
Clocking Epilepsies: A Chronomodulated Strategy-Based Therapy for Rhythmic Seizures

Abstract
:1. Introduction
2. Epilepsy Genes
2.1. Compilation of 661 Epilepsy-Related Genes from 2 Public Databases
2.2. Epileptic Driver Genes, Passenger Genes, and Undetermined Genes
2.2.1. Ion Channel Genes
2.2.2. Genes Involved in the mTOR Pathway
2.2.3. Genes Encoding Synaptic Support Proteins
2.2.4. Transcriptional Regulators
2.2.5. Effects of Epileptic Genes on Circadian Rhythms and the Sleep–Wake Cycle
3. Circadian Rhythms in Human Epilepsies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Seizures | Peak in 24-h Cycle | Subjects No. (Seizure No.) | References |
|---|---|---|---|
| TLE | 11:00–17:00 11:00–19:00 11:00–15:00 | 176 (808) 26 (90) 1 (694) | Hofstra et al. (2009) [118] Pavlova et al. (2004) [18] Quigg et al. (2000) [15] |
| LTLE | Morning | 8 (48) | Quigg et al. (1998) [127] |
| MTLE | 05:00–11:00 and 11:00–17:00 15:00 07:00–10:00 and 16:00–19:00 06:00–08:00 and 15:00–17:00 03:00 and 17:00–20:00 | 33 (450) 64 (774) 131 (669) 60 (694) 72 (No mention) | Hofstra et al. (2009) [117] Quigg et al. (1998) [127] Durazzo et al. (2008) [20] Karafin et al. (2010) [119] Spencer et al. (2016) [120] |
| NTLE | 11:00–17:00 03:00–07:00 | 33 (450) 18 (No mention) | Hofstra et al. (2009) [117] Spencer et al. (2016) [120] |
| XTLE | Morning | 26 (465) | Quigg et al. (1998) [127] |
| FLE | 23:00–05:00 19:00–23:00 04:00–07:00 around 03:00 | 33 (450) 26 (90) 131 (669) 17 (No mention) | Hofstra et al. (2009) [117] Pavlova et al. (2004) [18] Durazzo et al. (2008) [20] Spencer et al. (2016) [120] |
| PLE | 05:00–11:00 and 17:00–23:00 04:00–07:00 01:00–06:00 | 33 (450) 131 (669) 1 (315) | Hofstra et al. (2009) [117] Durazzo et al. (2008) [20] Quigg et al. (2000) [15] |
| OLE | 19:00–23:00 16:00–19:00 | 26 (90) 131 (669) | Pavlova et al. (2004) [18] Durazzo et al. (2008) [20] |
| GED | Morning | 29 (No mention) | Labate et al. (2007) [116] |
4. The Circadian Clock
5. The Roles of Circadian Clock Genes in Epilepsies
6. Mutual Effects between Epilepsy and Sleep
7. Animal Models for Epilepsies
7.1. Pharmacological Models
7.2. Genetic Models
7.3. Circadian Rhythms of Epileptic Animal Models
7.4. Advantages and Challenges of Epileptic Animal Models
8. A Chronomodulated Strategy for Epilepsy Therapy
8.1. Circadian Mechanisms Underlying Epileptogenesis
8.2. Pharmacokinetic and Pharmacodynamic Studies of AEDs
8.3. Epileptic Chronotherapy
9. Discussion
10. Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Leonardi, M.; Ustun, T.B. The global burden of epilepsy. Epilepsia 2002, 43, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Brodie, M.J. Effectiveness of First Antiepileptic Drug. Epilepsia 2001, 42, 1255–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, D.; Löscher, W. Drug Resistance in Epilepsy: Putative Neurobiologic and Clinical Mechanisms. Epilepsia 2005, 46, 858–877. [Google Scholar] [CrossRef] [PubMed]
- Semah, F.; Picot, M.C.; Adam, C.; Broglin, D.; Arzimanoglou, A.; Bazin, B.; Cavalcanti, D.; Baulac, M. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology 1998, 51, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Vogel, F. Moderne Probleme der Humangenetik; Springer: Berlin/Heidelberg, Germany, 1959; pp. 52–125. [Google Scholar]
- Tate, S.K.; Sisodiya, S.M. Multidrug resistance in epilepsy: A pharmacogenomic update. Expert Opin. Pharmacother. 2007, 8, 1441–1449. [Google Scholar] [CrossRef]
- Ott, J. Association of genetic loci: Replication or not, that is the question. Neurology 2004, 63, 955–958. [Google Scholar] [CrossRef]
- Löscher, W.; Klotz, U.; Zimprich, F.; Schmidt, D. The clinical impact of pharmacogenetics on the treatment of epilepsy. Epilepsia 2009, 50, 1–23. [Google Scholar] [CrossRef]
- Chouchi, M.; Klaa, H.; Ben-Youssef Turki, I.; Hila, L. ABCB1 Polymorphisms and Drug-Resistant Epilepsy in a Tunisian Population. Dis. Markers 2019, 2019, 1343650. [Google Scholar] [CrossRef] [Green Version]
- Ihtisham, K.; Ramanujam, B.; Srivastava, S.; Mehra, N.K.; Kaur, G.; Khanna, N.; Jain, S.; Kumar, S.; Kaul, B.; Samudrala, R.; et al. Association of cutaneous adverse drug reactions due to antiepileptic drugs with HLA alleles in a North Indian population. Seizure 2019, 66, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Langdon-Down, M.; Brain, W.R. Time of day in relation to convulsions in epilepsy. Lancet 1929, 213, 1029–1032. [Google Scholar] [CrossRef]
- Gowers, W.R. Epilepsy and Other Chronic Convulsive Diseases: Their Causes, Symptoms & Treatment; William Wood & Company: West Chester, PA, USA, 1885. [Google Scholar]
- Fisher, R.S.; Boas, W.V.E.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J., Jr. Epileptic seizures and epilepsy: Definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005, 46, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Horne, J.A.; Östberg, O. A self-assessment questionnaire to determine morningness-eveningness in human circadian rhythms. Int. J. Chronobiol. 1976, 4, 97–110. [Google Scholar] [PubMed]
- Quigg, M. Circadian rhythms: Interactions with seizures and epilepsy. Epilepsy Res. 2000, 42, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, M.K.; Shea, S.A.; Scheer, F.A.; Bromfield, E.B. Is there a circadian variation of epileptiform abnormalities in idiopathic generalized epilepsy? Epilepsy Behav. 2009, 16, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Hofstra, W.A. The circadian rhythm and its interaction with human epilepsy: A review of literature. Sleep Med. Rev. 2009, 13, 413–420. [Google Scholar] [CrossRef]
- Pavlova, M.K.; Shea, S.A.; Bromfield, E.B. Day/night patterns of focal seizures. Epilepsy Behav. 2004, 5, 44–49. [Google Scholar] [CrossRef]
- Pung, T.; Schmitz, B. Circadian rhythm and personality profile in juvenile myoclonic epilepsy. Epilepsia 2006, 47, 111–114. [Google Scholar] [CrossRef]
- Durazzo, T.; Spencer, S.; Duckrow, R.; Novotny, E.; Spencer, D.; Zaveri, H. Temporal distributions of seizure occurrence from various epileptogenic regions. Neurology 2008, 70, 1265–1271. [Google Scholar] [CrossRef]
- Lemmer, B.; Labrecque, G. Chronopharmacology and chronotherapeutics: Definitions and concepts. Chronobiol. Int. 1987, 4, 319–329. [Google Scholar] [CrossRef]
- Manganaro, S.; Loddenkemper, T.; Rotenberg, A. The Need for Antiepileptic Drug Chronotherapy to Treat Selected Childhood Epilepsy Syndromes and Avert the Harmful Consequences of Drug Resistance. J. Cent. Nerv. Syst. Dis. 2017, 9, 1179573516685883. [Google Scholar] [CrossRef] [Green Version]
- Loddenkemper, T.; Vendrame, M.; Zarowski, M.; Gregas, M.; Alexopoulos, A.V.; Wyllie, E.; Kothare, S.V. Circadian patterns of pediatric seizures. Neurology 2011, 76, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Guilhoto, L.M.; Loddenkemper, T.; Vendrame, M.; Bergin, A.; Bourgeois, B.F.; Kothare, S.V. Higher evening antiepileptic drug dose for nocturnal and early-morning seizures. Epilepsy Behav. 2011, 20, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Kalilani, L.; Sun, X.; Pelgrims, B.; Noack-Rink, M.; Villanueva, V. The epidemiology of drug-resistant epilepsy: A systematic review and meta-analysis. Epilepsia 2018, 59, 2179–2193. [Google Scholar] [CrossRef] [Green Version]
- Kwan, P.; Brodie, M.J. Early identification of refractory epilepsy. N. Engl. J. Med. 2000, 342, 314–319. [Google Scholar] [CrossRef]
- Sillanpää, M.; Schmidt, D. Natural history of treated childhood-onset epilepsy: Prospective, long-term population-based study. Brain 2006, 129, 617–624. [Google Scholar] [CrossRef] [Green Version]
- Wolking, S.; Moreau, C.; McCormack, M.; Krause, R.; Krenn, M.; Berkovic, S.; Cavalleri, G.L.; Delanty, N.; Depondt, C.; Johnson, M.R.; et al. Assessing the role of rare genetic variants in drug-resistant, non-lesional focal epilepsy. Ann. Clin. Transl. Neurol. 2021, 8, 1376–1387. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Tao, H.; Wang, Y.; Liu, Z.; Xu, Z.; Zhou, H.; Cai, Y.; Yao, L.; Chen, B.; Liang, W.; et al. A functional polymorphism of the microRNA-146a gene is associated with susceptibility to drug-resistant epilepsy and seizures frequency. Seizure 2015, 27, 60–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balestrini, S.; Sisodiya, S.M. Pharmacogenomics in epilepsy. Neurosci. Lett. 2018, 667, 27–39. [Google Scholar] [CrossRef]
- Božina, N.; Sporiš, I.; Božina, T.; Klarica-Domjanović, I.; Tvrdeić, A.; Sporiš, D. Pharmacogenetics and the treatment of epilepsy: What do we know? Pharmacogenomics 2019, 20, 1093–1101. [Google Scholar] [CrossRef]
- Vadlamudi, L.; Milne, R.L.; Lawrence, K.; Heron, S.E.; Eckhaus, J.; Keay, D.; Connellan, M.; Torn-Broers, Y.; Howell, R.A.; Mulley, J.C.; et al. Genetics of epilepsy: The testimony of twins in the molecular era. Neurology 2014, 83, 1042–1048. [Google Scholar] [CrossRef] [Green Version]
- Berkovic, S.F.; Howell, R.A.; Hay, D.A.; Hopper, J.L. Epilepsies in twins: Genetics of the major epilepsy syndromes. Ann. Neurol. 1998, 43, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Steinlein, O.K.; Mulley, J.C.; Propping, P.; Wallace, R.H.; Phillips, H.A.; Sutherland, G.R.; Scheffer, I.E.; Berkovic, S.F. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 1995, 11, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Lindstrom, J. Chromosomal localization of seven neuronal nicotinic acetylcholine receptor subunit genes in humans. Genomics 1992, 13, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Conti, V.; Aracri, P.; Chiti, L.; Brusco, S.; Mari, F.; Marini, C.; Albanese, M.; Marchi, A.; Liguori, C.; Placidi, F.; et al. Nocturnal frontal lobe epilepsy with paroxysmal arousals due to CHRNA2 loss of function. Neurology 2015, 84, 1520–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, C.A.; Petrovski, S.; Berkovic, S.F. Epilepsy genetics: Clinical impacts and biological insights. Lancet Neurol. 2020, 19, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Dunn, P.; Albury, C.L.; Maksemous, N.; Benton, M.C.; Sutherland, H.G.; Smith, R.A.; Haupt, L.M.; Griffiths, L.R. Next Generation Sequencing Methods for Diagnosis of Epilepsy Syndromes. Front. Genet. 2018, 9, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epilepsy Genetics Initiative. The Epilepsy Genetics Initiative: Systematic reanalysis of diagnostic exomes increases yield. Epilepsia 2019, 60, 797–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, D.; Cetica, V.; Marini, C.; Guerrini, R. Dravet syndrome as part of the clinical and genetic spectrum of sodium channel epilepsies and encephalopathies. Epilepsia 2019, 60 (Suppl. 3), S2–S7. [Google Scholar] [CrossRef]
