
Potential Novel Role of Membrane-Associated Carbonic Anhydrases in the Kidney


Abstract
:1. Introduction
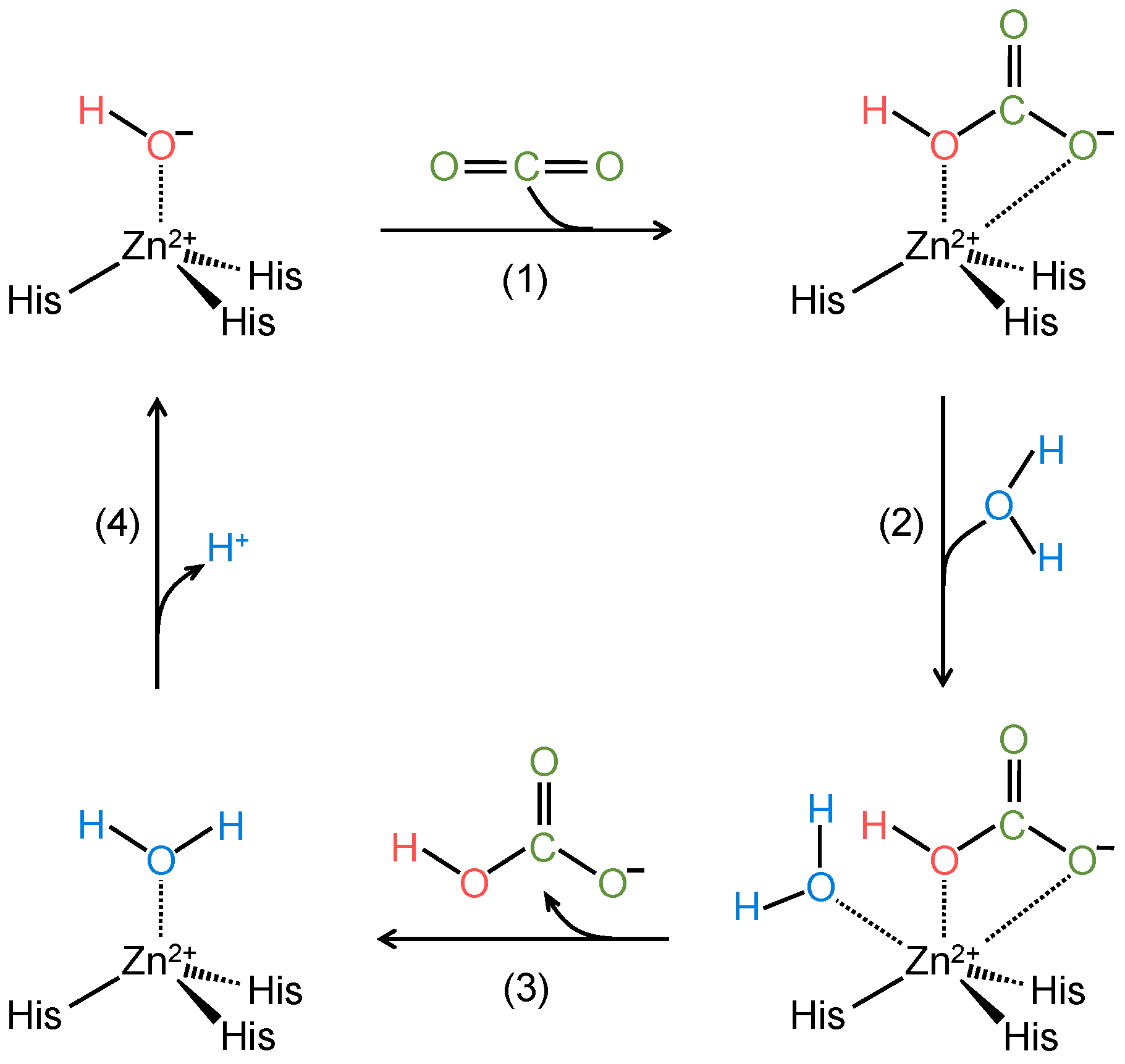
2. Catalytic Mechanism of α-Carbonic Anhydrases
3. Carbonic Anhydrases in Nanodomains Adjacent to the Cell Membrane
4. Carbonic Anhydrases and the Identification of Substrates of SLC4 Family Members
4.1. Brief Overview of the SLC4 Family Members
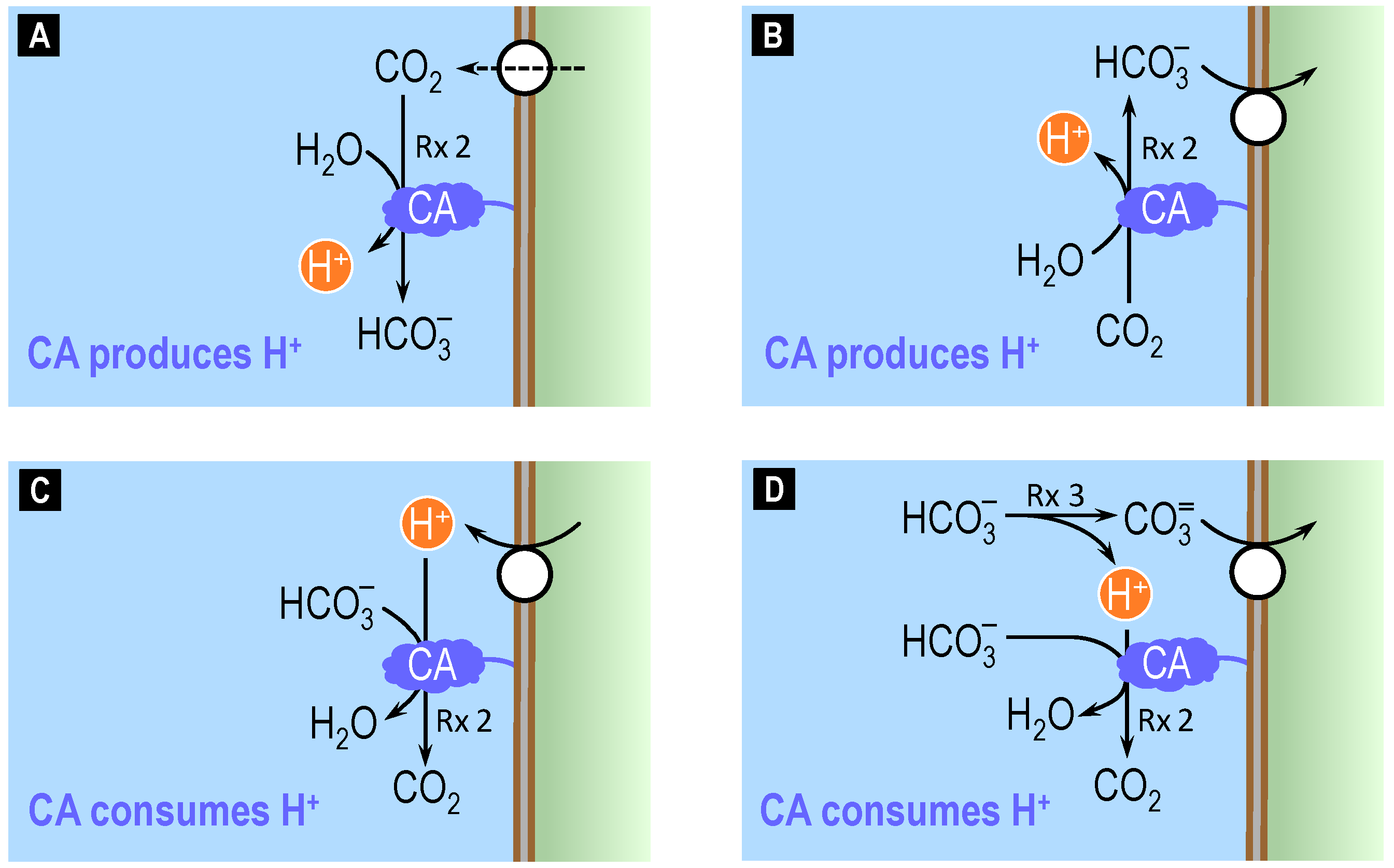
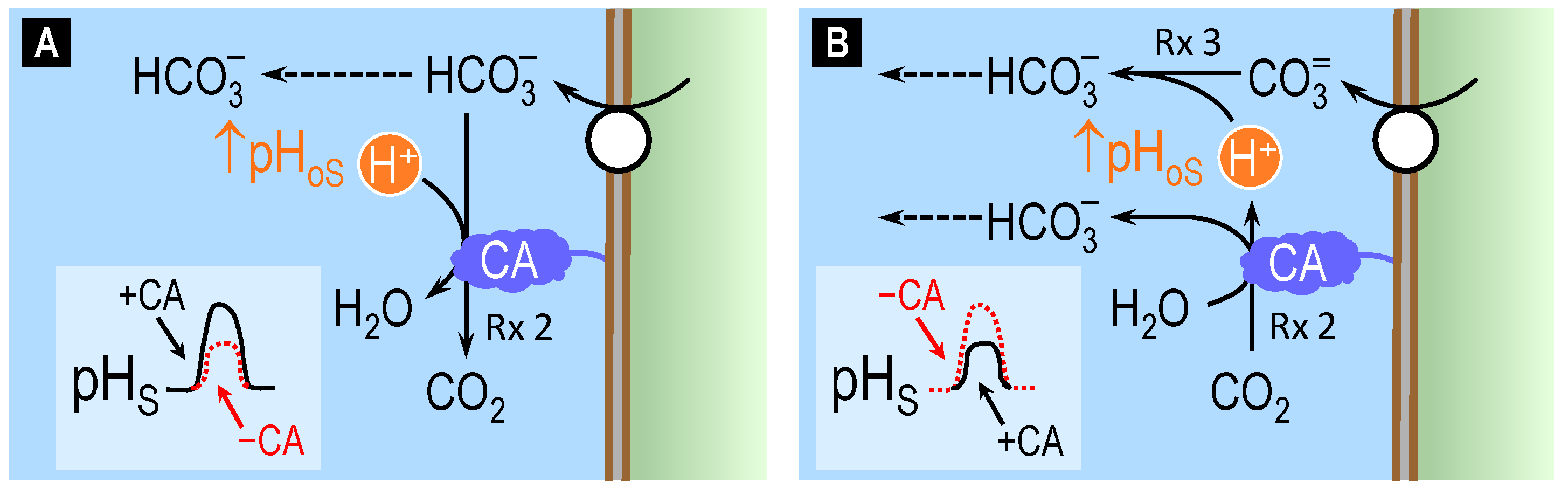
4.2. Theoretical Role of Carbonic Anhydrase in Distinguishing Bicarbonate versus Carbonate versus Proton Transport across Cell Membranes
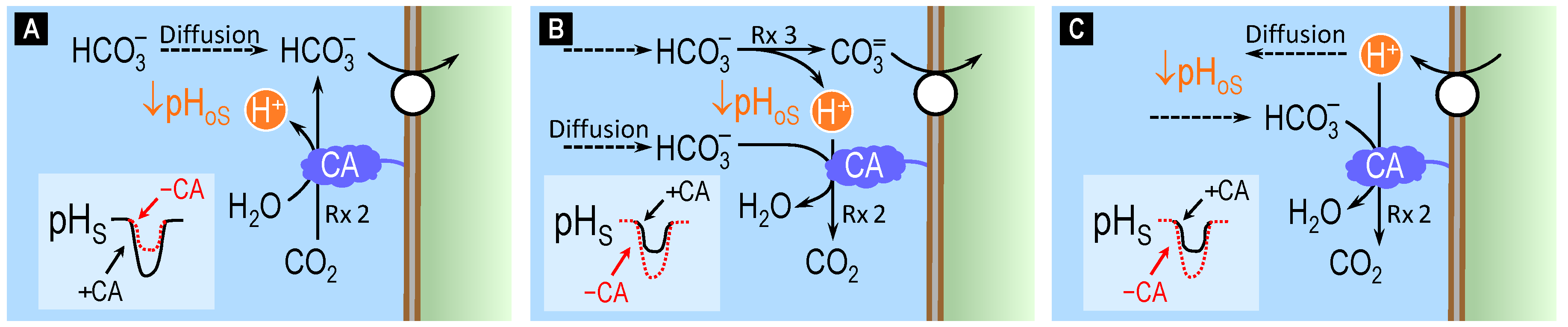
4.3. Surface pH Studies Supporting Carbonate as the Substrate of NCBTs
5. Relative Abundance of Members of the SLC4 and α-CA Families in Human Kidney
5.1. SLC4 Family Members
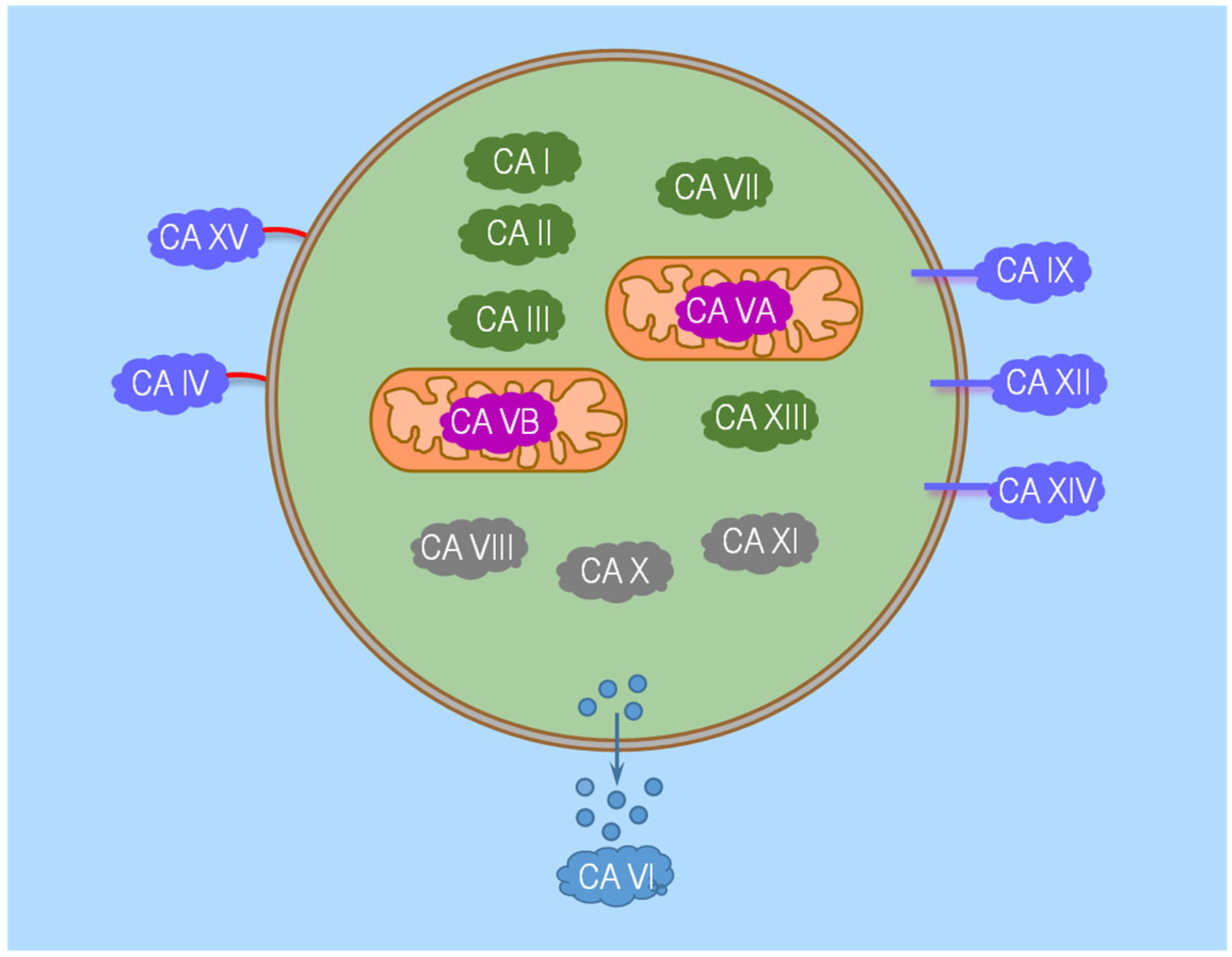
5.2. α-CA Family Members
6. Carbonic Anhydrases along the Nephron
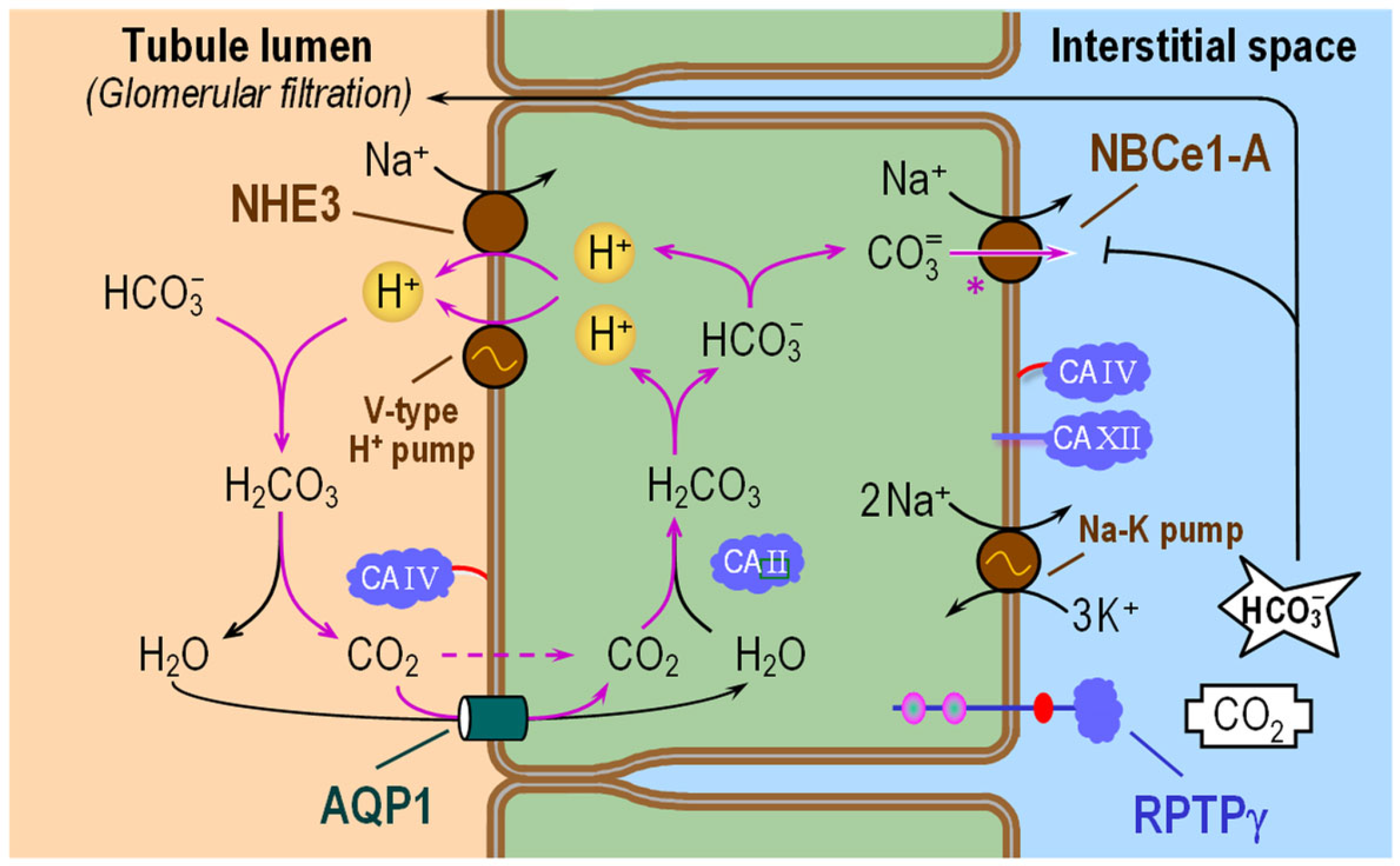
6.1. Proximal Tubule
6.1.1. Subcellular Localizations of CA Isozymes
6.1.2. Functional Interactions with Acid–Base Transporters
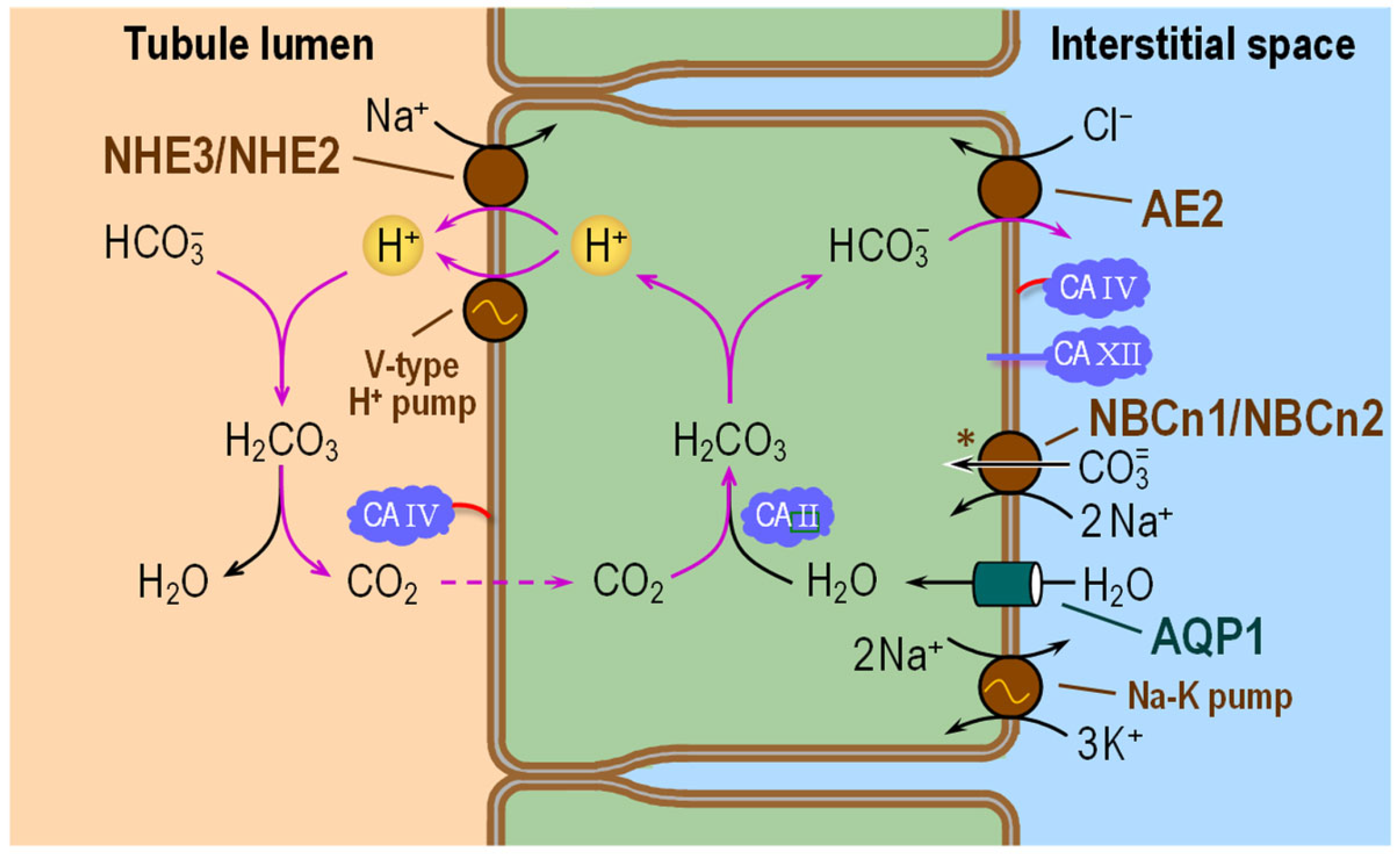
6.2. Thick Ascending Limb
6.2.1. Subcellular Localizations of CA Isozymes
6.2.2. Functional Interactions with Acid–Base Transporters
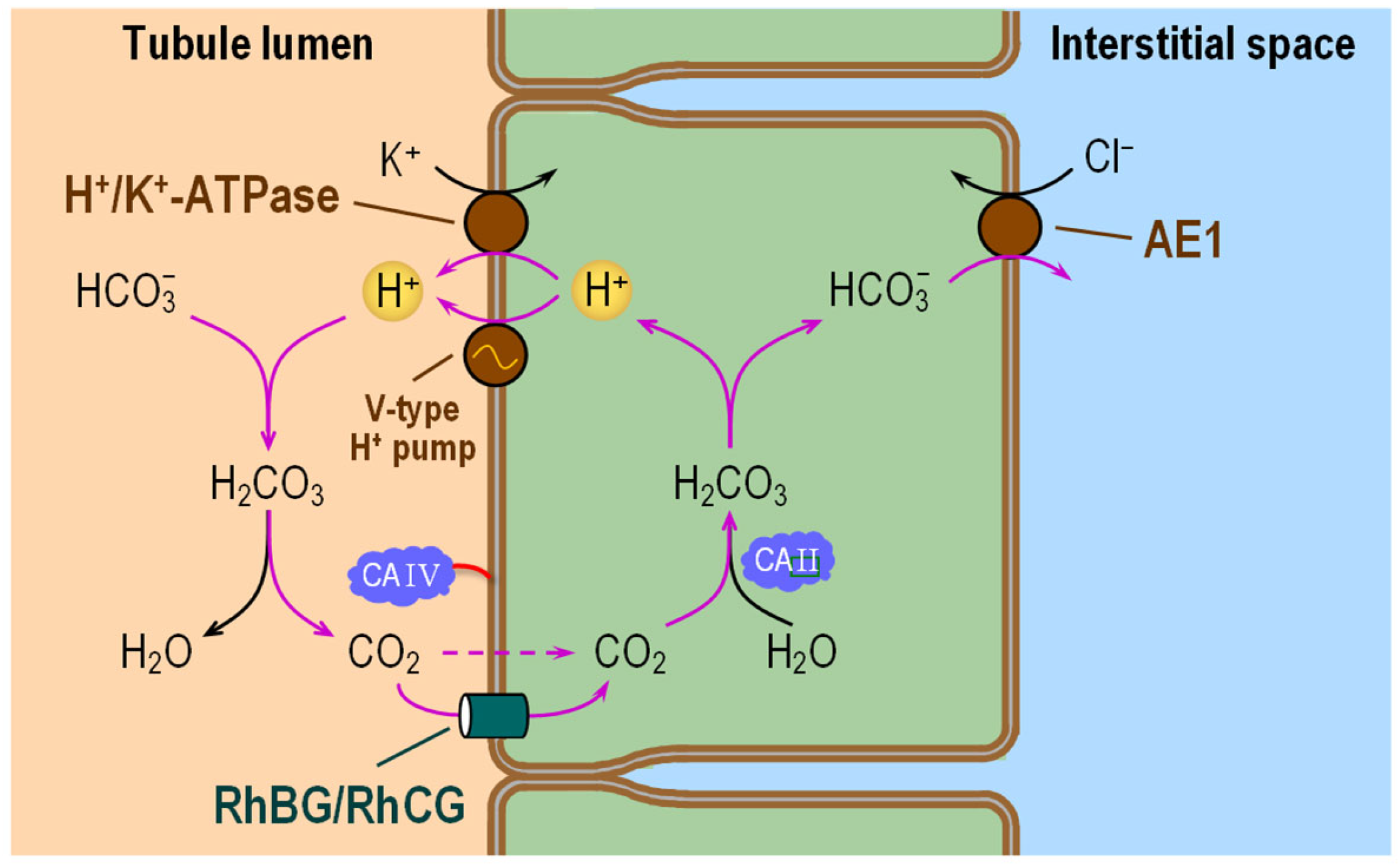
6.3. Tubules Distal to the Thick Ascending Limb
6.3.1. Subcellular Localizations of CA Isozymes
6.3.2. Functional Interactions with Acid–Base Transporters
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Supuran, C.T. Carbonic Anhydrases and Metabolism. Metabolites 2018, 8, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspatwar, A.; Tolvanen, M.E.E.; Barker, H.; Syrjänen, L.; Valanne, S.; Purmonen, S.; Waheed, A.; Sly, W.S.; Parkkila, S. Carbonic Anhydrases in Metazoan Model Organisms: Molecules, Mechanisms, and Physiology. Physiol. Rev. 2022, 102, 1327–1383. [Google Scholar] [CrossRef] [PubMed]
- Imtaiyaz Hassan, M.; Shajee, B.; Waheed, A.; Ahmad, F.; Sly, W.S. Structure, Function and Applications of Carbonic Anhydrase Isozymes. Bioorg. Med. Chem. 2013, 21, 1570–1582. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Structure and Function of Carbonic Anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef]
- Occhipinti, R.; Boron, W.F. Role of Carbonic Anhydrases and Inhibitors in Acid-Base Physiology: Insights from Mathematical Modeling. Int. J. Mol. Sci. 2019, 20, 3841. [Google Scholar] [CrossRef] [Green Version]
- Sly, W.S.; Hewett-Emmett, D.; Whyte, M.P.; Yu, Y.S.; Tashian, R.E. Carbonic Anhydrase II Deficiency Identified as the Primary Defect in the Autosomal Recessive Syndrome of Osteopetrosis with Renal Tubular Acidosis and Cerebral Calcification. Proc. Natl. Acad. Sci. USA 1983, 80, 2752–2756. [Google Scholar] [CrossRef] [Green Version]
- Mori, S.; Kou, I.; Sato, H.; Emi, M.; Ito, H.; Hosoi, T.; Ikegawa, S. Nucleotide Variations in Genes Encoding Carbonic Anhydrase 8 and 10 Associated with Femoral Bone Mineral Density in Japanese Female with Osteoporosis. J. Bone Miner. Metab. 2009, 27, 213–216. [Google Scholar] [CrossRef]
- Ivanov, S.V.; Kuzmin, I.; Wei, M.H.; Pack, S.; Geil, L.; Johnson, B.E.; Stanbridge, E.J.; Lerman, M.I. Down-Regulation of Transmembrane Carbonic Anhydrases in Renal Cell Carcinoma Cell Lines by Wild-Type von Hippel-Lindau Transgenes. Proc. Natl. Acad. Sci. USA 1998, 95, 12596–12601. [Google Scholar] [CrossRef] [Green Version]
- Parkkila, S.; Parkkila, A.K.; Saarnio, J.; Kivelä, J.; Karttunen, T.J.; Kaunisto, K.; Waheed, A.; Sly, W.S.; Türeci, O.; Virtanen, I.; et al. Expression of the Membrane-Associated Carbonic Anhydrase Isozyme XII in the Human Kidney and Renal Tumors. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2000, 48, 1601–1608. [Google Scholar] [CrossRef] [Green Version]
- Pastorekova, S.; Parkkila, S.; Zavada, J. Tumor-Associated Carbonic Anhydrases and Their Clinical Significance. Adv. Clin. Chem. 2006, 42, 167–216. [Google Scholar]
- Haapasalo, J.; Hilvo, M.; Nordfors, K.; Haapasalo, H.; Parkkila, S.; Hyrskyluoto, A.; Rantala, I.; Waheed, A.; Sly, W.S.; Pastorekova, S.; et al. Identification of an Alternatively Spliced Isoform of Carbonic Anhydrase XII in Diffusely Infiltrating Astrocytic Gliomas. Neuro-oncology 2008, 10, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Watson, P.H.; Chia, S.K.; Wykoff, C.C.; Han, C.; Leek, R.D.; Sly, W.S.; Gatter, K.C.; Ratcliffe, P.; Harris, A.L. Carbonic Anhydrase XII Is a Marker of Good Prognosis in Invasive Breast Carcinoma. Br. J. Cancer 2003, 88, 1065–1070. [Google Scholar] [CrossRef] [Green Version]
- Supuran, C.T. Emerging Role of Carbonic Anhydrase Inhibitors. Clin. Sci. Lond. Engl. 2021, 135, 1233–1249. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic Anhydrase Inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3467–3474. [Google Scholar] [CrossRef]
- Supuran, C.T.; Scozzafava, A.; Casini, A. Carbonic Anhydrase Inhibitors. Med. Res. Rev. 2003, 23, 146–189. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic Anhydrase Inhibitors as Emerging Agents for the Treatment and Imaging of Hypoxic Tumors. Expert Opin. Investig. Drugs 2018, 27, 963–970. [Google Scholar] [CrossRef]
- Courcier, J.; de la Taille, A.; Nourieh, M.; Leguerney, I.; Lassau, N.; Ingels, A. Carbonic Anhydrase IX in Renal Cell Carcinoma, Implications for Disease Management. Int. J. Mol. Sci. 2020, 21, 7146. [Google Scholar] [CrossRef]
