Scholar] [CrossRef]
- Schuldt, M.; van Driel, B.; Algül, S.; Parbhudayal, R.Y.; Barge-Schaapveld, D.Q.C.M.; Güçlü, A.; Jansen, M.; Michels, M.; Baas, A.F.; van de Wiel, M.A.; et al. Distinct Metabolomic Signatures in Preclinical and Obstructive Hypertrophic Cardiomyopathy. Cells 2021, 10, 2950. [Google Scholar] [CrossRef]
- Deidda, M.; Noto, A.; Pasqualucci, D.; Fattuoni, C.; Barberini, L.; Piras, C.; Bassareo, P.P.; Porcu, M.; Mercuro, G.; Dessalvi, C.C. The Echocardiographic Parameters of Systolic Function Are Associated with Specific Metabolomic Fingerprints in Obstructive and Non-Obstructive Hypertrophic Cardiomyopathy. Metabolites 2021, 11, 787. [Google Scholar] [CrossRef]
- Du, W.; Piek, A.; Schouten, E.M.; van de Kolk, C.W.A.; Mueller, C.; Mebazaa, A.; Voors, A.A.; de Boer, R.A.; Silljé, H.H.W. Plasma levels of heart failure biomarkers are primarily a reflection of extracardiac production. Theranostics 2018, 8, 4155–4169. [Google Scholar] [CrossRef]
- Vittorini, S.; Clerico, A. Cardiovascular biomarkers: Increasing impact of laboratory medicine in cardiology practice. Clin. Chem. Lab. Med. 2008, 46, 748–763. [Google Scholar] [CrossRef]
- Jansen, M.; Christiaans, I.; van der Crabben, S.N.; Michels, M.; Huurman, R.; Hoedemaekers, Y.M.; Dooijes, D.; Jongbloed, J.D.H.; Boven, L.G.; Deprez, R.H.L.; et al. BIO FOr CARE: Biomarkers of hypertrophic cardiomyopathy development and progression in carriers of Dutch founder truncating MYBPC3 variants-design and status. Neth. Heart J. 2021, 29, 318–329. [Google Scholar] [CrossRef]
- Haijes, H.A.; Willemsen, M.; Van der Ham, M.; Gerrits, J.; Pras-Raves, M.L.; Prinsen, H.; Van Hasselt, P.M.; De Sain-van der Velden, M.G.M.; Verhoeven-Duif, N.M.; Jans, J.J.M. Direct Infusion Based Metabolomics Identifies Metabolic Disease in Patients’ Dried Blood Spots and Plasma. Metabolites 2019, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.-A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA; 2016; pp. 785–794. [Google Scholar]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 26. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.H.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 22. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kolde, R. pheatmap: Pretty Heatmaps, Version 1.0.12. 2019. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 14 February 2023).



{kind=link}
{kind=link}
| Severe Phenotype | No/Mild Phenotype | Genotype-Negative | p-Value | |
|---|---|---|---|---|
| (n = 30) | (n = 30) | (n = 10) | ||
| Age (years) | 56.3 (38.2–71.7) | 58.4 (38.4–68.4) | 55.5 (41.5–64.5) | 0.835 |
| Male sex | 17 (56.7) | 17 (56.7) | 6 (60.0) | 1.00 |
| Index patient | 18 (60.0) | 5 (16.7) | 0.001 | |
| Body surface area (m2) | 2.0 (1.8–2.1) | 1.9 (1.8–2.1) | 0.437 | |
| Syncope | 8 (27.6) | 2 (6.7) | 0.042 | |
| Family history of SCD | 10 (37.0) | 7 (25.9) | 0.559 | |
| Non-sustained VT | 16 (61.5) | 8 (42.1) | 0.237 | |
| Maximum wall thickness (mm) | 21 (16–23) | 11 (9–13) | <0.001 | |
| Left atrial diameter (mm) | 43 (40–50) | 37 (35–42) | 0.062 | |
| LVOT gradient (mmHg) | 4 (3–80) | 5 (4–6) | 0.721 | |
| LV ejection fraction (%) | 58 (50–60) | 60 (58–65) | 0.010 | |
| Atrial fibrillation | 12 (41.4) | 2 (6.7) | 0.002 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jansen, M.; Schuldt, M.; van Driel, B.O.; Schmidt, A.F.; Christiaans, I.; van der Crabben, S.N.; Hoedemaekers, Y.M.; Dooijes, D.; Jongbloed, J.D.H.; Boven, L.G.; et al. Untargeted Metabolomics Identifies Potential Hypertrophic Cardiomyopathy Biomarkers in Carriers of MYBPC3 Founder Variants. Int. J. Mol. Sci. 2023, 24, 4031. https://doi.org/10.3390/ijms24044031
Jansen M, Schuldt M, van Driel BO, Schmidt AF, Christiaans I, van der Crabben SN, Hoedemaekers YM, Dooijes D, Jongbloed JDH, Boven LG, et al. Untargeted Metabolomics Identifies Potential Hypertrophic Cardiomyopathy Biomarkers in Carriers of MYBPC3 Founder Variants. International Journal of Molecular Sciences. 2023; 24(4):4031. https://doi.org/10.3390/ijms24044031
Chicago/Turabian StyleJansen, Mark, Maike Schuldt, Beau O. van Driel, Amand F. Schmidt, Imke Christiaans, Saskia N. van der Crabben, Yvonne M. Hoedemaekers, Dennis Dooijes, Jan D. H. Jongbloed, Ludolf G. Boven, and et al. 2023. "Untargeted Metabolomics Identifies Potential Hypertrophic Cardiomyopathy Biomarkers in Carriers of MYBPC3 Founder Variants" International Journal of Molecular Sciences 24, no. 4: 4031. https://doi.org/10.3390/ijms24044031
APA StyleJansen, M., Schuldt, M., van Driel, B. O., Schmidt, A. F., Christiaans, I., van der Crabben, S. N., Hoedemaekers, Y. M., Dooijes, D., Jongbloed, J. D. H., Boven, L. G., Deprez, R. H. L., Wilde, A. A. M., Jans, J. J. M., van der Velden, J., de Boer, R. A., van Tintelen, J. P., Asselbergs, F. W., & Baas, A. F. (2023). Untargeted Metabolomics Identifies Potential Hypertrophic Cardiomyopathy Biomarkers in Carriers of MYBPC3 Founder Variants. International Journal of Molecular Sciences, 24(4), 4031. https://doi.org/10.3390/ijms24044031