- Costain, G.; Cordeiro, D.; Matviychuk, D.; Mercimek-Andrews, S. Clinical Application of Targeted Next-Generation Sequencing Panels and Whole Exome Sequencing in Childhood Epilepsy. Neuroscience 2019, 418, 291–310. [Google Scholar] [CrossRef]
- Heinzen, E.L.; Yoon, W.; Tate, S.K.; Sen, A.; Wood, N.W.; Sisodiya, S.M.; Goldstein, D.B. Nova2 interacts with a cis-acting polymorphism to influence the proportions of drug-responsive splice variants of SCN1A. Am. J. Hum. Genet. 2007, 80, 876–883. [Google Scholar] [CrossRef] [Green Version]
- Fricke-Galindo, I.; Jung-Cook, H.; Llerena, A.; López-López, M. Pharmacogenetics of adverse reactions to antiepileptic drugs. Neurologia 2018, 33, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Zaccara, G.; Franciotta, D.; Perucca, E. Idiosyncratic Adverse Reactions to Antiepileptic Drugs. Epilepsia 2007, 48, 1223–1244. [Google Scholar] [CrossRef] [PubMed]
- Perucca, P.; Bahlo, M.; Berkovic, S.F. The Genetics of Epilepsy. Annu. Rev. Genom. Hum. Genet. 2020, 21, 205–230. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lin, Z.J.; Liu, L.; Xu, H.Q.; Shi, Y.W.; Yi, Y.H.; He, N.; Liao, W.P. Epilepsy-associated genes. Seizure 2017, 44, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Yin, N.; Li, M.O. SZT2 dictates GATOR control of mTORC1 signalling. Nature 2017, 543, 433–437. [Google Scholar] [CrossRef] [Green Version]
- Naseer, M.I.; Alwasiyah, M.K.; Abdulkareem, A.A.; Bajammal, R.A.; Trujillo, C.; Abu-Elmagd, M.; Jafri, M.A.; Chaudhary, A.G.; Al-Qahtani, M.H. A novel homozygous mutation in SZT2 gene in Saudi family with developmental delay, macrocephaly and epilepsy. Genes Genom. 2018, 40, 1149–1155. [Google Scholar] [CrossRef]
- Feng, H.; Sjögren, B.; Karaj, B.; Shaw, V.; Gezer, A.; Neubig, R.R. Movement disorder in GNAO1 encephalopathy associated with gain-of-function mutations. Neurology 2017, 89, 762–770. [Google Scholar] [CrossRef]
- Mazzola, L.; Oliver, K.L.; Labalme, A.; Baykan, B.; Muona, M.; Joensuu, T.H.; Courage, C.; Chatron, N.; Borsani, G.; Alix, E.; et al. Progressive Myoclonus Epilepsy Caused by a Homozygous Splicing Variant of SLC7A6OS. Ann. Neurol. 2021, 89, 402–407. [Google Scholar] [CrossRef]
- Jonker, J.W.; Schinkel, A.H. Pharmacological and physiological functions of the polyspecific organic cation transporters: OCT1, 2, and 3 (SLC22A1-3). J. Pharmacol. Exp. Ther. 2004, 308, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Vázquez, A.; Fricke-Galindo, I.; Dorado, P.; Jung-Cook, H.; Martínez-Juárez, I.E.; Monroy-Jaramillo, N.; Rojas-Tomé, I.S.; Peñas-Lledó, E.; Llerena, A.; López-López, M. Influence of genetic variants and antiepileptic drug co-treatment on lamotrigine plasma concentration in Mexican Mestizo patients with epilepsy. Pharm. J. 2020, 20, 845–856. [Google Scholar] [CrossRef]
- De Fusco, M.; Becchetti, A.; Patrignani, A.; Annesi, G.; Gambardella, A.; Quattrone, A.; Ballabio, A.; Wanke, E.; Casari, G. The nicotinic receptor beta 2 subunit is mutant in nocturnal frontal lobe epilepsy. Nat. Genet. 2000, 26, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.A.; Favre, I.; Kirkpatrick, M.; Zuberi, S.M.; Goudie, D.; Heron, S.E.; Scheffer, I.E.; Sutherland, G.R.; Berkovic, S.F.; Bertrand, D.; et al. CHRNB2 is the second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. Am. J. Hum. Genet. 2001, 68, 225–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aridon, P.; Marini, C.; Di Resta, C.; Brilli, E.; De Fusco, M.; Politi, F.; Parrini, E.; Manfredi, I.; Pisano, T.; Pruna, D.; et al. Increased sensitivity of the neuronal nicotinic receptor alpha 2 subunit causes familial epilepsy with nocturnal wandering and ictal fear. Am. J. Hum. Genet. 2006, 79, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Steinlein, O.K. Genetics and epilepsy. Dialogues Clin. Neurosci. 2008, 10, 29–38. [Google Scholar] [CrossRef]
- Quik, M.; Polonskaya, Y.; Gillespie, A.; Jakowec, M.; Lloyd, G.K.; Langston, J.W. Localization of nicotinic receptor subunit mRNAs in monkey brain by in situ hybridization. J. Comp. Neurol. 2000, 425, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Zoli, M.; Pistillo, F.; Gotti, C. Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology 2015, 96, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Hilscher, M.M.; Leão, R.N.; Edwards, S.J.; Leão, K.E.; Kullander, K. Chrna2-Martinotti Cells Synchronize Layer 5 Type A Pyramidal Cells via Rebound Excitation. PLoS Biol. 2017, 15, e2001392. [Google Scholar] [CrossRef]
- Son, J.H.; Winzer-Serhan, U.H. Postnatal expression of alpha2 nicotinic acetylcholine receptor subunit mRNA in developing cortex and hippocampus. J. Chem. Neuroanat. 2006, 32, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Becchetti, A.; Grandi, L.C.; Colombo, G.; Meneghini, S.; Amadeo, A. Nicotinic Receptors in Sleep-Related Hypermotor Epilepsy: Pathophysiology and Pharmacology. Brain Sci. 2020, 10, 907. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.W.; Avoli, M. GABA and epileptogenesis. Epilepsia 1997, 38, 399–407. [Google Scholar] [CrossRef]
- Riban, V.; Fitzsimons, H.L.; During, M.J. Gene therapy in epilepsy. Epilepsia 2009, 50, 24–32. [Google Scholar] [CrossRef]
- Gernert, M.; Thompson, K.W.; Löscher, W.; Tobin, A.J. Genetically engineered GABA-producing cells demonstrate anticonvulsant effects and long-term transgene expression when transplanted into the central piriform cortex of rats. Exp. Neurol. 2002, 176, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Asada, H.; Kawamura, Y.; Maruyama, K.; Kume, H.; Ding, R.G.; Kanbara, N.; Kuzume, H.; Sanbo, M.; Yagi, T.; Obata, K. Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc. Natl. Acad. Sci. USA 1997, 94, 6496–6499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, D.L.; Houser, C.R.; Tobin, A.J. Two forms of the gamma-aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions. J. Neurochem. 1991, 56, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Lynex, C.N.; Carr, I.M.; Leek, J.P.; Achuthan, R.; Mitchell, S.; Maher, E.R.; Woods, C.G.; Bonthon, D.T.; Markham, A.F. Homozygosity for a missense mutation in the 67 kDa isoform of glutamate decarboxylase in a family with autosomal recessive spastic cerebral palsy: Parallels with Stiff-Person Syndrome and other movement disorders. BMC Neurol. 2004, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Ruzicka, W.B.; Subburaju, S.; Benes, F.M. Circuit- and Diagnosis-Specific DNA Methylation Changes at γ-Aminobutyric Acid-Related Genes in Postmortem Human Hippocampus in Schizophrenia and Bipolar Disorder. JAMA Psychiatry 2015, 72, 541–551. [Google Scholar] [CrossRef]
- Neuray, C.; Maroofian, R.; Scala, M.; Sultan, T.; Pai, G.S.; Mojarrad, M.; Khashab, H.E.; Deholl, L.; Yue, W.; Alsaif, H.S.; et al. Early-infantile onset epilepsy and developmental delay caused by bi-allelic GAD1 variants. Brain 2020, 143, 2388–2397. [Google Scholar] [CrossRef]
- Wang, Y.; Zhan, L.; Zeng, W.; Li, K.; Sun, W.; Xu, Z.C.; Xu, E. Downregulation of hippocampal GABA after hypoxia-induced seizures in neonatal rats. Neurochem. Res. 2011, 36, 2409–2416. [Google Scholar] [CrossRef]
- Jacob, T.C.; Moss, S.J.; Jurd, R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Reviews. Neurosci. 2008, 9, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Sieghart, W.; Sperk, G. Subunit composition, distribution and function of GABA(A) receptor subtypes. Curr. Top. Med. Chem. 2002, 2, 795–816. [Google Scholar] [CrossRef]
- Cossette, P.; Liu, L.; Brisebois, K.; Dong, H.; Lortie, A.; Vanasse, M.; Saint-Hilaire, J.M.; Carmant, L.; Verner, A.; Lu, W.Y.; et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat. Genet. 2002, 31, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Naylor, D.E.; Liu, H.; Wasterlain, C.G. Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J. Neurosci. 2005, 25, 7724–7733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terunuma, M.; Xu, J.; Vithlani, M.; Sieghart, W.; Kittler, J.; Pangalos, M.; Haydon, P.G.; Coulter, D.A.; Moss, S.J. Deficits in phosphorylation of GABA(A) receptors by intimately associated protein kinase C activity underlie compromised synaptic inhibition during status epilepticus. J. Neurosci. 2008, 28, 376–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouilleret, V.; Loup, F.; Kiener, T.; Marescaux, C.; Fritschy, J.M. Early loss of interneurons and delayed subunit-specific changes in GABA(A)-receptor expression in a mouse model of mesial temporal lobe epilepsy. Hippocampus 2000, 10, 305–324. [Google Scholar] [CrossRef] [PubMed]
- Oyrer, J.; Maljevic, S.; Scheffer, I.E.; Berkovic, S.F.; Petrou, S.; Reid, C.A. Ion Channels in Genetic Epilepsy: From Genes and Mechanisms to Disease-Targeted Therapies. Pharmacol. Rev. 2018, 70, 142–173. [Google Scholar] [CrossRef] [PubMed]
- Brunklaus, A.; Du, J.; Steckler, F.; Ghanty, I.I.; Johannesen, K.M.; Fenger, C.D.; Schorge, S.; Baez-Nieto, D.; Wang, H.R.; Allen, A.; et al. Biological concepts in human sodium channel epilepsies and their relevance in clinical practice. Epilepsia 2020, 61, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Zuberi, S.M.; Brunklaus, A.; Birch, R.; Reavey, E.; Duncan, J.; Forbes, G.H. Genotype-phenotype associations in SCN1A-related epilepsies. Neurology 2011, 76, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Nabbout, R.; Gennaro, E.; Dalla Bernardina, B.; Dulac, O.; Madia, F.; Bertini, E.; Capovilla, G.; Chiron, C.; Cristofori, G.; Elia, M.; et al. Spectrum of SCN1A mutations in severe myoclonic epilepsy of infancy. Neurology 2003, 60, 1961–1967. [Google Scholar] [CrossRef]