- Ozensoy Guler, O.; Supuran, C.T.; Capasso, C. Carbonic Anhydrase IX as a Novel Candidate in Liquid Biopsy. J. Enzyme Inhib. Med. Chem. 2020, 35, 255–260. [Google Scholar] [CrossRef] [Green Version]
- Haapasalo, J.; Nordfors, K.; Haapasalo, H.; Parkkila, S. The Expression of Carbonic Anhydrases II, IX and XII in Brain Tumors. Cancers 2020, 12, 1723. [Google Scholar] [CrossRef]
- Meldrum, N.U.; Roughton, F.J. Carbonic Anhydrase. Its Preparation and Properties. J. Physiol. 1933, 80, 113–142. [Google Scholar] [CrossRef]
- Rickli, E.E.; Ghazanfar, S.A.; Gibbons, B.H.; Edsall, J.T. Carbonic Anhydrases from Human Erythrocytes: Preparation and Properties of Two Enzymes. J. Biol. Chem. 1964, 239, 1065–1078. [Google Scholar] [CrossRef] [PubMed]
- Nyman, P.O. Purification and Properties of Carbonic Anhydrase from Human Erythrocytes. Biochim. Biophys. Acta 1961, 52, 1–12. [Google Scholar] [CrossRef]
- Lindskog, S. Purification and Properties of Bovine Erythrocyte Carbonic Anhydrase. Biochim. Biophys. Acta 1960, 39, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Dobyan, D.C.; Bulger, R.E. Renal Carbonic Anhydrase. Am. J. Physiol. 1982, 243, F311–F324. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.L.; Clement, R.; Kosta, A.; Maberly, S.C.; Gontero, B. A New Widespread Subclass of Carbonic Anhydrase in Marine Phytoplankton. ISME J. 2019, 13, 2094–2106. [Google Scholar] [CrossRef] [Green Version]
- Del Prete, S.; Nocentini, A.; Supuran, C.T.; Capasso, C. Bacterial ι-Carbonic Anhydrase: A New Active Class of Carbonic Anhydrase Identified in the Genome of the Gram-Negative Bacterium Burkholderia Territorii. J. Enzyme Inhib. Med. Chem. 2020, 35, 1060–1068. [Google Scholar] [CrossRef] [Green Version]
- Nocentini, A.; Supuran, C.T.; Capasso, C. An Overview on the Recently Discovered Iota-Carbonic Anhydrases. J. Enzyme Inhib. Med. Chem. 2021, 36, 1988–1995. [Google Scholar] [CrossRef]
- Mboge, M.Y.; Mahon, B.P.; McKenna, R.; Frost, S.C. Carbonic Anhydrases: Role in pH Control and Cancer. Metabolites 2018, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Lomelino, C.L.; Andring, J.T.; McKenna, R. Crystallography and Its Impact on Carbonic Anhydrase Research. Int. J. Med. Chem. 2018, 2018, 9419521. [Google Scholar] [CrossRef] [Green Version]
- Syrjänen, L.; Tolvanen, M.; Hilvo, M.; Olatubosun, A.; Innocenti, A.; Scozzafava, A.; Leppiniemi, J.; Niederhauser, B.; Hytönen, V.P.; Gorr, T.A.; et al. Characterization of the First Beta-Class Carbonic Anhydrase from an Arthropod (Drosophila Melanogaster) and Phylogenetic Analysis of Beta-Class Carbonic Anhydrases in Invertebrates. BMC Biochem. 2010, 11, 28. [Google Scholar] [CrossRef] [Green Version]
- Zolfaghari Emameh, R.; Barker, H.; Tolvanen, M.E.E.; Ortutay, C.; Parkkila, S. Bioinformatic Analysis of Beta Carbonic Anhydrase Sequences from Protozoans and Metazoans. Parasit. Vectors 2014, 7, 38. [Google Scholar] [CrossRef] [Green Version]
- Supuran, C.T. Carbonic Anhydrases: Novel Therapeutic Applications for Inhibitors and Activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef]
- Del Prete, S.; Vullo, D.; Fisher, G.M.; Andrews, K.T.; Poulsen, S.-A.; Capasso, C.; Supuran, C.T. Discovery of a New Family of Carbonic Anhydrases in the Malaria Pathogen Plasmodium Falciparum—The η-Carbonic Anhydrases. Bioorg. Med. Chem. Lett. 2014, 24, 4389–4396. [Google Scholar] [CrossRef] [Green Version]
- Kikutani, S.; Nakajima, K.; Nagasato, C.; Tsuji, Y.; Miyatake, A.; Matsuda, Y. Thylakoid Luminal θ-Carbonic Anhydrase Critical for Growth and Photosynthesis in the Marine Diatom Phaeodactylum Tricornutum. Proc. Natl. Acad. Sci. USA 2016, 113, 9828–9833. [Google Scholar] [CrossRef] [Green Version]
- Ferry, J.G. The Gamma Class of Carbonic Anhydrases. Biochim. Biophys. Acta 2010, 1804, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Alber, B.E.; Colangelo, C.M.; Dong, J.; Stålhandske, C.M.; Baird, T.T.; Tu, C.; Fierke, C.A.; Silverman, D.N.; Scott, R.A.; Ferry, J.G. Kinetic and Spectroscopic Characterization of the Gamma-Carbonic Anhydrase from the Methanoarchaeon Methanosarcina Thermophila. Biochemistry 1999, 38, 13119–13128. [Google Scholar] [CrossRef]
- Amata, O.; Marino, T.; Russo, N.; Toscano, M. Catalytic Activity of a ζ-Class Zinc and Cadmium Containing Carbonic Anhydrase. Compared Work Mechanisms. Phys. Chem. Chem. Phys. PCCP 2011, 13, 3468–3477. [Google Scholar] [CrossRef]
- Hirakawa, Y.; Senda, M.; Fukuda, K.; Yu, H.Y.; Ishida, M.; Taira, M.; Kinbara, K.; Senda, T. Characterization of a Novel Type of Carbonic Anhydrase That Acts without Metal Cofactors. BMC Biol. 2021, 19, 105. [Google Scholar] [CrossRef]
- Jewell, D.A.; Tu, C.K.; Paranawithana, S.R.; Tanhauser, S.M.; LoGrasso, P.V.; Laipis, P.J.; Silverman, D.N. Enhancement of the Catalytic Properties of Human Carbonic Anhydrase III by Site-Directed Mutagenesis. Biochemistry 1991, 30, 1484–1490. [Google Scholar] [CrossRef]
- Jackman, J.E.; Merz, K.M.; Fierke, C.A. Disruption of the Active Site Solvent Network in Carbonic Anhydrase II Decreases the Efficiency of Proton Transfer. Biochemistry 1996, 35, 16421–16428. [Google Scholar] [CrossRef]
- Khalifah, R.G. The Carbon Dioxide Hydration Activity of Carbonic Anhydrase. I. Stop-Flow Kinetic Studies on the Native Human Isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [CrossRef] [PubMed]