- Begemann, A.; Acuña, M.A.; Zweier, M.; Vincent, M.; Steindl, K.; Bachmann-Gagescu, R.; Hackenberg, A.; Abela, L.; Plecko, B.; Kroell-Seger, J.; et al. Further corroboration of distinct functional features in SCN2A variants causing intellectual disability or epileptic phenotypes. Mol. Med. 2019, 25, 6. [Google Scholar] [CrossRef]
- Wallace, R.H.; Wang, D.W.; Singh, R.; Scheffer, I.E.; George, A.L., Jr.; Phillips, H.A.; Saar, K.; Reis, A.; Johnson, E.W.; Sutherland, G.R.; et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat. Genet. 1998, 19, 366–370. [Google Scholar] [CrossRef]
- Wimmer, V.C.; Reid, C.A.; Mitchell, S.; Richards, K.L.; Scaf, B.B.; Leaw, B.T.; Hill, E.L.; Royeck, M.; Horstmann, M.T.; Cromer, B.A.; et al. Axon initial segment dysfunction in a mouse model of genetic epilepsy with febrile seizures plus. J. Clin. Investig. 2010, 120, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Maljevic, S.; Lerche, H. Potassium channel genes and benign familial neonatal epilepsy. Prog. Brain Res. 2014, 213, 17–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.S.; Pan, Z.; Shi, W.; Brown, B.S.; Wymore, R.S.; Cohen, I.S.; Dixon, J.E.; McKinnon, D. KCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M-channel. Science 1998, 282, 1890–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, H.; Nagata, E.; Kosakai, A.; Nakamura, M.; Yokoyama, M.; Tanaka, K.; Sasai, H. Disruption of the epilepsy KCNQ2 gene results in neural hyperexcitability. J. Neurochem. 2000, 75, 28–33. [Google Scholar] [CrossRef]
- Zamponi, G.W.; Lory, P.; Perez-Reyes, E. Role of voltage-gated calcium channels in epilepsy. Pflug. Arch. Eur. J. Physiol. 2010, 460, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Carpintero Abad, R.; Sanmartí Vilaplana, F.X.; Serratosa Fernández, J.M. Genetic causes of epilepsy. Neurologist 2007, 13, S47–S51. [Google Scholar] [CrossRef]
- Citraro, R.; Leo, A.; Constanti, A.; Russo, E.; De Sarro, G. mTOR pathway inhibition as a new therapeutic strategy in epilepsy and epileptogenesis. Pharmacol. Res. 2016, 107, 333–343. [Google Scholar] [CrossRef] [Green Version]
- Hodges, S.L.; Lugo, J.N. Therapeutic role of targeting mTOR signaling and neuroinflammation in epilepsy. Epilepsy Res. 2020, 161, 106282. [Google Scholar] [CrossRef]
- Crino, P.B. mTOR: A pathogenic signaling pathway in developmental brain malformations. Trends Mol. Med. 2011, 17, 734–742. [Google Scholar] [CrossRef]
- Baulac, S.; Ishida, S.; Marsan, E.; Miquel, C.; Biraben, A.; Nguyen, D.K.; Nordli, D.; Cossette, P.; Nguyen, S.; Lambrecq, V.; et al. Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann. Neurol. 2015, 77, 675–683. [Google Scholar] [CrossRef] [Green Version]
- Sim, J.C.; Scerri, T.; Fanjul-Fernández, M.; Riseley, J.R.; Gillies, G.; Pope, K.; van Roozendaal, H.; Heng, J.I.; Mandelstam, S.A.; McGillivray, G.; et al. Familial cortical dysplasia caused by mutation in the mammalian target of rapamycin regulator NPRL3. Ann. Neurol. 2016, 79, 132–137. [Google Scholar] [CrossRef]
- Baldassari, S.; Picard, F.; Verbeek, N.E.; Van Kempen, M.; Brilstra, E.H.; Lesca, G.; Conti, V.; Guerrini, R.; Bisulli, F.; Licchetta, L.; et al. The landscape of epilepsy-related GATOR1 variants. Genet. Med. 2019, 21, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Weckhuysen, S.; Marsan, E.; Lambrecq, V.; Marchal, C.; Morin-Brureau, M.; An-Gourfinkel, I.; Baulac, M.; Fohlen, M.; Kallay Zetchi, C.; Seeck, M.; et al. Involvement of GATOR complex genes in familial focal epilepsies and focal cortical dysplasia. Epilepsia 2016, 57, 994–1003. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, S.M.; De Camilli, P. Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol. 2012, 13, 75–88. [Google Scholar] [CrossRef] [Green Version]
- Von Spiczak, S.; Helbig, K.L.; Shinde, D.N.; Huether, R.; Pendziwiat, M.; Lourenço, C.; Nunes, M.E.; Sarco, D.P.; Kaplan, R.A.; Dlugos, D.J.; et al. DNM1 encephalopathy: A new disease of vesicle fission. Neurology 2017, 89, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Cali, E.; Rocca, C.; Salpietro, V.; Houlden, H. Epileptic Phenotypes Associated with SNAREs and Related Synaptic Vesicle Exocytosis Machinery. Front. Neurol. 2021, 12, 806506. [Google Scholar] [CrossRef] [PubMed]
- Wolking, S.; May, P.; Mei, D.; Møller, R.S.; Balestrini, S.; Helbig, K.L.; Altuzarra, C.D.; Chatron, N.; Kaiwar, C.; Stöhr, K.; et al. Clinical spectrum of STX1B-related epileptic disorders. Neurology 2019, 92, e1238–e1249. [Google Scholar] [CrossRef] [Green Version]
- Strømme, P.; Mangelsdorf, M.E.; Shaw, M.A.; Lower, K.M.; Lewis, S.M.; Bruyere, H.; Lütcherath, V.; Gedeon, A.K.; Wallace, R.H.; Scheffer, I.E.; et al. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat. Genet. 2002, 30, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Poeta, L.; Fusco, F.; Drongitis, D.; Shoubridge, C.; Manganelli, G.; Filosa, S.; Paciolla, M.; Courtney, M.; Collombat, P.; Lioi, M.B.; et al. A regulatory path associated with X-linked intellectual disability and epilepsy links KDM5C to the polyalanine expansions in ARX. Am. J. Hum. Genet. 2013, 92, 114–125. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Saitoh, S.; Kamei, A.; Shiraishi, H.; Ueda, Y.; Akasaka, M.; Tohyama, J.; Akasaka, N.; Hayasaka, K. A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome). Am. J. Hum. Genet. 2007, 81, 361–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Esch, H. MECP2 Duplication Syndrome. Mol. Syndromol. 2012, 2, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignoli, A.; Borgatti, R.; Peron, A.; Zucca, C.; Ballarati, L.; Bonaglia, C.; Bellini, M.; Giordano, L.; Romaniello, R.; Bedeschi, M.F.; et al. Electroclinical pattern in MECP2 duplication syndrome: Eight new reported cases and review of literature. Epilepsia 2012, 53, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.E.A.; Bussi, I.L.; Ben-Hamo, M.; Caldart, C.S.; Catterall, W.A.; De La Iglesia, H.O. Circadian regulation of sleep in a pre-clinical model of Dravet syndrome: Dynamics of sleep stage and siesta re-entrainment. Sleep 2019, 42, zsz173. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Eaton, M.; Liu, Y.; Zhang, J.; Chen, X.; Tu, X.; Shi, Y.; Que, Z.; Wettschurack, K.; Zhang, Z.; et al. Deficiency of autism-related Scn2a gene in mice disrupts sleep patterns and circadian rhythms. Neurobiol. Dis. 2022, 168, 105690. [Google Scholar] [CrossRef]
- Wallace, E.; Wright, S.; Schoenike, B.; Roopra, A.; Rho, J.M.; Maganti, R.K. Altered circadian rhythms and oscillation of clock genes and sirtuin 1 in a model of sudden unexpected death in epilepsy. Epilepsia 2018, 59, 1527–1539. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, S.; Campusano, J.M.; Hodge, J.J.L. The Drosophila ortholog of the schizophrenia-associated CACNA1A and CACNA1B voltage-gated calcium channels regulate memory, sleep and circadian rhythms. Neurobiol. Dis. 2021, 155, 105394. [Google Scholar] [CrossRef]
- Martínez de Paz, A.; Sanchez-Mut, J.V.; Samitier-Martí, M.; Petazzi, P.; Sáez, M.; Szczesna, K.; Huertas, D.; Esteller, M.; Ausió, J. Circadian cycle-dependent MeCP2 and brain chromatin changes. PLoS ONE 2015, 10, e0123693. [Google Scholar] [CrossRef] [Green Version]
- Autret, A.; Lucas, B.; Laffont, F.; Bertrand, P.; Degiovanni, E.; De Toffol, B. Two distinct classifications of adult epilepsies: By time of seizures and by sensitivity of the interictal paroxysmal activities to sleep and waking. Electroencephalogr. Clin. Neurophysiol. 1987, 66, 211–218. [Google Scholar] [CrossRef]
- Karoly, P.; Goldenholz, D.; Freestone, D.; Moss, R.; Grayden, D.; Theodore, W.; Cook, M. Circadian and circaseptan rhythms in human epilepsy: A retrospective cohort study. Lancet Neurol. 2018, 17, 977–985. [Google Scholar] [CrossRef]
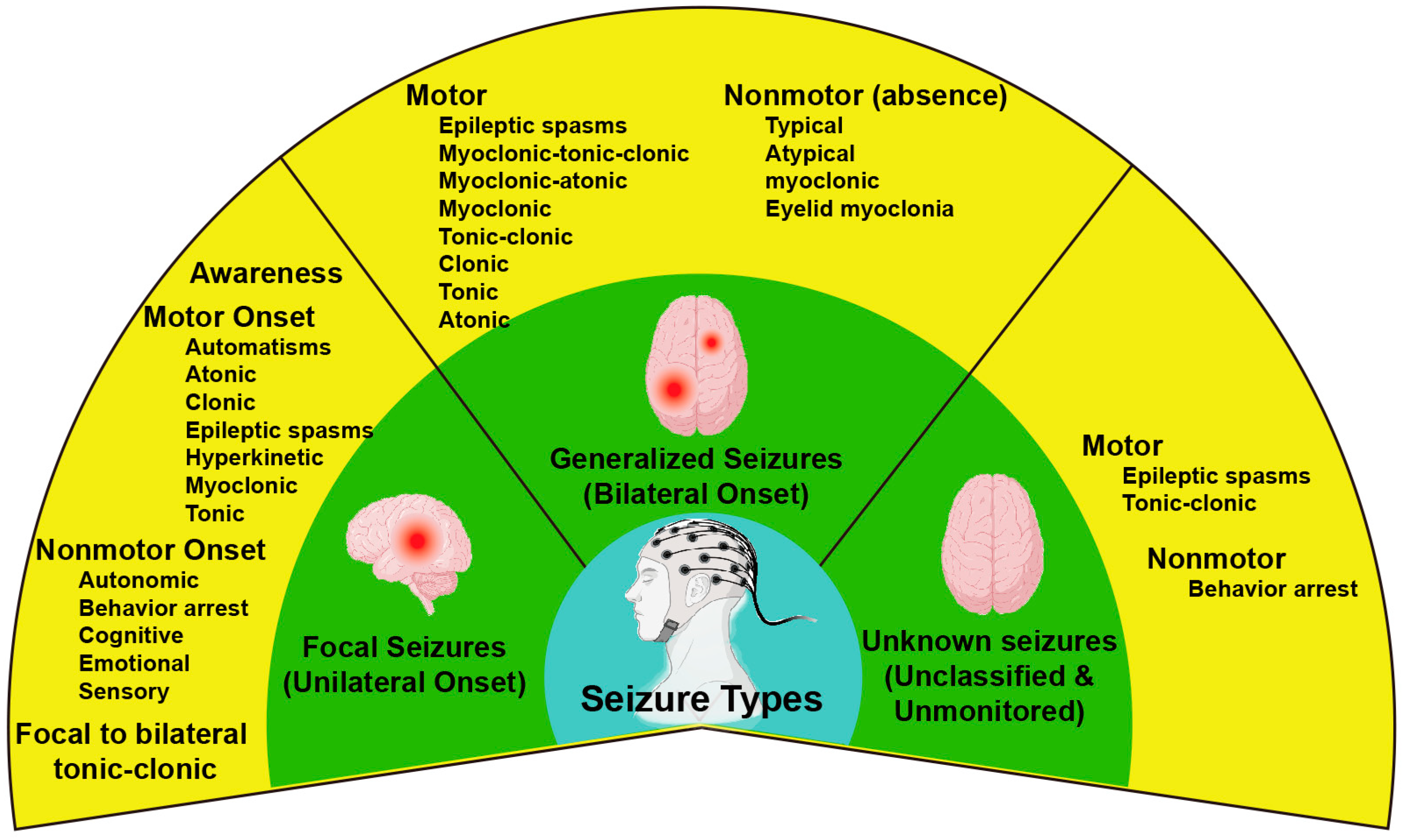
- Berg, A.T.; Berkovic, S.F.; Brodie, M.J.; Buchhalter, J.; Cross, J.H.; Van Emde Boas, W.; Engel, J.; French, J.; Glauser, T.A.; Mathern, G.W. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010, 51, 676–685. [Google Scholar] [CrossRef]
- Mirzoev, A.; Bercovici, E.; Stewart, L.S.; Cortez, M.A.; Snead, O.C., 3rd; Desrocher, M. Circadian profiles of focal epileptic seizures: A need for reappraisal. Seizure 2012, 21, 412–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, R.S.; Cross, J.H.; French, J.A.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L.; Peltola, J.; Roulet Perez, E.; et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 522–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labate, A.; Ambrosio, R.; Gambardella, A.; Sturniolo, M.; Pucci, F.; Quattrone, A. Usefulness of a morning routine EEG recording in patients with juvenile myoclonic epilepsy. Epilepsy Res. 2007, 77, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Hofstra, W.A.; Spetgens, W.P.; Leijten, F.S.; Van Rijen, P.C.; Gosselaar, P.; Van Der Palen, J.; De Weerd, A.W. Diurnal rhythms in seizures detected by intracranial electrocorticographic monitoring: An observational study. Epilepsy Behav. 2009, 14, 617–621. [Google Scholar] [CrossRef]
- Hofstra, W.A.; Grootemarsink, B.E.; Dieker, R.; Van Der Palen, J.; De Weerd, A.W. Temporal distribution of clinical seizures over the 24-h day: A retrospective observational study in a tertiary epilepsy clinic. Epilepsia 2009, 50, 2019–2026. [Google Scholar] [CrossRef]
- Karafin, M.; St Louis, E.K.; Zimmerman, M.B.; Sparks, J.D.; Granner, M.A. Bimodal ultradian seizure periodicity in human mesial temporal lobe epilepsy. Seizure 2010, 19, 347–351. [Google Scholar] [CrossRef] [Green Version]
- Spencer, D.C.; Sun, F.T.; Brown, S.N.; Jobst, B.C.; Fountain, N.B.; Wong, V.S.; Mirro, E.A.; Quigg, M. Circadian and ultradian patterns of epileptiform discharges differ by seizure-onset location during long-term ambulatory intracranial monitoring. Epilepsia 2016, 57, 1495–1502. [Google Scholar] [CrossRef] [Green Version]
- Daley, J.T.; DeWolfe, J.L. Sleep, Circadian Rhythms, and Epilepsy. Curr. Treat. Options Neurol. 2018, 20, 47. [Google Scholar] [CrossRef]
- Leguia, M.G.; Andrzejak, R.G.; Rummel, C.; Fan, J.M.; Mirro, E.A.; Tcheng, T.K.; Rao, V.R.; Baud, M.O. Seizure Cycles in Focal Epilepsy. JAMA Neurol. 2021, 78, 454–463. [Google Scholar] [CrossRef]
- Baud, M.O.; Kleen, J.K.; Mirro, E.A.; Andrechak, J.C.; King-Stephens, D.; Chang, E.F.; Rao, V.R. Multi-day rhythms modulate seizure risk in epilepsy. Nat. Commun. 2018, 9, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregg, N.M.; Nasseri, M.; Kremen, V.; Patterson, E.E.; Sturges, B.K.; Denison, T.J.; Brinkmann, B.H.; Worrell, G.A. Circadian and multiday seizure periodicities, and seizure clusters in canine epilepsy. Brain Commun. 2020, 2, fcaa008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronkainen, E.; Ansakorpi, H.; Huikuri, H.V.; Myllylä, V.V.; Isojärvi, J.I.; Korpelainen, J.T. Suppressed circadian heart rate dynamics in temporal lobe epilepsy. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1382–1386. [Google Scholar] [CrossRef] [PubMed]
- Schapel, G.J.; Beran, R.G.; Kennaway, D.L.; McLoughney, J.; Matthews, C.D. Melatonin response in active epilepsy. Epilepsia 1995, 36, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Quigg, M.; Straume, M.; Menaker, M.; Bertram, E.H., 3rd. Temporal distribution of partial seizures: Comparison of an animal model with human partial epilepsy. Ann. Neurol. 1998, 43, 748–755. [Google Scholar] [CrossRef]
- Crnko, S.; Du Pré, B.C.; Sluijter, J.P.G.; Van Laake, L.W. Circadian rhythms and the molecular clock in cardiovascular biology and disease. Nat. Rev. Cardiol. 2019, 16, 437–447. [Google Scholar] [CrossRef]
- Wang, H. Perfect timing: A Nobel Prize in Physiology or Medicine for circadian clocks. Sci. Bull. 2018, 63, 398–401. [Google Scholar] [CrossRef] [Green Version]
- Fagiani, F.; Di Marino, D.; Romagnoli, A.; Travelli, C.; Voltan, D.; Di Cesare Mannelli, L.; Racchi, M.; Govoni, S.; Lanni, C. Molecular regulations of circadian rhythm and implications for physiology and diseases. Signal Transduct. Target. Ther. 2022, 7, 41. [Google Scholar] [CrossRef]
- Lee, Y.; Field, J.M.; Sehgal, A. Circadian Rhythms, Disease and Chronotherapy. J. Biol. Rhythm. 2021, 36, 503–531. [Google Scholar] [CrossRef]
- Buijs, F.N.; León-Mercado, L.; Guzmán-Ruiz, M.; Guerrero-Vargas, N.N.; Romo-Nava, F.; Buijs, R.M. The Circadian System: A Regulatory Feedback Network of Periphery and Brain. Physiology 2016, 31, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Shin-ichi, T.I.; Kawamura, H. Characteristics of a circadian pacemaker in the suprachiasmatic nucleus. J. Comp. Physiol. 1982, 146, 153–160. [Google Scholar]
- Schwartz, W.J.; Reppert, S.M.; Eagan, S.M.; Moore-Ede, M.C. In vivo metabolic activity of the suprachiasmatic nuclei: A comparative study. Brain Res. 1983, 274, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.J.; Peirson, S.N.; Berson, D.M.; Brown, T.M.; Cooper, H.M.; Czeisler, C.A.; Figueiro, M.G.; Gamlin, P.D.; Lockley, S.W.; O’Hagan, J.B.; et al. Measuring and using light in the melanopsin age. Trends Neurosci. 2014, 37, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bilu, C.; Kronfeld-Schor, N. Effects of circadian phase and melatonin injection on anxiety-like behavior in nocturnal and diurnal rodents. Chronobiol. Int. 2013, 30, 828–836. [Google Scholar] [CrossRef]
- Vivanco, P.; Ortiz, V.; Rol, M.A.; Madrid, J.A. Looking for the keys to diurnality downstream from the circadian clock: Role of melatonin in a dual-phasing rodent, Octodon degus. J. Pineal Res. 2007, 42, 280–290. [Google Scholar] [CrossRef]
- Partch, C.L.; Green, C.B.; Takahashi, J.S. Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 2014, 24, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.A.; Azzi, A. Peripheral circadian oscillators in mammals. Handb. Exp. Pharmacol. 2013, 217, 45–66. [Google Scholar] [CrossRef]
- Gerstner, J.R.; Smith, G.G.; Lenz, O.; Perron, I.J.; Buono, R.J.; Ferraro, T.N. BMAL1 controls the diurnal rhythm and set point for electrical seizure threshold in mice. Front. Syst. Neurosci. 2014, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Fu, X.; Smith, N.A.; Ziobro, J.; Curiel, J.; Tenga, M.J.; Martin, B.; Freedman, S.; Cea-Del Rio, C.A.; Oboti, L.; et al. Loss of CLOCK Results in Dysfunction of Brain Circuits Underlying Focal Epilepsy. Neuron 2017, 96, 387–401. [Google Scholar] [CrossRef] [Green Version]
- Gekakis, N.; Staknis, D.; Nguyen, H.B.; Davis, F.C.; Wilsbacher, L.D.; King, D.P.; Takahashi, J.S.; Weitz, C.J. Role of the CLOCK protein in the mammalian circadian mechanism. Science 1998, 280, 1564–1569. [Google Scholar] [CrossRef]
- Sato, T.K.; Yamada, R.G.; Ukai, H.; Baggs, J.E.; Miraglia, L.J.; Kobayashi, T.J.; Welsh, D.K.; Kay, S.A.; Ueda, H.R.; Hogenesch, J.B. Feedback repression is required for mammalian circadian clock function. Nat. Genet. 2006, 38, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Gachon, F.; Fonjallaz, P.; Damiola, F.; Gos, P.; Kodama, T.; Zakany, J.; Duboule, D.; Petit, B.; Tafti, M.; Schibler, U. The loss of circadian PAR bZip transcription factors results in epilepsy. Genes Dev. 2004, 18, 1397–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, H.R.; Hayashi, S.; Chen, W.; Sano, M.; Machida, M.; Shigeyoshi, Y.; Iino, M.; Hashimoto, S. System-level identification of transcriptional circuits underlying mammalian circadian clocks. Nat. Genet. 2005, 37, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Ukai-Tadenuma, M.; Yamada, R.G.; Xu, H.; Ripperger, J.A.; Liu, A.C.; Ueda, H.R. Delay in feedback repression by cryptochrome 1 is required for circadian clock function. Cell 2011, 144, 268–281. [Google Scholar] [CrossRef] [Green Version]
- Minami, Y.; Ode, K.L.; Ueda, H.R. Mammalian circadian clock: The roles of transcriptional repression and delay. Handb. Exp. Pharmacol. 2013, 217, 359–377. [Google Scholar] [CrossRef]
- Lipton, J.O.; Yuan, E.D.; Boyle, L.M.; Ebrahimi-Fakhari, D.; Kwiatkowski, E.; Nathan, A.; Guttler, T.; Davis, F.; Asara, J.M.; Sahin, M. The Circadian Protein BMAL1 Regulates Translation in Response to S6K1-Mediated Phosphorylation. Cell 2015, 161, 1138–1151. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, C.; Kathale, N.D.; Liu, D.; Lee, C.; Freeman, D.A.; Hogenesch, J.B.; Cao, R. mTOR signaling regulates central and peripheral circadian clock function. PLoS Genet. 2018, 14, e1007369. [Google Scholar] [CrossRef]
- Liu, D.; Stowie, A.; De Zavalia, N.; Leise, T.; Pathak, S.S.; Drewes, L.R.; Davidson, A.J.; Amir, S.; Sonenberg, N.; Cao, R. mTOR signaling in VIP neurons regulates circadian clock synchrony and olfaction. Proc. Natl. Acad. Sci. USA 2018, 115, E3296–E3304. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Nobili, L.; Khatami, R.; Loddenkemper, T.; Cajochen, C.; Dijk, D.J.; Eriksson, S.H. Circadian rhythm and epilepsy. Lancet Neurol. 2018, 17, 1098–1108. [Google Scholar] [CrossRef]
- Sahar, S.; Zocchi, L.; Kinoshita, C.; Borrelli, E.; Sassone-Corsi, P. Regulation of BMAL1 protein stability and circadian function by GSK3beta-mediated phosphorylation. PLoS ONE 2010, 5, e8561. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.K.; Dakshinamurti, K. Seizure activity in pyridoxine-deficient adult rats. Epilepsia 1992, 33, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liu, Y.; Liu, L.; Meng, Q.; Du, C.; Li, K.; Dong, S.; Zhang, Y.; Li, H.; Zhang, H. Decreased expression of the clock gene Bmal1 is involved in the pathogenesis of temporal lobe epilepsy. Mol Brain 2021, 14, 113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Yu, F.; Xu, H.; Chen, M.; Chen, X.; Guo, L.; Zhou, C.; Xu, Y.; Wang, F.; Yu, J.; et al. Dysregulation of REV-ERBα impairs GABAergic function and promotes epileptic seizures in preclinical models. Nat. Commun. 2021, 12, 1216. [Google Scholar] [CrossRef]
- Ng, M.; Pavlova, M. Why are seizures rare in rapid eye movement sleep? Review of the frequency of seizures in different sleep stages. Epilepsy Res. Treat. 2013, 2013, 932790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frauscher, B.; Von Ellenrieder, N.; Dubeau, F.; Gotman, J. EEG desynchronization during phasic REM sleep suppresses interictal epileptic activity in humans. Epilepsia 2016, 57, 879–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saper, C.B.; Scammell, T.E.; Lu, J. Hypothalamic regulation of sleep and circadian rhythms. Nature 2005, 437, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Bazhenov, M.; Timofeev, I.; Steriade, M.; Sejnowski, T. Spiking-bursting activity in the thalamic reticular nucleus initiates sequences of spindle oscillations in thalamic networks. J. Neurophysiol. 2000, 84, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Minecan, D.; Natarajan, A.; Marzec, M.; Malow, B. Relationship of epileptic seizures to sleep stage and sleep depth. Sleep 2002, 25, 899–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, B.; Hu, W.; Ye, L.; Krishnan, B.; Aung, T.; Jones, S.E.; Najm, I.M.; Alexopoulos, A.V.; Zhang, K.; Zhu, J.; et al. Small Lesion Size Is Associated with Sleep-Related Epilepsy in Focal Cortical Dysplasia Type II. Front. Neurol. 2018, 9, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva Lourenço, C.; Tjepkema-Cloostermans, M.C.; Van Putten, M. Machine learning for detection of interictal epileptiform discharges. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2021, 132, 1433–1443. [Google Scholar] [CrossRef] [PubMed]
- De Berardis, D.; Orsolini, L.; Serroni, N.; Girinelli, G.; Iasevoli, F.; Tomasetti, C.; Mazza, M.; Valchera, A.; Fornaro, M.; Perna, G. The role of melatonin in mood disorders. ChronoPhysiology Ther. 2015, 5, 65. [Google Scholar] [CrossRef] [Green Version]