- Dodgson, S.J.; Shank, R.P.; Maryanoff, B.E. Topiramate as an Inhibitor of Carbonic Anhydrase Isoenzymes. Epilepsia 2000, 41, 35–39. [Google Scholar] [CrossRef]
- Fujikawa-Adachi, K.; Nishimori, I.; Taguchi, T.; Onishi, S. Human Mitochondrial Carbonic Anhydrase VB: cDNA cloning, mRNA expression, subcellular localization, and mapping to chromosome X. J. Biol. Chem. 1999, 274, 21228–21233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagao, Y.; Batanian, J.R.; Clemente, M.F.; Sly, W.S. Genomic Organization of the Human Gene (CA5) and Pseudogene for Mitochondrial Carbonic Anhydrase V and Their Localization to Chromosomes 16q and 16p. Genomics 1995, 28, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Silverman, D.N.; Lindskog, S. The Catalytic Mechanism of Carbonic Anhydrase: Implications of a Rate-Limiting Protolysis of Water. Acc. Chem. Res. 1988, 21, 30–36. [Google Scholar] [CrossRef]
- Liljas, A.; Carlsson, M.; Håkansson, K.; Lindahl, M.; Svensson, L.A.; Wehnert, A.; Cruickshank, D.W.J.; Helliwell, J.R.; Johnson, L.N.; Moffat, K.; et al. Laue and Monochromatic Crystallography on Carbonic Anhydrase. Philos. Trans. R. Soc. Lond. Ser. Phys. Eng. Sci. 1992, 340, 301–309. [Google Scholar] [CrossRef]
- Bertini, I.; Luchinat, C.; Rosi, M.; Sgamellotti, A.; Tarantelli, F. pKa of Zinc-Bound Water and Nucleophilicity of Hydroxo-Containing Species. Ab Initio Calculations on Models for Zinc Enzymes. Inorg. Chem. 1990, 29, 1460–1463. [Google Scholar] [CrossRef]
- Ataie, N.J.; Hoang, Q.Q.; Zahniser, M.P.D.; Tu, Y.; Milne, A.; Petsko, G.A.; Ringe, D. Zinc Coordination Geometry and Ligand Binding Affinity: The Structural and Kinetic Analysis of the Second-Shell Serine 228 Residue and the Methionine 180 Residue of the Aminopeptidase from Vibrio Proteolyticus. Biochemistry 2008, 47, 7673–7683. [Google Scholar] [CrossRef] [Green Version]
- Goto, M.; Takahashi, T.; Yamashita, F.; Koreeda, A.; Mori, H.; Ohta, M.; Arakawa, Y. Inhibition of the Metallo-Beta-Lactamase Produced from Serratia Marcescens by Thiol Compounds. Biol. Pharm. Bull. 1997, 20, 1136–1140. [Google Scholar] [CrossRef] [Green Version]
- Chegwidden, W.W.R.; Carter, N.N.D.; Edwards, Y.Y.H. The Carbonic Anhydrases; Springer: Berlin/Heidelberg, Germany, 2000; ISBN 3-7643-5670-7. [Google Scholar]
- Tu, C.K.; Silverman, D.N.; Forsman, C.; Jonsson, B.H.; Lindskog, S. Role of Histidine 64 in the Catalytic Mechanism of Human Carbonic Anhydrase II Studied with a Site-Specific Mutant. Biochemistry 1989, 28, 7913–7918. [Google Scholar] [CrossRef]
- Engstrand, C.; Forsman, C.; Liang, Z.; Lindskog, S. Proton Transfer Roles of Lysine 64 and Glutamic Acid 64 Replacing Histidine 64 in the Active Site of Human Carbonic Anhydrase II. Biochim. Biophys. Acta 1992, 1122, 321–326. [Google Scholar] [CrossRef]
- Krebs, J.F.; Ippolito, J.A.; Christianson, D.W.; Fierke, C.A. Structural and Functional Importance of a Conserved Hydrogen Bond Network in Human Carbonic Anhydrase II. J. Biol. Chem. 1993, 268, 27458–27466. [Google Scholar] [CrossRef]
- Liang, Z.; Xue, Y.; Behravan, G.; Jonsson, B.H.; Lindskog, S. Importance of the Conserved Active-Site Residues Tyr7, Glu106 and Thr199 for the Catalytic Function of Human Carbonic Anhydrase II. Eur. J. Biochem. 1993, 211, 821–827. [Google Scholar] [CrossRef]
- Aspatwar, A.; Tolvanen, M.E.; Parkkila, S. Phylogeny and Expression of Carbonic Anhydrase-Related Proteins. BMC Mol. Biol. 2010, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Musa-Aziz, R.; Occhipinti, R.; Boron, W.F. Evidence from Simultaneous Intracellular- and Surface-pH Transients That Carbonic Anhydrase II Enhances CO₂ Fluxes across Xenopus Oocytes Plasma Membranes. Am. J. Physiol. Cell Physiol. 2014, 307, C791–C813. [Google Scholar] [CrossRef] [Green Version]
- Musa-Aziz, R.; Occhipinti, R.; Boron, W.F. Evidence from Simultaneous Intracellular- and Surface-pH Transients That Carbonic Anhydrase IV Enhances CO₂ Fluxes across Xenopus Oocyte Plasma Membranes. Am. J. Physiol. Cell Physiol. 2014, 307, C814–C840. [Google Scholar] [CrossRef] [Green Version]
- Occhipinti, R.; Musa-Aziz, R.; Boron, W.F. Evidence from Mathematical Modeling That Carbonic Anhydrase II and IV Enhance CO₂ Fluxes across Xenopus Oocytes Plasma Membranes. Am. J. Physiol. Cell Physiol. 2014, 307, C841–C858. [Google Scholar] [CrossRef] [Green Version]
- Gutknecht, J.; Bisson, M.A.; Tosteson, F.C. Diffusion of Carbon Dioxide through Lipid Bilayer Membranes: Effects of Carbonic Anhydrase, Bicarbonate, and Unstirred Layers. J. Gen. Physiol. 1977, 69, 779–794. [Google Scholar] [CrossRef] [Green Version]
- Boron, W.F. Intracellular pH-Regulating Mechanism of the Squid Axon. Relation between the External Na+ and HCO₃− Dependences. J. Gen. Physiol. 1985, 85, 325–345. [Google Scholar] [CrossRef] [Green Version]
- Jentsch, T.J.; Schwartz, P.; Schill, B.S.; Langner, B.; Lepple, A.P.; Keller, S.K.; Wiederholt, M. Kinetic Properties of the Sodium Bicarbonate (Carbonate) Symport in Monkey Kidney Epithelial Cells (BSC-1). Interactions between Na+, HCO₃−, and pH. J. Biol. Chem. 1986, 261, 10673–10679. [Google Scholar] [CrossRef]