- Bazil, C.W.; Short, D.; Crispin, D.; Zheng, W. Patients with intractable epilepsy have low melatonin, which increases following seizures. Neurology 2000, 55, 1746–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Carballo, A.; Acuna-Castroviejo, D.; Rodriguez-Cabezas, T.; Munoz-Hoyos, A. Effects of febrile and epileptic convulsions on daily variations in plasma melatonin concentration in children. J. Pineal Res. 1994, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kothare, S.V.; Kaleyias, J. Sleep and epilepsy in children and adolescents. Sleep Med. 2010, 11, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Guo, D.; Liu, Y.Y.; Qiao, D.D.; Ye, J.Y.; Xue, R. Juvenile myoclonic epilepsy and sleep. Epilepsy Behav. 2018, 80, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Malow, B.A.; Levy, K.; Maturen, K.; Bowes, R. Obstructive sleep apnea is common in medically refractory epilepsy patients. Neurology 2000, 55, 1002–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dell, K.L.; Payne, D.E.; Kremen, V.; Maturana, M.I.; Gerla, V.; Nejedly, P.; Worrell, G.A.; Lenka, L.; Mivalt, F.; Boston, R.C.; et al. Seizure likelihood varies with day-to-day variations in sleep duration in patients with refractory focal epilepsy: A longitudinal electroencephalography investigation. EClinicalMedicine 2021, 37, 100934. [Google Scholar] [CrossRef]
- Staniszewska, A.; Maka, A.; Religioni, U.; Olejniczak, D. Sleep disturbances among patients with epilepsy. Neuropsychiatr. Dis. Treat. 2017, 13, 1797–1803. [Google Scholar] [CrossRef] [Green Version]
- Leschziner, G. Seizures and Sleep: Not such strange bedfellows. Adv. Clin. Neurosci. Rehabil. 2022, 21, 19–21. [Google Scholar] [CrossRef]
- Bazil, C.W.; Castro, L.H.; Walczak, T.S. Reduction of rapid eye movement sleep by diurnal and nocturnal seizures in temporal lobe epilepsy. Arch. Neurol. 2000, 57, 363–368. [Google Scholar] [CrossRef]
- Gelinas, J.N.; Khodagholy, D.; Thesen, T.; Devinsky, O.; Buzsáki, G. Interictal epileptiform discharges induce hippocampal-cortical coupling in temporal lobe epilepsy. Nat. Med. 2016, 22, 641–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekky, J.F.; Elbhrawy, S.M.; Boraey, M.F.; Omar, H.M. Sleep architecture in patients with Juvenile Myoclonic Epilepsy. Sleep Med. 2017, 38, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Shvarts, V.; Chung, S. Epilepsy, antiseizure therapy, and sleep cycle parameters. Epilepsy Res. Treat. 2013, 2013, 670682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
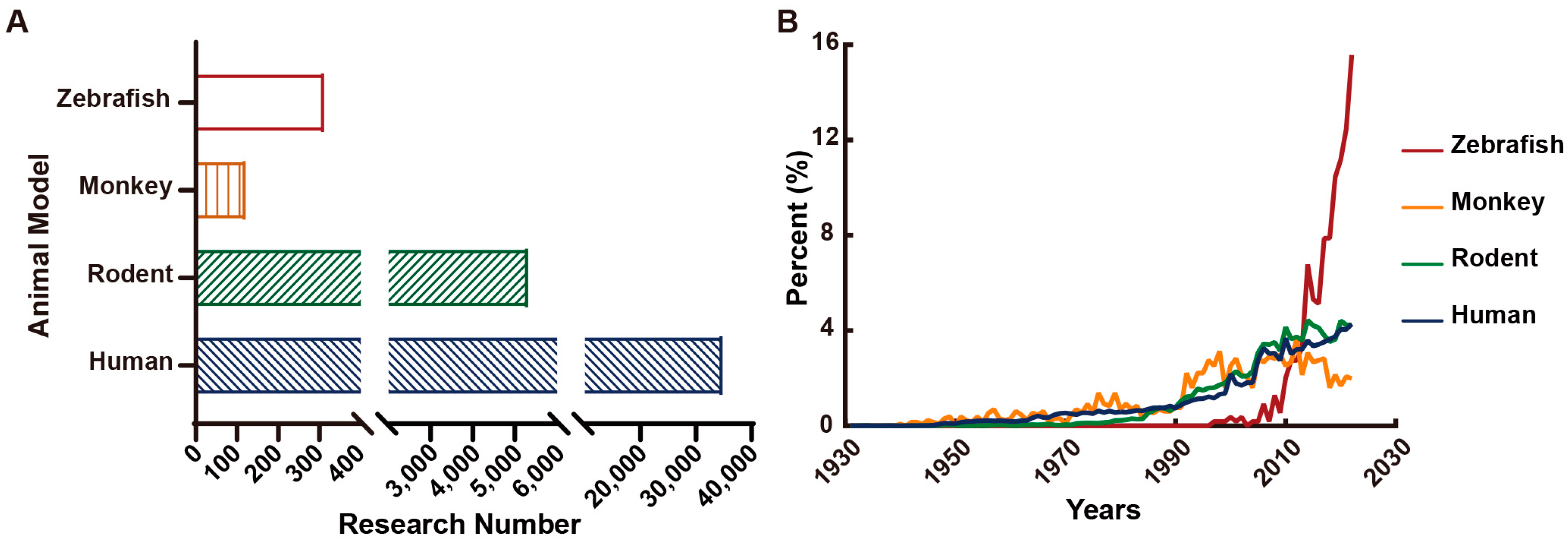
- Löscher, W. Animal Models of Seizures and Epilepsy: Past, Present, and Future Role for the Discovery of Antiseizure Drugs. Neurochem. Res. 2017, 42, 1873–1888. [Google Scholar] [CrossRef]
- Marshall, G.F.; Gonzalez-Sulser, A.; Abbott, C.M. Modelling epilepsy in the mouse: Challenges and solutions. Dis. Model. Mech. 2021, 14, dmm047449. [Google Scholar] [CrossRef]
- Porter, R.J.; Cereghino, J.J.; Gladding, G.D.; Hessie, B.; Kupferberg, H.J.; Scoville, B.; White, B.G. Antiepileptic Drug Development Program. Clevel. Clin. J. Med. 1984, 51, 293–305. [Google Scholar] [CrossRef]
- Leclercq, K.; Afrikanova, T.; Langlois, M.; De Prins, A.; Buenafe, O.E.; Rospo, C.C.; Van Eeckhaut, A.; de Witte, P.A.; Crawford, A.D.; Smolders, I.; et al. Cross-species pharmacological characterization of the allylglycine seizure model in mice and larval zebrafish. Epilepsy Behav. 2015, 45, 53–63. [Google Scholar] [CrossRef]
- Baxendale, S.; Holdsworth, C.J.; Meza Santoscoy, P.L.; Harrison, M.R.M.; Fox, J.; Parkin, C.A.; Ingham, P.W.; Cunliffe, V.T. Identification of compounds with anti-convulsant properties in a zebrafish model of epileptic seizures. Dis. Model. Mech. 2012, 5, 773–784. [Google Scholar] [CrossRef] [Green Version]
- Reza, H.M.; Mohammad, H.; Golnaz, E.; Gholamreza, S. Effect of methanolic extract of Hyoscymus niger L. on the seizure induced by picritoxin in mice. Pak. J. Pharm. Sci. 2009, 22, 308–312. [Google Scholar]
- Alfaro, J.M.; Ripoll-Gómez, J.; Burgos, J.S. Kainate administered to adult zebrafish causes seizures similar to those in rodent models. Eur. J. Neurosci. 2011, 33, 1252–1255. [Google Scholar] [CrossRef]
- Lévesque, M.; Avoli, M. The kainic acid model of temporal lobe epilepsy. Neurosci. Biobehav. Rev. 2013, 37, 2887–2899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debski, K.J.; Ceglia, N.; Ghestem, A.; Ivanov, A.I.; Brancati, G.E.; Broer, S.; Bot, A.M.; Muller, J.A.; Schoch, S.; Becker, A.; et al. The circadian dynamics of the hippocampal transcriptome and proteome is altered in experimental temporal lobe epilepsy. Sci. Adv. 2020, 6, eaat5979. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Lee, P.; Baraban, S.C.; Lee, L.P. A Novel Long-term, Multi-Channel and Non-invasive Electrophysiology Platform for Zebrafish. Sci. Rep. 2016, 6, 28248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gawel, K.; Turski, W.A.; Van Der Ent, W.; Mathai, B.J.; Kirstein-Smardzewska, K.J.; Simonsen, A.; Esguerra, C.V. Phenotypic Characterization of Larval Zebrafish (Danio rerio) with Partial Knockdown of the cacna1a Gene. Mol. Neurobiol. 2020, 57, 1904–1916. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.-H.; Sung, S.-Y.; Chang, W.-N.; Kao, T.-T.; Du, H.-C.; Hsiao, T.-H.; Safo, M.K.; Fu, T.-F. Zebrafish larvae exposed to ginkgotoxin exhibit seizure-like behavior that is relieved by pyridoxal-5′-phosphate, GABA and anti-epileptic drugs. Dis. Model. Mech. 2012, 5, 785–795. [Google Scholar] [CrossRef] [Green Version]
- Benke, T.A.; Swann, J. The tetanus toxin model of chronic epilepsy. Adv. Exp. Med. Biol. 2004, 548, 226–238. [Google Scholar] [CrossRef]
- Mitchell, J.; Gatherer, M.; Sundstrom, L.E. Loss of hilar somatostatin neurons following tetanus toxin-induced seizures. Acta Neuropathol. 1995, 89, 425–430. [Google Scholar] [CrossRef]
- Chrościńska-Krawczyk, M.; Jargiełło-Baszak, M.; Wałek, M.; Tylus, B.; Czuczwar, S.J. Caffeine and the anticonvulsant potency of antiepileptic drugs: Experimental and clinical data. Pharmacol. Rep. 2011, 63, 12–18. [Google Scholar] [CrossRef]
- Garba, K.; Yaro, A.H.; Ya’u, J. Anticonvulsant effects of ethanol stem bark extract of Lannea barteri (Anacardiaceae) in mice and chicks. J. Ethnopharmacol. 2015, 172, 227–231. [Google Scholar] [CrossRef]
- White, H.S. Clinical Significance of Animal Seizure Models and Mechanism of Action Studies of Potential Antiepileptic Drugs. Epilepsia 1997, 38, S9–S17. [Google Scholar] [CrossRef]
- Stöhr, T.; Kupferberg, H.J.; Stables, J.P.; Choi, D.; Harris, R.H.; Kohn, H.; Walton, N.; White, H.S. Lacosamide, a novel anti-convulsant drug, shows efficacy with a wide safety margin in rodent models for epilepsy. Epilepsy Res. 2007, 74, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Scholl, U.I.; Choi, M.; Liu, T.; Ramaekers, V.T.; Häusler, M.G.; Grimmer, J.; Tobe, S.W.; Farhi, A.; Nelson-Williams, C.; Lifton, R.P. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc. Natl. Acad. Sci. USA 2009, 106, 5842–5847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Shekaili, H.H.; Petkau, T.L.; Pena, I.; Lengyell, T.C.; Verhoeven-Duif, N.M.; Ciapaite, J.; Bosma, M.; van Faassen, M.; Kema, I.P.; Horvath, G.; et al. A novel mouse model for pyridoxine-dependent epilepsy due to antiquitin deficiency. Hum. Mol. Genet. 2020, 29, 3266–3284. [Google Scholar] [CrossRef] [PubMed]
- Wither, R.G.; Colic, S.; Bardakjian, B.L.; Snead, O.C., 3rd; Zhang, L.; Eubanks, J.H. Electrographic and pharmacological characterization of a progressive epilepsy phenotype in female MeCP2-deficient mice. Epilepsy Res. 2018, 140, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.H.; Mantegazza, M.; Westenbroek, R.E.; Robbins, C.A.; Kalume, F.; Burton, K.A.; Spain, W.J.; McKnight, G.S.; Scheuer, T.; Catterall, W.A. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 2006, 9, 1142–1149. [Google Scholar] [CrossRef]
- Okuda, K.; Kobayashi, S.; Fukaya, M.; Watanabe, A.; Murakami, T.; Hagiwara, M.; Sato, T.; Ueno, H.; Ogonuki, N.; Komano-Inoue, S.; et al. CDKL5 controls postsynaptic localization of GluN2B-containing NMDA receptors in the hippocampus and regulates seizure susceptibility. Neurobiol. Dis. 2017, 106, 158–170. [Google Scholar] [CrossRef]
- Gonzalez-Sulser, A. Rodent genetic models of neurodevelopmental disorders and epilepsy. Eur. J. Paediatr. Neurol. 2020, 24, 66–69. [Google Scholar] [CrossRef]
- Chabrol, E.; Navarro, V.; Provenzano, G.; Cohen, I.; Dinocourt, C.; Rivaud-Péchoux, S.; Fricker, D.; Baulac, M.; Miles, R.; LeGuern, E.; et al. Electroclinical characterization of epileptic seizures in leucine-rich, glioma-inactivated 1-deficient mice. Brain 2010, 133, 2749–2762. [Google Scholar] [CrossRef]
- Miura, K.; Kishino, T.; Li, E.; Webber, H.; Dikkes, P.; Holmes, G.L.; Wagstaff, J. Neurobehavioral and Electroencephalographic Abnormalities in Ube3aMaternal-Deficient Mice. Neurobiol. Dis. 2002, 9, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Shin, W.; Kweon, H.; Kang, R.; Kim, D.; Kim, K.; Kang, M.; Kim, S.Y.; Hwang, S.N.; Kim, J.Y.; Yang, E.; et al. Scn2a Haploinsufficiency in Mice Suppresses Hippocampal Neuronal Excitability, Excitatory Synaptic Drive, and Long-Term Potentiation, and Spatial Learning and Memory. Front. Mol. Neurosci. 2019, 12, 145. [Google Scholar] [CrossRef] [Green Version]
- Wagnon, J.L.; Korn, M.J.; Parent, R.; Tarpey, T.A.; Jones, J.M.; Hammer, M.F.; Murphy, G.G.; Parent, J.M.; Meisler, M.H. Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Hum. Mol. Genet. 2015, 24, 506–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Malley, H.A.; Hull, J.M.; Clawson, B.C.; Chen, C.; Owens-Fiestan, G.; Jameson, M.B.; Aton, S.J.; Parent, J.M.; Isom, L.L. Scn1b deletion in adult mice results in seizures and SUDEP. Ann. Clin. Transl. Neurol. 2019, 6, 1121–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.A.; Otto, J.F.; Dahle, E.J.; Pappas, C.; Leslie, J.D.; Vilaythong, A.; Noebels, J.L.; White, H.S.; Wilcox, K.S.; Leppert, M.F. Mouse models of human KCNQ2 and KCNQ3 mutations for benign familial neonatal convulsions show seizures and neuronal plasticity without synaptic reorganization. J. Physiol. 2008, 586, 3405–3423. [Google Scholar] [CrossRef] [PubMed]