- Boron, W.F.; Knakal, R.C. Intracellular pH-Regulating Mechanism of the Squid Axon. Interaction between DNDS and Extracellular Na+ and HCO₃−. J. Gen. Physiol. 1989, 93, 123–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boron, W.F.; Knakal, R.C. Na+-Dependent Cl−-HCO₃− Exchange in the Squid Axon. Dependence on Extracellular pH. J. Gen. Physiol. 1992, 99, 817–837. [Google Scholar] [CrossRef] [PubMed]
- Boron, W.F.; Russell, J.M. Stoichiometry and Ion Dependencies of the Intracellular-pH-Regulating Mechanism in Squid Giant Axons. J. Gen. Physiol. 1983, 81, 373–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soleimani, M.; Aronson, P.S. Ionic Mechanism of Na+-HCO3− Cotransport in Rabbit Renal Basolateral Membrane Vesicles. J. Biol. Chem. 1989, 264, 18302–18308. [Google Scholar] [CrossRef]
- Zhu, Q.; Shao, X.M.; Kao, L.; Azimov, R.; Weinstein, A.M.; Newman, D.; Liu, W.; Kurtz, I. Missense Mutation T485S Alters NBCe1-A Electrogenicity Causing Proximal Renal Tubular Acidosis. Am. J. Physiol. Cell Physiol. 2013, 305, C392–C405. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-K.; Boron, W.F.; Parker, M.D. Substrate Specificity of the Electrogenic Sodium/Bicarbonate Cotransporter NBCe1-A (SLC4A4, Variant A) from Humans and Rabbits. Am. J. Physiol. Renal Physiol. 2013, 304, F883–F899. [Google Scholar] [CrossRef] [Green Version]
- Seki, G.; Coppola, S.; Yoshitomi, K.; Burckhardt, B.C.; Samarzija, I.; Müller-Berger, S.; Frömter, E. On the Mechanism of Bicarbonate Exit from Renal Proximal Tubular Cells. Kidney Int. 1996, 49, 1671–1677. [Google Scholar] [CrossRef] [Green Version]
- Boron, W.F. Evaluating the Role of Carbonic Anhydrases in the Transport of HCO₃−-Related Species. Biochim. Biophys. Acta 2010, 1804, 410–421. [Google Scholar] [CrossRef] [Green Version]
- Grichtchenko, I.I.; Chesler, M. Depolarization-Induced Acid Secretion in Gliotic Hippocampal Slices. Neuroscience 1994, 62, 1057–1070. [Google Scholar] [CrossRef]
- Occhipinti, R.; Lu, J.; Boron, W.F. Is the Electrogenic Na/HCO₃ Cotransporter a CO₂ Channel? FASEB J. 2016, 30, 971.2. [Google Scholar] [CrossRef]
- Moss, F.J.; Occhipinti, R.; Zeise, B.; Zhao, P.; Wang, D.-K.; Lu, J.; Boron, W.F. The Electrogenic Sodium Bicarbonate Cotransporter NBCe1 as a Conduit for CO2. FASEB J. 2019, 33, 544.3. [Google Scholar] [CrossRef]
- Lee, S.-K.; Occhipinti, R.; Moss, F.J.; Parker, M.D.; Grichtchenko, I.I.; Boron, W.F. Distinguishing among HCO₃−, CO₃=, and H+ as Substrates of Proteins That Appear to Be “Bicarbonate” Transporters. J. Am. Soc. Nephrol. 2023, 34, 40–54. [Google Scholar] [CrossRef]
- Alper, S.L.; Sharma, A.K. The SLC26 Gene Family of Anion Transporters and Channels. Mol. Asp. Med. 2013, 34, 494–515. [Google Scholar] [CrossRef] [Green Version]
- Amlal, H.; Petrovic, S.; Xu, J.; Wang, Z.; Sun, X.; Barone, S.; Soleimani, M. Deletion of the Anion Exchanger Slc26a4 (Pendrin) Decreases Apical Cl(−)/HCO3(−) Exchanger Activity and Impairs Bicarbonate Secretion in Kidney Collecting Duct. Am. J. Physiol. Cell Physiol. 2010, 299, C33–C41. [Google Scholar] [CrossRef] [Green Version]
- Royaux, I.E.; Wall, S.M.; Karniski, L.P.; Everett, L.A.; Suzuki, K.; Knepper, M.A.; Green, E.D. Pendrin, Encoded by the Pendred Syndrome Gene, Resides in the Apical Region of Renal Intercalated Cells and Mediates Bicarbonate Secretion. Proc. Natl. Acad. Sci. USA 2001, 98, 4221–4226. [Google Scholar] [CrossRef] [Green Version]
- Cordat, E.; Casey, J.R. Bicarbonate Transport in Cell Physiology and Disease. Biochem. J. 2009, 417, 423. [Google Scholar] [CrossRef] [Green Version]
- Tsuganezawa, H.; Kobayashi, K.; Iyori, M.; Araki, T.; Koizumi, A.; Watanabe, S.; Kaneko, A.; Fukao, T.; Monkawa, T.; Yoshida, T.; et al. A New Member of the HCO3− Transporter Superfamily Is an Apical Anion Exchanger of Beta-Intercalated Cells in the Kidney. J. Biol. Chem. 2001, 276, 8180–8189. [Google Scholar] [CrossRef] [Green Version]
- Ko, S.B.H.; Luo, X.; Hager, H.; Rojek, A.; Choi, J.Y.; Licht, C.; Suzuki, M.; Muallem, S.; Nielsen, S.; Ishibashi, K. AE4 Is a DIDS-Sensitive Cl−/HCO3− Exchanger in the Basolateral Membrane of the Renal CCD and the SMG Duct. Am. J. Physiol. Cell Physiol. 2002, 283, C1206–C1218. [Google Scholar] [CrossRef]
- Xu, J.; Barone, S.; Petrovic, S.; Wang, Z.; Seidler, U.; Riederer, B.; Ramaswamy, K.; Dudeja, P.K.; Shull, G.E.; Soleimani, M. Identification of an Apical Cl−/HCO3− Exchanger in Gastric Surface Mucous and Duodenal Villus Cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G1225–G1234. [Google Scholar] [CrossRef] [Green Version]
- Chambrey, R.; Kurth, I.; Peti-Peterdi, J.; Houillier, P.; Purkerson, J.M.; Leviel, F.; Hentschke, M.; Zdebik, A.A.; Schwartz, G.J.; Hübner, C.A.; et al. Renal Intercalated Cells Are Rather Energized by a Proton than a Sodium Pump. Proc. Natl. Acad. Sci. USA 2013, 110, 7928–7933. [Google Scholar] [CrossRef] [Green Version]
- Peña-Münzenmayer, G.; Catalán, M.A.; Kondo, Y.; Jaramillo, Y.; Liu, F.; Shull, G.E.; Melvin, J.E. Ae4 (Slc4a9) Anion Exchanger Drives Cl− Uptake-Dependent Fluid Secretion by Mouse Submandibular Gland Acinar Cells. J. Biol. Chem. 2015, 290, 10677–10688. [Google Scholar] [CrossRef] [Green Version]
- Myers, E.J.; Marshall, A.; Jennings, M.L.; Parker, M.D. Mouse Slc4a11 Expressed in Xenopus Oocytes Is an Ideally Selective H+/OH− Conductance Pathway That Is Stimulated by Rises in Intracellular and Extracellular PH. Am. J. Physiol.-Cell Physiol. 2016, 311, C945–C959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, L.; Azimov, R.; Abuladze, N.; Newman, D.; Kurtz, I. Human SLC4A11-C Functions as a DIDS-Stimulatable H+(OH−) Permeation Pathway: Partial Correction of R109H Mutant Transport. Am. J. Physiol. Cell Physiol. 2015, 308, C176–C188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, L.J.; Tanner, M.J. Erythroid Band 3 Variants and Disease. Baillieres Best Pract. Res. Clin. Haematol. 1999, 12, 637–654. [Google Scholar] [CrossRef] [PubMed]
- Alloisio, N.; Texier, P.; Vallier, A.; Ribeiro, M.L.; Morlé, L.; Bozon, M.; Bursaux, E.; Maillet, P.; Gonçalves, P.; Tanner, M.J.; et al. Modulation of Clinical Expression and Band 3 Deficiency in Hereditary Spherocytosis. Blood 1997, 90, 414–420. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, M.L.; Alloisio, N.; Almeida, H.; Gomes, C.; Texier, P.; Lemos, C.; Mimoso, G.; Morlé, L.; Bey-Cabet, F.; Rudigoz, R.C.; et al. Severe Hereditary Spherocytosis and Distal Renal Tubular Acidosis Associated with the Total Absence of Band 3. Blood 2000, 96, 1602–1604. [Google Scholar]
- Igarashi, T.; Inatomi, J.; Sekine, T.; Cha, S.H.; Kanai, Y.; Kunimi, M.; Tsukamoto, K.; Satoh, H.; Shimadzu, M.; Tozawa, F.; et al. Mutations in SLC4A4 Cause Permanent Isolated Proximal Renal Tubular Acidosis with Ocular Abnormalities. Nat. Genet. 1999, 23, 264–266. [Google Scholar] [CrossRef]
- Demirci, F.Y.K.; Chang, M.-H.; Mah, T.S.; Romero, M.F.; Gorin, M.B. Proximal Renal Tubular Acidosis and Ocular Pathology: A Novel Missense Mutation in the Gene (SLC4A4) for Sodium Bicarbonate Cotransporter Protein (NBCe1). Mol. Vis. 2006, 12, 324–330. [Google Scholar]
- Suzuki, M.; Van Paesschen, W.; Stalmans, I.; Horita, S.; Yamada, H.; Bergmans, B.A.; Legius, E.; Riant, F.; De Jonghe, P.; Li, Y.; et al. Defective Membrane Expression of the Na+-HCO₃− Cotransporter NBCe1 Is Associated with Familial Migraine. Proc. Natl. Acad. Sci. USA 2010, 107, 15963–15968. [Google Scholar] [CrossRef] [Green Version]
- Alka, K.; Casey, J.R. Bicarbonate Transport in Health and Disease. IUBMB Life 2014, 66, 596–615. [Google Scholar] [CrossRef]
- Romero, M.F.; Chen, A.-P.; Parker, M.D.; Boron, W.F. The SLC4 Family of Bicarbonate (HCO₃−) Transporters. Mol. Asp. Med. 2013, 34, 159–182. [Google Scholar] [CrossRef] [Green Version]
- Parker, M.D.; Boron, W.F. The Divergence, Actions, Roles, and Relatives of Sodium-Coupled Bicarbonate Transporters. Physiol. Rev. 2013, 93, 803–959. [Google Scholar] [CrossRef] [Green Version]
- Pushkin, A.; Kurtz, I. SLC4 Base (HCO3−, CO32−) Transporters: Classification, Function, Structure, Genetic Diseases, and Knockout Models. Am. J. Physiol. Renal Physiol. 2006, 290, F580–F599. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-K.; Boron, W.F.; Parker, M.D. Monitoring Ion Activities in and around Cells Using Ion-Selective Liquid-Membrane Microelectrodes. Sensors 2013, 13, 984–1003. [Google Scholar] [CrossRef] [Green Version]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-Specific Expression by Genome-Wide Integration of Transcriptomics and Antibody-Based Proteomics. Mol. Cell. Proteomics MCP 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Hilvo, M.; Tolvanen, M.; Clark, A.; Shen, B.; Shah, G.N.; Waheed, A.; Halmi, P.; Hänninen, M.; Hämäläinen, J.M.; Vihinen, M.; et al. Characterization of CA XV, a New GPI-Anchored Form of Carbonic Anhydrase. Biochem. J. 2005, 392, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Davenport, H.W.; Wilhelmi, A.E. Renal Carbonic Anhydrase. Proc. Soc. Exp. Biol. Med. 1941, 48, 53–56. [Google Scholar] [CrossRef]
- Hober, R. Effect of Some Sulfonamides on Renal Secretion. Proc. Soc. Exp. Biol. Med. 1942, 49, 87–90. [Google Scholar] [CrossRef]
- Pitts, R.F.; Alexander, R.S. The Renal Reabsorptive Mechanism for Inorganic Phosphate in Normal and Acidotic Dogs. Am. J. Physiol. 1944, 142, 648–662. [Google Scholar] [CrossRef] [Green Version]
- Pitts, R.F.; Alexander, R.S. The Nature of the Renal Tubular Mechanism for Acidifying the Urine. Am. J. Physiol.-Leg. Content 1945, 144, 239–254. [Google Scholar] [CrossRef]
- Pitts, R.F. The Renal Regulation of Acid Base Balance with Special Reference to the Mechanism for Acidifying the Urine. Science 1945, 102, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Davenport, H.W. Carbonic Anhydrase in Tissues Other than Blood. Physiol. Rev. 1946, 26, 560–573. [Google Scholar] [CrossRef] [PubMed]
- Berliner, R.W.; Orloff, J. Carbonic Anhydrase Inhibitors. Pharmacol. Rev. 1956, 8, 137–174. [Google Scholar] [PubMed]
- Smith, H.W. The Physiology of the Kidney; Oxford University Press: Oxford, UK, 1937. [Google Scholar]
- Gottschalk, C.W.; Berliner, R.W.; Giebisch, G.H. Renal Physiology: People and Ideas; Springer: Berlin/Heidelberg, Germany, 2013; ISBN 978-1-4614-7545-3. [Google Scholar]
- Nicholson, T.F. The Site of Acidification of the Urine in the Dog’s Kidney. Can. J. Biochem. Physiol. 1957, 35, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.M.; Bott, P.A.; Oliver, J.; MacDowell, M.C. The Collection and Analysis of Fluid from Single Nephrons of the Mammalian Kidney. Am. J. Physiol.-Leg. Content 1941, 134, 580–595. [Google Scholar] [CrossRef] [Green Version]
- Berliner, R.W. Some aspects of ion exchange in electrolyte transport by the renal tubules. In Metabolic Aspects of Transport Across Cell Membranes; University of Wisconsin Press: Madison, WI, USA, 1957; pp. 203–220. [Google Scholar]