- Simeone, K.A.; Hallgren, J.; Bockman, C.S.; Aggarwal, A.; Kansal, V.; Netzel, L.; Iyer, S.H.; Matthews, S.A.; Deodhar, M.; Oldenburg, P.J.; et al. Respiratory dysfunction progresses with age in Kcna1-null mice, a model of sudden unexpected death in epilepsy. Epilepsia 2018, 59, 345–357. [Google Scholar] [CrossRef] [Green Version]
- Robbins, C.A.; Tempel, B.L. Kv1.1 and Kv1.2: Similar channels, different seizure models. Epilepsia 2012, 53 (Suppl. 1), 134–141. [Google Scholar] [CrossRef]
- Whitmire, L.E.; Ling, L.; Bugay, V.; Carver, C.M.; Timilsina, S.; Chuang, H.H.; Jaffe, D.B.; Shapiro, M.S.; Cavazos, J.E.; Brenner, R. Downregulation of KCNMB4 expression and changes in BK channel subtype in hippocampal granule neurons following seizure activity. PLoS ONE 2017, 12, e0188064. [Google Scholar] [CrossRef] [Green Version]
- Mark, M.D.; Maejima, T.; Kuckelsberg, D.; Yoo, J.W.; Hyde, R.A.; Shah, V.; Gutierrez, D.; Moreno, R.L.; Kruse, W.; Noebels, J.L.; et al. Delayed postnatal loss of P/Q-type calcium channels recapitulates the absence epilepsy, dyskinesia, and ataxia phenotypes of genomic Cacna1a mutations. J. Neurosci. 2011, 31, 4311–4326. [Google Scholar] [CrossRef] [Green Version]
- Machnes, Z.M.; Huang, T.C.; Chang, P.K.; Gill, R.; Reist, N.; Dezsi, G.; Ozturk, E.; Charron, F.; O’Brien, T.J.; Jones, N.C.; et al. DNA methylation mediates persistent epileptiform activity in vitro and in vivo. PLoS ONE 2013, 8, e76299. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Okada, M.; Yoshida, S.; Ueno, S.; Mori, F.; Takahara, T.; Saito, R.; Miura, Y.; Kishi, A.; Tomiyama, M.; et al. Rats harboring S284L Chrna4 mutation show attenuation of synaptic and extrasynaptic GABAergic transmission and exhibit the nocturnal frontal lobe epilepsy phenotype. J. Neurosci. 2008, 28, 12465–12476. [Google Scholar] [CrossRef] [Green Version]
- Warner, T.A.; Liu, Z.; Macdonald, R.L.; Kang, J.Q. Heat induced temperature dysregulation and seizures in Dravet Syndrome/GEFS+ Gabrg2(+/Q390X) mice. Epilepsy Res. 2017, 134, 1–8. [Google Scholar] [CrossRef]
- Puranam, R.S.; He, X.P.; Yao, L.; Le, T.; Jang, W.; Rehder, C.W.; Lewis, D.V.; McNamara, J.O. Disruption of Fgf13 causes synaptic excitatory-inhibitory imbalance and genetic epilepsy and febrile seizures plus. J. Neurosci. 2015, 35, 8866–8881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, D.L.; Thomas, D.; Galvan, V.; Bredesen, D.E.; Lamb, B.T.; Pimplikar, S.W. Abnormal neuronal networks and seizure susceptibility in mice overexpressing the APP intracellular domain. Neurobiol. Aging 2011, 32, 1725–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grier, M.D.; Carson, R.P.; Lagrange, A.H. Toward a Broader View of Ube3a in a Mouse Model of Angelman Syndrome: Expression in Brain, Spinal Cord, Sciatic Nerve and Glial Cells. PLoS ONE 2015, 10, e0124649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.; Zhang, Y.; Kim, S.; Kim, Y.; Lee, Y.; Han, K. Spontaneous seizure and partial lethality of juvenile Shank3-overexpressing mice in C57BL/6 J background. Mol. Brain 2018, 11, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peñagarikano, O.; Abrahams, B.S.; Herman, E.I.; Winden, K.D.; Gdalyahu, A.; Dong, H.; Sonnenblick, L.I.; Gruver, R.; Almajano, J.; Bragin, A.; et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 2011, 147, 235–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesh, S.; Delgado-Escueta, A.V.; Sakamoto, T.; Avila, M.R.; Machado-Salas, J.; Hoshii, Y.; Akagi, T.; Gomi, H.; Suzuki, T.; Amano, K.; et al. Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum. Mol. Genet. 2002, 11, 1251–1262. [Google Scholar] [CrossRef] [Green Version]
- Wagnon, J.L.; Mahaffey, C.L.; Sun, W.; Yang, Y.; Chao, H.T.; Frankel, W.N. Etiology of a genetically complex seizure disorder in Celf4 mutant mice. Genes Brain Behav. 2011, 10, 765–777. [Google Scholar] [CrossRef] [Green Version]
- Acampora, D.; Mazan, S.; Avantaggiato, V.; Barone, P.; Tuorto, F.; Lallemand, Y.; Brûlet, P.; Simeone, A. Epilepsy and brain abnormalities in mice lacking the Otx1 gene. Nat. Genet. 1996, 14, 218–222. [Google Scholar] [CrossRef]
- Menten-Dedoyart, C.; Serrano Navacerrada, M.E.; Bartholome, O.; Sánchez Gil, J.; Neirinckx, V.; Wislet, S.; Becker, G.; Plenevaux, A.; Van den Ackerveken, P.; Rogister, B. Development and Validation of a New Mouse Model to Investigate the Role of SV2A in Epilepsy. PLoS ONE 2016, 11, e0166525. [Google Scholar] [CrossRef]
- Hu, H.; Zhu, T.; Gong, L.; Zhao, Y.; Shao, Y.; Li, S.; Sun, Z.; Ling, Y.; Tao, Y.; Ying, Y.; et al. Transient receptor potential melastatin 2 contributes to neuroinflammation and negatively regulates cognitive outcomes in a pilocarpine-induced mouse model of epilepsy. Int. Immunopharmacol. 2020, 87, 106824. [Google Scholar] [CrossRef]
- Zhang, D.; Yuan, C.; Liu, M.; Zhou, X.; Ge, S.; Wang, X.; Luo, G.; Hou, M.; Liu, Z.; Wang, Q.K.; et al. Deficiency of SCAMP5 leads to pediatric epilepsy and dysregulation of neurotransmitter release in the brain. Hum. Genet. 2020, 139, 545–555. [Google Scholar] [CrossRef]
- Mota Vieira, M.; Nguyen, T.A.; Wu, K.; Badger, J.D., 2nd; Collins, B.M.; Anggono, V.; Lu, W.; Roche, K.W. An Epilepsy-Associated GRIN2A Rare Variant Disrupts CaMKIIα Phosphorylation of GluN2A and NMDA Receptor Trafficking. Cell Rep. 2020, 32, 108104. [Google Scholar] [CrossRef] [PubMed]
- Klofas, L.K.; Short, B.P.; Zhou, C.; Carson, R.P. Prevention of premature death and seizures in a Depdc5 mouse epilepsy model through inhibition of mTORC1. Hum. Mol. Genet. 2020, 29, 1365–1377. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.; Ren, S.; Gao, P.; Wan, D.; Rong, S.; Li, X.; Liu, S.; Xu, S.; Sun, K.; Guo, B.; et al. ALG13 participates in epileptogenesis via regulation of GABA(A) receptors in mouse models. Cell Death Discov. 2020, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, A.; Kunisawa, N.; Sugimura, T.; Sato, K.; Yoshida, Y.; Suzuki, T.; Sakuma, T.; Yamamoto, T.; Asano, M.; Saito, Y.; et al. Loss of HCN1 subunits causes absence epilepsy in rats. Brain Res. 2019, 1706, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Baraban, S.; Taylor, M.; Castro, P.; Baier, H. Pentylenetetrazole induced changes in zebrafish behavior, neural activity and c-fos expression. Neuroscience 2005, 131, 759–768. [Google Scholar] [CrossRef]
- Lopes, M.W.; Sapio, M.R.; Leal, R.B.; Fricker, L.D. Knockdown of Carboxypeptidase A6 in Zebrafish Larvae Reduces Response to Seizure-Inducing Drugs and Causes Changes in the Level of mRNAs Encoding Signaling Molecules. PLoS ONE 2016, 11, e0152905. [Google Scholar] [CrossRef]
- Vermoesen, K.; Serruys, A.-S.K.; Loyens, E.; Afrikanova, T.; Massie, A.; Schallier, A.; Michotte, Y.; Crawford, A.D.; Esguerra, C.V.; de Witte, P.A.M.; et al. Assessment of the convulsant liability of antidepressants using zebrafish and mouse seizure models. Epilepsy Behav. 2011, 22, 450–460. [Google Scholar] [CrossRef]
- Gawel, K.; Langlois, M.; Martins, T.; van der Ent, W.; Tiraboschi, E.; Jacmin, M.; Crawford, A.D.; Esguerra, C.V. Seizing the moment: Zebrafish epilepsy models. Neurosci. Biobehav. Rev. 2020, 116, 1–20. [Google Scholar] [CrossRef]
- Baraban, S.C.; Dinday, M.T.; Hortopan, G.A. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat. Commun. 2013, 4, 2410. [Google Scholar] [CrossRef] [Green Version]
- Samarut, E.; Swaminathan, A.; Riche, R.; Liao, M.; Hassan-Abdi, R.; Renault, S.; Allard, M.; Dufour, L.; Cossette, P.; Soussi-Yanicostas, N.; et al. gamma-Aminobutyric acid receptor alpha 1 subunit loss of function causes genetic generalized epilepsy by impairing inhibitory network neurodevelopment. Epilepsia 2018, 59, 2061–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, M.; Kundap, U.; Rosch, R.E.; Burrows, D.R.W.; Meyer, M.P.; Bencheikh, B.O.A.; Cossette, P.; Samarut, É. Targeted knockout of GABA receptor gamma 2 subunit provokes transient light-induced reflex seizures in zebrafish larvae. Dis. Model. Mech. 2019, 12, dmm.040782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmood, F.; Mozere, M.; Zdebik, A.A.; Stanescu, H.C.; Tobin, J.; Beales, P.L.; Kleta, R.; Bockenhauer, D.; Russell, C. Generation and validation of a zebrafish model of EAST (epilepsy, ataxia, sensorineural deafness and tubulopathy) syndrome. Dis. Model. Mech. 2013, 6, 652–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chege, S.W.; Hortopan, G.A.; Dinday, M.T.; Baraban, S.C. Expression and function of KCNQ channels in larval zebrafish. Dev. Neurobiol. 2012, 72, 186–198. [Google Scholar] [CrossRef]
- Schubert, J.; Siekierska, A.; Langlois, M.; May, P.; Huneau, C.; Becker, F.; Muhle, H.; Suls, A.; Lemke, J.R.; de Kovel, C.G.F.; et al. Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat. Genet. 2014, 46, 1327–1332. [Google Scholar] [CrossRef]
- Carvill, G.L.; Heavin, S.B.; Yendle, S.C.; McMahon, J.M.; O’Roak, B.J.; Cook, J.; Khan, A.; Dorschner, M.O.; Weaver, M.; Calvert, S.; et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet. 2013, 45, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Griffin, A.; Carpenter, C.; Liu, J.; Paterno, R.; Grone, B.; Hamling, K.; Moog, M.; Dinday, M.T.; Figueroa, F.; Anvar, M.; et al. Phenotypic analysis of catastrophic childhood epilepsy genes. Commun. Biol. 2021, 4, 680. [Google Scholar] [CrossRef]