- Gottschalk, C.W.; Lassiter, W.E.; Mylle, M. Localization of Urine Acidification in the Mammalian Kidney. Am. J. Physiol.-Leg. Content 1960, 198, 581–585. [Google Scholar] [CrossRef]
- DuBose, T.D., Jr. Carbonic Anhydrase-Dependent Bicarbonate Transport in the Kidney. Ann. N. Y. Acad. Sci. 1984, 429, 528–537. [Google Scholar] [CrossRef]
- Lucci, M.S.; Tinker, J.P.; Weiner, I.M.; DuBose, T.D. Function of Proximal Tubule Carbonic Anhydrase Defined by Selective Inhibition. Am. J. Physiol.-Ren. Physiol. 1983, 245, F443–F449. [Google Scholar] [CrossRef]
- Lucci, M.S.; Pucacco, L.R.; DuBose, T.D.; Kokko, J.P.; Carter, N.W. Direct Evaluation of Acidification by Rat Proximal Tubule: Role of Carbonic Anhydrase. Am. J. Physiol. 1980, 238, F372–F379. [Google Scholar] [CrossRef]
- Rector, F.C.; Carter, N.W.; Seldin, D.W. The Mechanism of Bicarbonate Reabsorption in the Proximal and Distal Tubules of the Kidney. J. Clin. Investig. 1965, 44, 278–290. [Google Scholar] [CrossRef]
- DuBose, T.D.; Pucacco, L.R.; Carter, N.W. Determination of Disequilibrium pH in the Rat Kidney in Vivo: Evidence of Hydrogen Secretion. Am. J. Physiol. 1981, 240, F138–F146. [Google Scholar] [CrossRef]
- McKinney, T.D.; Burg, M.B. Bicarbonate Transport by Rabbit Cortical Collecting Tubules. Effect of Acid and Alkali Loads in Vivo on Transport in Vitro. J. Clin. Investig. 1977, 60, 766–768. [Google Scholar] [CrossRef]
- Burg, M.; Green, N. Bicarbonate Transport by Isolated Perfused Rabbit Proximal Convoluted Tubules. Am. J. Physiol. 1977, 233, F307–F314. [Google Scholar] [CrossRef]
- McKinney, T.D.; Burg, M.B. Bicarbonate and Fluid Absorption by Renal Proximal Straight Tubules. Kidney Int. 1977, 12, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Good, D.W.; Knepper, M.A.; Burg, M.B. Ammonia and Bicarbonate Transport by Thick Ascending Limb of Rat Kidney. Am. J. Physiol.-Ren. Physiol. 1984, 247, F35–F44. [Google Scholar] [CrossRef]
- Purkerson, J.M.; Schwartz, G.J. The Role of Carbonic Anhydrases in Renal Physiology. Kidney Int. 2007, 71, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Lönnerholm, G.; Wistrand, P.J. Carbonic Anhydrase in the Human Kidney: A Histochemical and Immunocytochemical Study. Kidney Int. 1984, 25, 886–898. [Google Scholar] [CrossRef] [Green Version]
- Wistrand, J.; Lindahl, S.; Wåhlstrand, T. Human Renal Carbonic Anhydrase. Purification and Properties. Eur. J. Biochem. FEBS 1975, 57, 189–195. [Google Scholar] [CrossRef]
- Lönnerholm, G.; Wistrand, P.J. Membrane-Bound Carbonic Anhydrase CA IV in the Human Kidney. Acta Physiol. Scand. 1991, 141, 231–234. [Google Scholar] [CrossRef]
- Wistrand, P.J. Human Renal Cytoplasmic Carbonic Anhydrase. Tissue Levels and Kinetic Properties under near Physiological Conditions. Acta Physiol. Scand. 1980, 109, 239–248. [Google Scholar] [CrossRef]
- Maren, T.H.; Ellison, A.C. A Study of Renal Carbonic Anhydrase. Mol. Pharmacol. 1967, 3, 503–508. [Google Scholar] [PubMed]
- Schwartz, G.J.; Kittelberger, A.M.; Barnhart, D.A.; Vijayakumar, S. Carbonic Anhydrase IV Is Expressed in H(+)-Secreting Cells of Rabbit Kidney. Am. J. Physiol. Ren. Physiol. 2000, 278, F894–F904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wistrand, P.J.; Kinne, R. Carbonic Anhydrase Activity of Isolated Brush Border and Basal-Lateral Membranes of Renal Tubular Cells. Pflugers Arch. 1977, 370, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Murer, H.; Hopfer, U.; Kinne, R. Sodium/Proton Antiport in Brush-Border-Membrane Vesicles Isolated from Rat Small Intestine and Kidney. Biochem. J. 1976, 154, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Kinsella, J.L.; Aronson, P.S. Properties of the Na+-H+ Exchanger in Renal Microvillus Membrane Vesicles. Am. J. Physiol. 1980, 238, F461–F469. [Google Scholar] [CrossRef] [Green Version]
- Boron, W.F.; Boulpaep, E.L. Intracellular pH Regulation in the Renal Proximal Tubule of the Salamander. Na+-H+ Exchange. J. Gen. Physiol. 1983, 81, 29–52. [Google Scholar] [CrossRef]
- Guo, Y.-M.; Liu, Y.; Liu, M.; Wang, J.-L.; Xie, Z.-D.; Chen, K.-J.; Wang, D.-K.; Occhipinti, R.; Boron, W.F.; Chen, L.-M. Na+/HCO₃− Cotransporter NBCn2 Mediates HCO₃− Reclamation in the Apical Membrane of Renal Proximal Tubules. J. Am. Soc. Nephrol. JASN 2017, 28, 2409–2419. [Google Scholar] [CrossRef] [Green Version]
- Nakhoul, N.L.; Davis, B.A.; Romero, M.F.; Boron, W.F. Effect of Expressing the Water Channel Aquaporin-1 on the CO₂ Permeability of Xenopus Oocytes. Am. J. Physiol. 1998, 274, C543–C548. [Google Scholar] [CrossRef]
- Zhou, Y.; Bouyer, P.; Boron, W.F. Evidence That AQP1 Is a Functional CO₂ Channel in Proximal Tubules. FASEB J. 2006, 20, A1225–A1226. [Google Scholar] [CrossRef]
- Cooper, G.J.; Boron, W.F. Effect of PCMBS on CO₂ Permeability of Xenopus Oocytes Expressing Aquaporin 1 or Its C189S Mutant. Am. J. Physiol. 1998, 275, C1481–C1486. [Google Scholar] [CrossRef]
- Preston, G.M.; Carroll, T.P.; Guggino, W.B.; Agre, P. Appearance of Water Channels in Xenopus Oocytes Expressing Red Cell CHIP28 Protein. Science 1992, 256, 385–387. [Google Scholar] [CrossRef] [Green Version]
- Boron, W.F.; Boulpaep, E.L. Intracellular pH Regulation in the Renal Proximal Tubule of the Salamander. Basolateral HCO₃− Transport. J. Gen. Physiol. 1983, 81, 53–94. [Google Scholar] [CrossRef]
- Zhou, Y.; Skelton, L.A.; Xu, L.; Chandler, M.P.; Berthiaume, J.M.; Boron, W.F. Role of Receptor Protein Tyrosine Phosphatase γ in Sensing Extracellular CO₂ and HCO₃−. J. Am. Soc. Nephrol. JASN 2016, 27, 2616–2621. [Google Scholar] [CrossRef] [Green Version]
- Skelton, L.A.; Boron, W.F. Effect of Acute Acid-Base Disturbances on ErbB1/2 Tyrosine Phosphorylation in Rabbit Renal Proximal Tubules. Am. J. Physiol. Ren. Physiol. 2013, 305, F1747–F1764. [Google Scholar] [CrossRef] [Green Version]
- Skelton, L.A.; Boron, W.F.; Zhou, Y. Acid-Base Transport by the Renal Proximal Tubule. J. Nephrol. 2010, 23 (Suppl. S16), S4–S18. [Google Scholar]
- Skelton, L.A.; Boron, W.F. Effect of Acute Acid-Base Disturbances on the Phosphorylation of Phospholipase C-Γ1 and Erk1/2 in the Renal Proximal Tubule. Physiol. Rep. 2015, 3, e12280. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.; Zhu, X.L.; Sly, W.S. Localization of Membrane-Associated Carbonic Anhydrase Type IV in Kidney Epithelial Cells. Proc. Natl. Acad. Sci. USA 1990, 87, 7457–7461. [Google Scholar] [CrossRef] [Green Version]
- Barnea, G.; Silvennoinen, O.; Shaanan, B.; Honegger, A.M.; Canoll, P.D.; D’Eustachio, P.; Morse, B.; Levy, J.B.; Laforgia, S.; Huebner, K. Identification of a Carbonic Anhydrase-like Domain in the Extracellular Region of RPTP Gamma Defines a New Subfamily of Receptor Tyrosine Phosphatases. Mol. Cell. Biol. 1993, 13, 1497–1506. [Google Scholar]
- Skelton, L.; Musa-Aziz, R.; Qin, X.; Boron, W.F. Mutations That “Restore” Enzymatic Activity to the Carbonic Anhydrase-like Domain (CALD) of Receptor Protein Tyrosine Phosphatase Gamma (RPTPγ): Importance of Shuttle Hits and CO2 Binding Site. J. Am. Soc. Nephrol. 2010, 21, 252A. [Google Scholar]
- Gross, E.; Pushkin, A.; Abuladze, N.; Fedotoff, O.; Kurtz, I. Regulation of the Sodium Bicarbonate Cotransporter KNBC1 Function: Role of Asp986, Asp988 and KNBC1-Carbonic Anhydrase II Binding. J. Physiol. 2002, 544, 679–685. [Google Scholar] [CrossRef]
- Pushkin, A.; Abuladze, N.; Gross, E.; Newman, D.; Tatishchev, S.; Lee, I.; Fedotoff, O.; Bondar, G.; Azimov, R.; Ngyuen, M.; et al. Molecular Mechanism of KNBC1-Carbonic Anhydrase II Interaction in Proximal Tubule Cells. J. Physiol. 2004, 559, 55–65. [Google Scholar] [CrossRef]
- Alvarez, B.V.; Loiselle, F.B.; Supuran, C.T.; Schwartz, G.J.; Casey, J.R. Direct Extracellular Interaction between Carbonic Anhydrase IV and the Human NBC1 Sodium/Bicarbonate Co-Transporter. Biochemistry 2003, 42, 12321–12329. [Google Scholar] [CrossRef] [PubMed]
- Piermarini, P.M.; Kim, E.Y.; Boron, W.F. Evidence against a Direct Interaction between Intracellular Carbonic Anhydrase II and Pure C-Terminal Domains of SLC4 Bicarbonate Transporters. J. Biol. Chem. 2007, 282, 1409–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Daly, C.M.; Parker, M.D.; Gill, H.S.; Piermarini, P.M.; Pelletier, M.F.; Boron, W.F. Effect of Human Carbonic Anhydrase II on the Activity of the Human Electrogenic Na/HCO3 Cotransporter NBCe1-A in Xenopus Oocytes. J. Biol. Chem. 2006, 281, 19241–19250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, F.J.; Boron, W.F. Carbonic Anhydrases Enhance Activity of Endogenous Na-H Exchangers and Not the Electrogenic Na/HCO₃ Cotransporter NBCe1-A, Expressed in Xenopus Oocytes. J. Physiol. 2020, 598, 5821–5856. [Google Scholar] [CrossRef]
- Zacchia, M.; Capolongo, G.; Rinaldi, L.; Capasso, G. The Importance of the Thick Ascending Limb of Henle’s Loop in Renal Physiology and Pathophysiology. Int. J. Nephrol. Renov. Dis. 2018, 11, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Vorum, H.; Kwon, T.H.; Fulton, C.; Simonsen, B.; Choi, I.; Boron, W.; Maunsbach, A.B.; Nielsen, S.; Aalkjaer, C. Immunolocalization of Electroneutral Na-HCO₃− Cotransporter in Rat Kidney. Am. J. Physiol. Ren. Physiol. 2000, 279, F901–F909. [Google Scholar] [CrossRef] [Green Version]
- Boedtkjer, E.; Praetorius, J.; Füchtbauer, E.-M.; Aalkjaer, C. Antibody-Independent Localization of the Electroneutral Na+-HCO₃− Cotransporter NBCn1 (Slc4a7) in Mice. Am. J. Physiol. Cell Physiol. 2008, 294, C591–C603. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-L.; Wang, X.-Y.; Wang, D.-K.; Parker, M.D.; Musa-Aziz, R.; Popple, J.; Guo, Y.-M.; Min, T.-X.; Xia, T.; Tan, M.; et al. Multiple Acid-Base and Electrolyte Disturbances Upregulate NBCn1, NBCn2, IRBIT and L-IRBIT in the MTAL. J. Physiol. 2020, 598, 3395–3415. [Google Scholar] [CrossRef]
- Choi, I.; Aalkjaer, C.; Boulpaep, E.L.; Boron, W.F. An Electroneutral Sodium/Bicarbonate Cotransporter NBCn1 and Associated Sodium Channel. Nature 2000, 405, 571–575. [Google Scholar] [CrossRef]
- Odgaard, E.; Jakobsen, J.K.; Frische, S.; Praetorius, J.; Nielsen, S.; Aalkjaer, C.; Leipziger, J. Basolateral Na+-Dependent HCO3− Transporter NBCn1-Mediated HCO3− Influx in Rat Medullary Thick Ascending Limb. J. Physiol. 2004, 555, 205–218. [Google Scholar] [CrossRef]
- Lee, S.; Lee, H.J.; Yang, H.S.; Thornell, I.M.; Bevensee, M.O.; Choi, I. Sodium-Bicarbonate Cotransporter NBCn1 in the Kidney Medullary Thick Ascending Limb Cell Line Is Upregulated under Acidic Conditions and Enhances Ammonium Transport. Exp. Physiol. 2010, 95, 926–937. [Google Scholar] [CrossRef] [Green Version]
- Attmane-Elakeb, A.; Mount, D.B.; Sibella, V.; Vernimmen, C.; Hebert, S.C.; Bichara, M. Stimulation by in Vivo and in Vitro Metabolic Acidosis of Expression of RBSC-1, the Na+-K+(NH4+)-2Cl- Cotransporter of the Rat Medullary Thick Ascending Limb. J. Biol. Chem. 1998, 273, 33681–33691. [Google Scholar] [CrossRef] [Green Version]