- Teng, Y.; Xie, X.; Walker, S.; Rempala, G.; Kozlowski, D.J.; Mumm, J.S.; Cowell, J.K. Knockdown of zebrafish Lgi1a results in abnormal development, brain defects and a seizure-like behavioral phenotype. Hum. Mol. Genet. 2010, 19, 4409–4420. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, A.; Hassan-Abdi, R.; Renault, S.; Siekierska, A.; Riché, R.; Liao, M.; de Witte, P.A.M.; Yanicostas, C.; Soussi-Yanicostas, N.; Drapeau, P.; et al. Non-canonical mTOR-Independent Role of DEPDC5 in Regulating GABAergic Network Development. Curr. Biol. 2018, 28, 1924–1937. [Google Scholar] [CrossRef] [Green Version]
- Weckhuysen, S.; Korff, C.M. Epilepsy: Old syndromes, new genes. Curr. Neurol. Neurosci. Rep. 2014, 14, 447. [Google Scholar] [CrossRef]
- Harkin, L.A.; McMahon, J.M.; Iona, X.; Dibbens, L.; Pelekanos, J.T.; Zuberi, S.M.; Sadleir, L.G.; Andermann, E.; Gill, D.; Farrell, K.; et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain 2007, 130, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, K. Molecular and cellular basis: Insights from experimental models of Dravet syndrome. Epilepsia 2011, 52 (Suppl. 2), 70–71. [Google Scholar] [CrossRef] [PubMed]
- Jansen, N.A.; Dehghani, A.; Breukel, C.; Tolner, E.A.; Van Den Maagdenberg, A. Focal and generalized seizure activity after local hippocampal or cortical ablation of Na(V) 1.1 channels in mice. Epilepsia 2020, 61, e30–e36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grone, B.P.; Qu, T.; Baraban, S.C. Behavioral Comorbidities and Drug Treatments in a Zebrafish scn1lab Model of Dravet Syndrome. eNeuro 2017, 4, eneuro.0066. [Google Scholar] [CrossRef] [Green Version]
- Calhoun, J.D.; Hawkins, N.A.; Zachwieja, N.J.; Kearney, J.A. Cacna1g is a genetic modifier of epilepsy caused by mutation of voltage-gated sodium channel Scn2a. Epilepsia 2016, 57, e103–e107. [Google Scholar] [CrossRef] [Green Version]
- Makinson, C.D.; Tanaka, B.S.; Sorokin, J.M.; Wong, J.C.; Christian, C.A.; Goldin, A.L.; Escayg, A.; Huguenard, J.R. Regulation of Thalamic and Cortical Network Synchrony by Scn8a. Neuron 2017, 93, 1165–1179. [Google Scholar] [CrossRef] [Green Version]
- Allen, N.M.; Mannion, M.; Conroy, J.; Lynch, S.A.; Shahwan, A.; Lynch, B.; King, M.D. The variable phenotypes of KCNQ-related epilepsy. Epilepsia 2014, 55, e99–e105. [Google Scholar] [CrossRef]
- Olsen, M.L.; Sontheimer, H. Functional implications for Kir4.1 channels in glial biology: From K+ buffering to cell differentiation. J. Neurochem. 2008, 107, 589–601. [Google Scholar] [CrossRef] [Green Version]
- Reichold, M.; Zdebik, A.A.; Lieberer, E.; Rapedius, M.; Schmidt, K.; Bandulik, S.; Sterner, C.; Tegtmeier, I.; Penton, D.; Baukrowitz, T.; et al. KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc. Natl. Acad. Sci. USA 2010, 107, 14490–14495. [Google Scholar] [CrossRef] [Green Version]
- Raol, Y.H.; Lund, I.V.; Bandyopadhyay, S.; Zhang, G.; Roberts, D.S.; Wolfe, J.H.; Russek, S.J.; Brooks-Kayal, A.R. Enhancing GABA(A) receptor alpha 1 subunit levels in hippocampal dentate gyrus inhibits epilepsy development in an animal model of temporal lobe epilepsy. J. Neurosci. 2006, 26, 11342–11346. [Google Scholar] [CrossRef] [Green Version]
- Gregg, N.M.; Sladky, V.; Nejedly, P.; Mivalt, F.; Kim, I.; Balzekas, I.; Sturges, B.K.; Crowe, C.; Patterson, E.E.; Van Gompel, J.J.; et al. Thalamic deep brain stimulation modulates cycles of seizure risk in epilepsy. Sci. Rep. 2021, 11, 24250. [Google Scholar] [CrossRef] [PubMed]
- Bajorat, R.; Wilde, M.; Sellmann, T.; Kirschstein, T.; Köhling, R. Seizure frequency in pilocarpine-treated rats is independent of circadian rhythm. Epilepsia 2011, 52, e118–e122. [Google Scholar] [CrossRef]
- Quigg, M.; Clayburn, H.; Straume, M.; Menaker, M.; Bertram, E.H., 3rd. Effects of circadian regulation and rest-activity state on spontaneous seizures in a rat model of limbic epilepsy. Epilepsia 2000, 41, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Quigg, M.; Straume, M.; Smith, T.; Menaker, M.; Bertram, E.H. Seizures induce phase shifts of rat circadian rhythms. Brain Res. 2001, 913, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Matzen, J.; Buchheim, K.; Holtkamp, M. Circadian dentate gyrus excitability in a rat model of temporal lobe epilepsy. Exp. Neurol. 2012, 234, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Strand, A.; Aragaki, A.; Baquet, Z.; Hodges, A.; Cunningham, P.; Holmans, P.; Jones, K.; Kooperberg, C.; Olson, J. Conservation of Regional Gene Expression in Mouse and Human Brain. PLoS Genet. 2007, 3, e59. [Google Scholar] [CrossRef] [Green Version]
- Waterston, R.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; An, P.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2003, 420, 520–562. [Google Scholar] [CrossRef] [Green Version]
- Amendola, E.; Zhan, Y.; Mattucci, C.; Castroflorio, E.; Calcagno, E.; Fuchs, C.; Lonetti, G.; Silingardi, D.; Vyssotski, A.; Farley, D.; et al. Mapping Pathological Phenotypes in a Mouse Model of CDKL5 Disorder. PLoS ONE 2014, 9, e91613. [Google Scholar] [CrossRef]
- Bunton-Stasyshyn, R.K.A.; Wagnon, J.L.; Wengert, E.R.; Barker, B.S.; Faulkner, A.; Wagley, P.K.; Bhatia, K.; Jones, J.M.; Maniaci, M.R.; Parent, J.M.; et al. Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain 2019, 142, 362–375. [Google Scholar] [CrossRef] [Green Version]
- Kerjan, G.; Koizumi, H.; Han, E.B.; Dubé, C.M.; Djakovic, S.N.; Patrick, G.N.; Baram, T.Z.; Heinemann, S.F.; Gleeson, J.G. Mice lacking doublecortin and doublecortin-like kinase 2 display altered hippocampal neuronal maturation and spontaneous seizures. Proc. Natl. Acad. Sci. USA 2009, 106, 6766–6771. [Google Scholar] [CrossRef] [Green Version]
- MacRae, C.A.; Peterson, R.T. Zebrafish as tools for drug discovery. Nat. Reviews. Drug Discov. 2015, 14, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Hortopan, G.A.; Dinday, M.T.; Baraban, S.C. Zebrafish as a model for studying genetic aspects of epilepsy. Dis. Model. Mech. 2010, 3, 144–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.; Stewart, A.; Gilder, T.; Wu, N.; Frank, K.; Gaikwad, S.; Suciu, C.; Dileo, J.; Utterback, E.; Chang, K.; et al. Modeling seizure-related behavioral and endocrine phenotypes in adult zebrafish. Brain Res. 2010, 1348, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Rosch, R.E.; Hunter, P.R.; Baldeweg, T.; Friston, K.J.; Meyer, M.P. Calcium imaging and dynamic causal modelling reveal brain-wide changes in effective connectivity and synaptic dynamics during epileptic seizures. PLoS Comput. Biol. 2018, 14, e1006375. [Google Scholar] [CrossRef]
- Randlett, O.; Wee, C.L.; Naumann, E.A.; Nnaemeka, O.; Schoppik, D.; Fitzgerald, J.E.; Portugues, R.; Lacoste, A.M.; Riegler, C.; Engert, F.; et al. Whole-brain activity mapping onto a zebrafish brain atlas. Nat. Methods 2015, 12, 1039–1046. [Google Scholar] [CrossRef] [Green Version]
- Brunal, A.A.; Clark, K.C.; Ma, M.; Woods, I.G.; Pan, Y.A. Effects of Constitutive and Acute Connexin 36 Deficiency on Brain-Wide Susceptibility to PTZ-Induced Neuronal Hyperactivity. Front. Mol. Neurosci. 2020, 13, 587978. [Google Scholar] [CrossRef]
- Stewart, A.M.; Desmond, D.; Kyzar, E.; Gaikwad, S.; Roth, A.; Riehl, R.; Collins, C.; Monnig, L.; Green, J.; Kalueff, A.V. Perspectives of zebrafish models of epilepsy: What, how and where next? Brain Res. Bull. 2012, 87, 135–143. [Google Scholar] [CrossRef]
- Baraban, S. Modeling Epilepsy and Seizures in Developing Zebrafish Larvae. In Models of Seizures and Epilepsy, 1st ed.; Asla Pitkänen, A., Schwartzkroin, P., Moshé, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; pp. 189–198. [Google Scholar] [CrossRef]
- Zhong, X.L.; Yu, J.T.; Zhang, Q.; Wang, N.D.; Tan, L. Deep brain stimulation for epilepsy in clinical practice and in animal models. Brain Res. Bull. 2011, 85, 81–88. [Google Scholar] [CrossRef]
- Yaksi, E.; Jamali, A.; Diaz Verdugo, C.; Jurisch-Yaksi, N. Past, present and future of zebrafish in epilepsy research. FEBS J. 2021, 288, 7243–7255. [Google Scholar] [CrossRef]
- Kalsbeek, A.; Van Der Spek, R.; Lei, J.; Endert, E.; Buijs, R.M.; Fliers, E. Circadian rhythms in the hypothalamo-pituitary-adrenal (HPA) axis. Mol. Cell. Endocrinol. 2012, 349, 20–29. [Google Scholar] [CrossRef]
- Androulakis, I.P. Circadian rhythms and the HPA axis: A systems view. WIREs Mech. Dis. 2021, 13, e1518. [Google Scholar] [CrossRef]
- Sen, A.; Sellix, M.T. The Circadian Timing System and Environmental Circadian Disruption: From Follicles to Fertility. Endocrinology 2016, 157, 3366–3373. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Zhao, H.; Lu, Z.; Lei, X.; Zhang, Y. Circadian Rhythms Within the Female HPG Axis: From Physiology to Etiology. Endocrinology 2021, 162, bqab117. [Google Scholar] [CrossRef] [PubMed]
- Fawley, J.A.; Pouliot, W.A.; Dudek, F.E. Epilepsy and reproductive disorders: The role of the gonadotropin-releasing hormone network. Epilepsy Behav. 2006, 8, 477–482. [Google Scholar] [CrossRef]
- Peng, B.W.; Li, X.J.; Wu, W.X.; Zeng, Y.R.; Liao, Y.T.; Hou, C.; Liang, H.C.; Zhang, W.; Wang, X.Y.; Chen, W.X. The Possible Role of Hypothalamus-Pituitary-Adrenal Dysfunction in Epileptic Spasms. Seizure 2020, 81, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Basu, T.; Maguire, J.; Salpekar, J.A. Hypothalamic-pituitary-adrenal axis targets for the treatment of epilepsy. Neurosci. Lett. 2021, 746, 135618. [Google Scholar] [CrossRef]
- Sun, S.; Wang, H. Disrupting the circadian dynamics of epileptic genes in mouse temporal lobe epilepsy. Front. Mol. Neurosci. 2022, 11, 751. [Google Scholar]
- Wen, Z.P.; Fan, S.S.; Du, C.; Yin, T.; Zhou, B.T.; Peng, Z.F.; Xie, Y.Y.; Zhang, W.; Chen, Y.; Tang, J.; et al. Influence of acylpeptide hydrolase polymorphisms on valproic acid level in Chinese epilepsy patients. Pharmacogenomics 2016, 17, 1219–1225. [Google Scholar] [CrossRef]
- Musiek, E.S.; Fitzgerald, G.A. Molecular clocks in pharmacology. Handb. Exp. Pharmacol. 2013, 217, 243–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.; Wang, Z.; Wu, B. Pharmacokinetics-based Chronotherapy. Curr. Drug. Metab. 2022, 23, 2–7. [Google Scholar] [CrossRef]
- Ruben, M.D.; Smith, D.F.; FitzGerald, G.A.; Hogenesch, J.B. Dosing time matters. Science 2019, 365, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, J.M.; Bouzom, F.; Hugues, C.; Ungell, A.L. Oral drug absorption in pediatrics: The intestinal wall, its developmental changes and current tools for predictions. Biopharm. Drug Dispos. 2017, 38, 209–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konturek, P.C.; Brzozowski, T.; Konturek, S.J. Gut clock: Implication of circadian rhythms in the gastrointestinal tract. J. Physiol. Pharmacol. 2011, 62, 139–150. [Google Scholar]
- Kurishima, A.; Hayashi, M.; Shimozato, R.; Isozaki, R.; Shioda, T.; Iijima, A. Successful Treatment of Symptomatic Epilepsy with Oral Valproic Acid and Levetiracetam in a Patient with Short-bowel Syndrome: A Case Report. Intern. Med. 2021, 61, 1457–1461. [Google Scholar] [CrossRef]
- Lee, K.; Kim, N.; Shim, J.O.; Kim, G.H. Gut Bacterial Dysbiosis in Children with Intractable Epilepsy. J. Clin. Med. 2020, 10, 5. [Google Scholar] [CrossRef]
- Patel, I.H.; Venkataramanan, R.; Levy, R.H.; Viswanathan, C.T.; Ojemann, L.M. Diurnal oscillations in plasma protein binding of valproic acid. Epilepsia 1982, 23, 283–290. [Google Scholar] [CrossRef]
- Riva, R.; Albani, F.; Ambrosetto, G.; Contin, M.; Cortelli, P.; Perucca, E.; Baruzzi, A. Diurnal fluctuations in free and total steady-state plasma levels of carbamazepine and correlation with intermittent side effects. Epilepsia 1984, 25, 476–481. [Google Scholar] [CrossRef]
- Ando, H.; Yanagihara, H.; Sugimoto, K.; Hayashi, Y.; Tsuruoka, S.; Takamura, T.; Kaneko, S.; Fujimura, A. Daily rhythms of P-glycoprotein expression in mice. Chronobiol. Int. 2005, 22, 655–665. [Google Scholar] [CrossRef]
- Pácha, J.; Balounová, K.; Soták, M. Circadian regulation of transporter expression and implications for drug disposition. Expert Opin. Drug Metab. Toxicol. 2021, 17, 425–439. [Google Scholar] [CrossRef]
- Zhao, M.; Xing, H.; Chen, M.; Dong, D.; Wu, B. Circadian clock-controlled drug metabolism and transport. Xenobiotica Fate Foreign Compd. Biol. Syst. 2020, 50, 495–505. [Google Scholar] [CrossRef]
- Gumz, M.L.; Stow, L.R.; Lynch, I.J.; Greenlee, M.M.; Rudin, A.; Cain, B.D.; Weaver, D.R.; Wingo, C.S. The circadian clock protein Period 1 regulates expression of the renal epithelial sodium channel in mice. J. Clin. Investig. 2009, 119, 2423–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuber, A.M.; Centeno, G.; Pradervand, S.; Nikolaeva, S.; Maquelin, L.; Cardinaux, L.; Bonny, O.; Firsov, D. Molecular clock is involved in predictive circadian adjustment of renal function. Proc. Natl. Acad. Sci. USA 2009, 106, 16523–16528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriksson, E.; Huber, A.L.; Soto, E.K.; Kriebs, A.; Vaughan, M.E.; Duglan, D.; Chan, A.B.; Papp, S.J.; Nguyen, M.; Afetian, M.E.; et al. The Liver Circadian Clock Modulates Biochemical and Physiological Responses to Metformin. J. Biol. Rhythm. 2017, 32, 345–358. [Google Scholar] [CrossRef]
- Fauteck, J.; Schmidt, H.; Lerchl, A.; Kurlemann, G.; Wittkowski, W. Melatonin in epilepsy: First results of replacement therapy and first clinical results. Biol. Signals Recept. 1999, 8, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Cederroth, C.R.; Albrecht, U.; Bass, J.; Brown, S.A.; Dyhrfjeld-Johnsen, J.; Gachon, F.; Green, C.B.; Hastings, M.H.; Helfrich-Forster, C.; Hogenesch, J.B.; et al. Medicine in the Fourth Dimension. Cell. Metab. 2019, 30, 238–250. [Google Scholar] [CrossRef]
- Allada, R.; Bass, J. Circadian Mechanisms in Medicine. N. Engl. J. Med. 2021, 384, 550–561. [Google Scholar] [CrossRef]
- Kramer, A.; Lange, T.; Spies, C.; Finger, A.M.; Berg, D.; Oster, H. Foundations of circadian medicine. PLoS Biol. 2022, 20, e3001567. [Google Scholar] [CrossRef]
- Selfridge, J.M.; Gotoh, T.; Schiffhauer, S.; Liu, J.; Stauffer, P.E.; Li, A.; Capelluto, D.G.; Finkielstein, C.V. Chronotherapy: Intuitive, Sound, Founded... But Not Broadly Applied. Drugs 2016, 76, 1507–1521. [Google Scholar] [CrossRef] [Green Version]
- Torres-Ruiz, R.; Rodriguez-Perales, S. CRISPR-Cas9 technology: Applications and human disease modelling. Brief. Funct. Genom. 2017, 16, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Fuller, P.M.; Gooley, J.J.; Saper, C.B. Neurobiology of the sleep-wake cycle: Sleep architecture, circadian regulation, and regulatory feedback. J. Biol. Rhythm. 2006, 21, 482–493. [Google Scholar] [CrossRef]
- Fountain, N.B.; Kim, J.S.; Lee, S.I. Sleep deprivation activates epileptiform discharges independent of the activating effects of sleep. J. Clin. Neurophysiol. 1998, 15, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Thijs, R.D.; Surges, R.; O’Brien, T.J.; Sander, J.W. Epilepsy in adults. Lancet 2019, 393, 689–701. [Google Scholar] [CrossRef]
- Yu, W.; Gwinn, M.; Dotson, W.D.; Green, R.F.; Clyne, M.; Wulf, A.; Bowen, S.; Kolor, K.; Khoury, M.J. A knowledge base for tracking the impact of genomics on population health. Genet. Med. 2016, 18, 1312–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
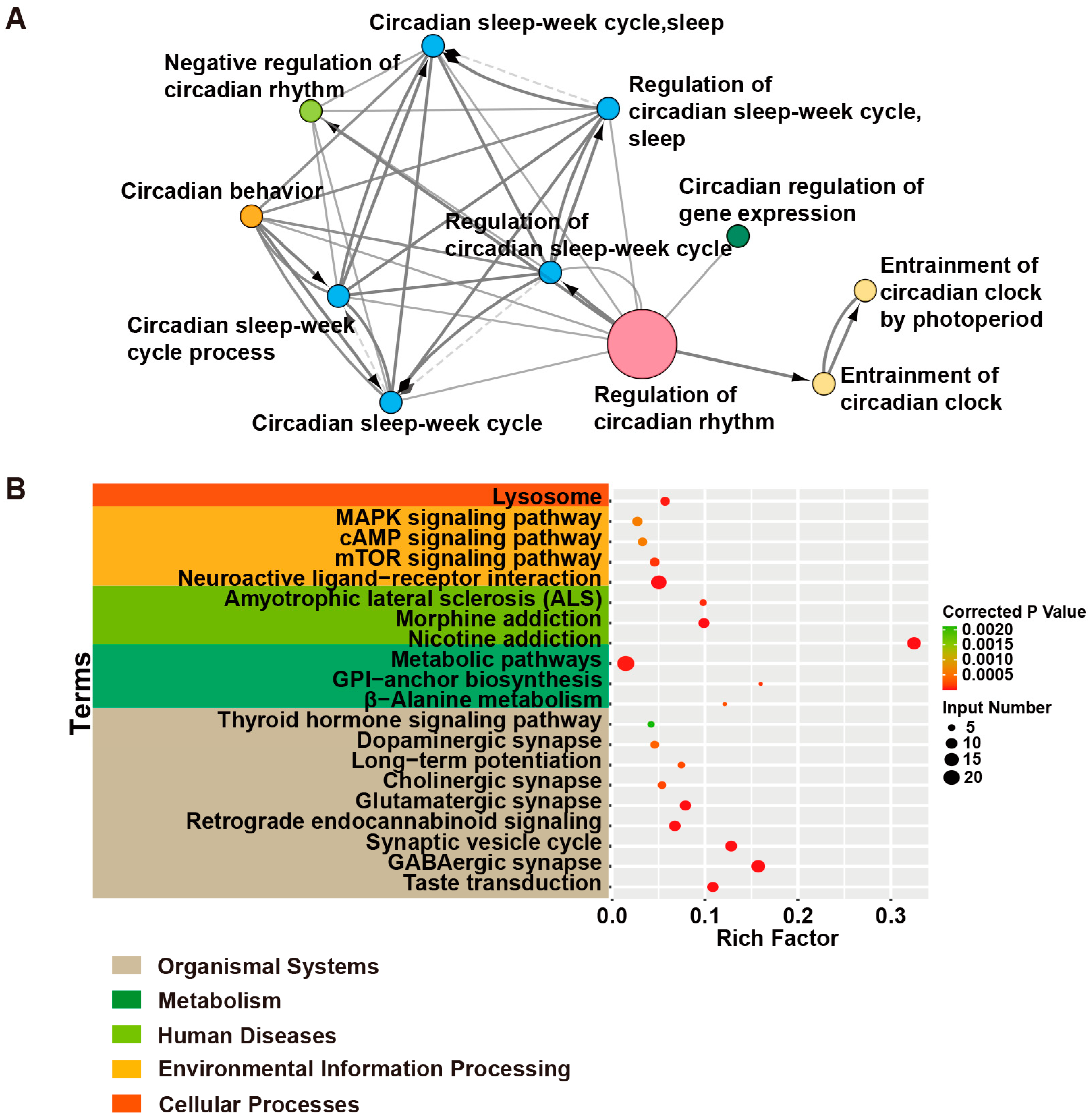
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]





| Terms | Input Genes |
|---|---|
| Taste transduction | CACNA1A, HCN4, GABRA5, SCN2A, GEFSP7, GABBR2, GABRA6, GABRA1, GABRA2 |
| GABAergic synapse | SLC6A1, GABRA6, SLC38A3, GABRG2, GAD1, SLC12A5, ABAT, CACNA1A, GABRA5, GABBR2, GABRB3, GABRA1, GABRB2, GABRA2 |
| Synaptic vesicle cycle | ATP6V0C, SLC6A1, STX1B, ATP6V1A, CACNA1A, STXBP1, CPLX1, DNM1, ATP6V0A1, SLC1A2 |
| Retrograde endocannabinoid signaling | PLCB1, GABRA6, GRIA2, GABRG2, CACNA1A, GABRA5, GABRB2, GABRB3, GABRA1, GABRA2 |
| Glutamatergic synapse | PLCB1, SLC38A3, GRIA2, CACNA1A, GRIK2, GRIN2A, PPP3CA, GRIN1, SLC1A2 |
| Cholinergic synapse | PLCB1, CACNA1A, CHRNB2, KCNQ2, KCNQ3, CHRNA4 |
| Long-term potentiation | GRIA2, PPP3CA, GRIN1, GRIN2A, PLCB1 |
| Dopaminergic synapse | PLCB1, GRIA2, CACNA1A, GRIN2A, SCN1A, PPP3CA |
| Thyroid hormone signaling pathway | PDPK1, NOTCH3, MTOR, SLC2A1, PLCB1 |
| β-Alanine metabolism | ABAT, GAD1, ALDH2, ALDH7A1 |
| Glycosylphosphatidylinositol (GPI)-anchor biosynthesis | PIGP, PIGQ, PIGS, PIGA |
| Metabolic pathways | ST3GAL3, PIGA, ATP6V0C, ATP6V1A, ASAH1, PNPO, PLCB1, ATP6V0C, ACP1, ALDH2, PIGP, PIGQ, PIGS, SYNJ1, MDH2, ABAT, ALDH7A1, CAD, ALG14, GAD1, UGP2 |
| Amyotrophic lateral sclerosis (ALS) | GRIA2, PPP3CA, GRIN2A, SLC1A2, GRIN1 |
| Nicotine addiction | GABRA6, GRIA2, GABRG2, CHRNB2, CHRNA4, GRIN2A, CACNA1A, GABRA5, GRIN1, GABRB3, GABRA1, GABRB2, GABRA2 |
| Morphine addiction | GABRA6, GABRG2, CACNA1A, GABRA5, GABBR2, GABRB3, GABRA1, GABRB2, GABRA2 |
| Neuroactive ligand–receptor interaction | GABRA6, GRIA2, GABRG2, GRIK2, CHRNB2, GABRB2, GLUD1, LEPR, CHRNA4, GRIN2A, CHRNA2, GABRA5, GRIN1, GABRB3, GABRA1, GABBR2, GABRA2 |
| mTOR signaling pathway | ATP6V1A, MTOR, DEPDC5, PDPK1, STRADA, NPRL3, NPRL2 |
| cAMP signaling pathway | GRIA2, HCN2, HCN4, GLI3, GRIN2A, GRIN1, GABBR2 |
| MAPK signaling pathway | RAPGEF2, CACNA1A, CACNA1E, NTRK2, CACNB4, MEF2C, EJM4, PPP3CA |
| Lysosome | MFSD8, AP3B2, ASAH1, ATP6V0C, SCARB2, ATP6V0A1, TPP1 |
| Others | SZT2, DOCK7, HNRNPU, GEFSP4, GEFSP8, GEFSP6, SLC25A12, SLC25A22, LNPK, EJM9, EJM3, ETL6, ETL3, TRAK1, P4HTM, DALRD3, UBA5, YEATS2, TNRC6A, MARCH6, EPPS, HWE1, HWE2, SPATA5, CYFIP2, PHACTR1, CNPY3, ICK, ACTL6B, RHOBTB2, PLPBP, OXR1, EIG1, EIG2, EIG3, EIG4, EIG5, EIG7, ETL4, ETL2, FRRS1L, KCNC1, DENND5A, TRAPPC4, PACS2, DMXL2, AARS1, SEMA6B, SYN1, SIK1, EEF1A2, SCN8A, LGI1, ARHGEF9, HCN1, NHLRC1, ADAM22, ADAM10, KIF3C, SMS, PDPK1(PDK1), CDYL, KCNV2, MECP2, NIPA1, SYNGAP1, EJM1, EJM2, CYFIP1, PCDH19, TRAPPC6B, SAMD12, LGI4, TBC1D24, SETD1A, CDKL5, EFHC1, POLG, NRXN1, CNTNAP2, EPM2A, ARX, KCNA2, FOXG1, CSTB, CHRNA7, SLC9A6, KCNAB1, ZEB2, SMC1A, REST, NR2F1, MYH1, KCNT2, ARV1, CASR, GUF1, PRDM8, YWHAG, NECAP1, SLC13A5, GOSR2, LMNB2, KCNB1, CLN8, FGF12, SATB2, KCNMA1, SCN1B, KCNE1, KCNMB3, FGF12, NCDN, FBXO28, YIPF5, MED23, CELF2, KCNC2 |
| Rodents | Zebrafish | ||
|---|---|---|---|
| Pharmacological Models | Genetic Models | Pharmacological Models | Genetic Models |
| Pentylenetetrazol (PTZ) [192], (D,L)-Allylglycine (AG) [179], Kainic acid (KA) [176,183], Picrotoxin [193], Bicuculline [193], Pilocarpine [184], Tetanus toxin [188], Caffeine [190], Strychnine [191] | Kcnj10 [194], Aldh7a1 [195], Mecp2 [196], Scn1a [197], Cdkl5 [198], Syngap1 [199], Lgi1 [200], Ube3a [201], Scn2a [202], Scn8a [203], Scn1b [204], Kcnq2/3 [205], Kcna1 [206], Kcna2 [207], Kcnmb4 [208], Cacna1a [209], Gria2 [210], Chma4 [211], Gabrg2 [212], Fgf13 [213], App [214], Ube3a [215], Shank3 [216], Cntnap2 [217], Epm2a [218], Celf4 [219], Otx1 [220], Sv2a [221], Trpm2 [222], Scamp5 [223], Grin2a [224], Depdc5 [225], Alg13 [226], Hcn1 [227] | Pentylenetetrazol (PTZ) [228], (D,L)-Allylglycine (AG) [179], Kainic acid (KA) [182], Picrotoxin [180], Pilocarpine [229,230], Ginkgotoxin [187,231] | scn1lab [232], gabra1 [233], gabrg2 [234], kcnj10 [235], kcnq2/3 [236], stx1b [237], chd2 [238], arxa [239], eef1a2 [239], gabrb3 [239], pnpo [239], strada [239], lgi1a [240], cacna1a/b [186], depdc5 [241] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, S.; Wang, H. Clocking Epilepsies: A Chronomodulated Strategy-Based Therapy for Rhythmic Seizures. Int. J. Mol. Sci. 2023, 24, 4223. https://doi.org/10.3390/ijms24044223
Sun S, Wang H. Clocking Epilepsies: A Chronomodulated Strategy-Based Therapy for Rhythmic Seizures. International Journal of Molecular Sciences. 2023; 24(4):4223. https://doi.org/10.3390/ijms24044223
Chicago/Turabian StyleSun, Sha, and Han Wang. 2023. "Clocking Epilepsies: A Chronomodulated Strategy-Based Therapy for Rhythmic Seizures" International Journal of Molecular Sciences 24, no. 4: 4223. https://doi.org/10.3390/ijms24044223
APA StyleSun, S., & Wang, H. (2023). Clocking Epilepsies: A Chronomodulated Strategy-Based Therapy for Rhythmic Seizures. International Journal of Molecular Sciences, 24(4), 4223. https://doi.org/10.3390/ijms24044223