- Jans, F.; Balut, C.; Ameloot, M.; Wouters, P.; Steels, P. Investigation of the Ba2+− Sensitive NH4+ Transport Pathways in the Apical Cell Membrane of Primary Cultured Rabbit MTAL Cells. Nephron Physiol. 2007, 106, p45–p53. [Google Scholar] [CrossRef]
- Cabral, P.D.; Herrera, M. Membrane-Associated Aquaporin-1 Facilitates Osmotically Driven Water Flux across the Basolateral Membrane of the Thick Ascending Limb. Am. J. Physiol. Ren. Physiol. 2012, 303, F621–F629. [Google Scholar] [CrossRef] [Green Version]
- Geyer, R.R.; Parker, M.D.; Toye, A.M.; Boron, W.F.; Musa-Aziz, R. Relative CO₂/NH₃ Permeabilities of Human RhAG, RhBG and RhCG. J. Membr. Biol. 2013, 246, 915–926. [Google Scholar] [CrossRef]
- Kwon, T.-H.; Fulton, C.; Wang, W.; Kurtz, I.; Frøkiaer, J.; Aalkjaer, C.; Nielsen, S. Chronic Metabolic Acidosis Upregulates Rat Kidney Na-HCO3− Cotransporters NBCn1 and NBC3 but Not NBC1. Am. J. Physiol. Ren. Physiol. 2002, 282, F341–F351. [Google Scholar] [CrossRef]
- Sterling, D.; Reithmeier, R.A.; Casey, J.R. A Transport Metabolon. Functional Interaction of Carbonic Anhydrase II and Chloride/Bicarbonate Exchangers. J. Biol. Chem. 2001, 276, 47886–47894. [Google Scholar] [CrossRef] [Green Version]
- Vince, J.W.; Reithmeier, R.A. Identification of the Carbonic Anhydrase II Binding Site in the Cl−/HCO3− Anion Exchanger AE1. Biochemistry 2000, 39, 5527–5533. [Google Scholar] [CrossRef]
- Vince, J.W.; Reithmeier, R.A. Carbonic Anhydrase II Binds to the Carboxyl Terminus of Human Band 3, the Erythrocyte Cl−/HCO3− Exchanger. J. Biol. Chem. 1998, 273, 28430–28437. [Google Scholar] [CrossRef] [Green Version]
- Vince, J.W.; Carlsson, U.; Reithmeier, R.A. Localization of the Cl−/HCO3− Anion Exchanger Binding Site to the Amino-Terminal Region of Carbonic Anhydrase II. Biochemistry 2000, 39, 13344–13349. [Google Scholar] [CrossRef] [PubMed]
- Sterling, D.; Alvarez, B.V.; Casey, J.R. The Extracellular Component of a Transport Metabolon. Extracellular Loop 4 of the Human AE1 Cl−/HCO3− Exchanger Binds Carbonic Anhydrase IV. J. Biol. Chem. 2002, 277, 25239–25246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Samir, S.; Papadopoulos, S.; Scheibe, R.J.; Meißner, J.D.; Cartron, J.-P.; Sly, W.S.; Alper, S.L.; Gros, G.; Endeward, V. Activity and Distribution of Intracellular Carbonic Anhydrase II and Their Effects on the Transport Activity of Anion Exchanger AE1/SLC4A1. J. Physiol. 2013, 591, 4963–4982. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; Kaissling, B. Chapter 20—Structural Organization of the Mammalian Kidney. In Seldin and Giebisch’s the Kidney, 5th ed.; Alpern, R.J., Moe, O.W., Caplan, M., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 595–691. ISBN 978-0-12-381462-3. [Google Scholar]
- Teng-umnuay, P.; Verlander, J.W.; Yuan, W.; Tisher, C.C.; Madsen, K.M. Identification of Distinct Subpopulations of Intercalated Cells in the Mouse Collecting Duct. J. Am. Soc. Nephrol. JASN 1996, 7, 260–274. [Google Scholar] [CrossRef]
- Kim, J.; Kim, Y.H.; Cha, J.H.; Tisher, C.C.; Madsen, K.M. Intercalated Cell Subtypes in Connecting Tubule and Cortical Collecting Duct of Rat and Mouse. J. Am. Soc. Nephrol. JASN 1999, 10, 1–12. [Google Scholar] [CrossRef]
- Wall, S.M.; Verlander, J.W.; Romero, C.A. The Renal Physiology of Pendrin-Positive Intercalated Cells. Physiol. Rev. 2020, 100, 1119–1147. [Google Scholar] [CrossRef]
- Chen, L.; Clark, J.Z.; Nelson, J.W.; Kaissling, B.; Ellison, D.H.; Knepper, M.A. Renal-Tubule Epithelial Cell Nomenclature for Single-Cell RNA-Sequencing Studies. J. Am. Soc. Nephrol. JASN 2019, 30, 1358–1364. [Google Scholar] [CrossRef]
- Loffing, J.; Pietri, L.; Aregger, F.; Bloch-Faure, M.; Ziegler, U.; Meneton, P.; Rossier, B.C.; Kaissling, B. Differential Subcellular Localization of ENaC Subunits in Mouse Kidney in Response to High- and Low-Na Diets. Am. J. Physiol. Ren. Physiol. 2000, 279, F252–F258. [Google Scholar] [CrossRef] [Green Version]
- Masilamani, S.; Kim, G.H.; Mitchell, C.; Wade, J.B.; Knepper, M.A. Aldosterone-Mediated Regulation of ENaC Alpha, Beta, and Gamma Subunit Proteins in Rat Kidney. J. Clin. Investig. 1999, 104, R19–R23. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Al-bataineh, M.M.; Pastor-Soler, N.M. Collecting Duct Intercalated Cell Function and Regulation. Clin. J. Am. Soc. Nephrol. CJASN 2015, 10, 305–324. [Google Scholar] [CrossRef] [Green Version]
- Alper, S.L.; Natale, J.; Gluck, S.; Lodish, H.F.; Brown, D. Subtypes of Intercalated Cells in Rat Kidney Collecting Duct Defined by Antibodies against Erythroid Band 3 and Renal Vacuolar H+-ATPase. Proc. Natl. Acad. Sci. USA 1989, 86, 5429–5433. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.; Gluck, S.; Hartwig, J. Structure of the Novel Membrane-Coating Material in Proton-Secreting Epithelial Cells and Identification as an H+ ATPase. J. Cell Biol. 1987, 105, 1637–1648. [Google Scholar] [CrossRef]
- Brown, D.; Weyer, P.; Orci, L. Nonclathrin-Coated Vesicles Are Involved in Endocytosis in Kidney Collecting Duct Intercalated Cells. Anat. Rec. 1987, 218, 237–242. [Google Scholar] [CrossRef]
- Brown, D.; Hirsch, S.; Gluck, S. An H+-ATPase in Opposite Plasma Membrane Domains in Kidney Epithelial Cell Subpopulations. Nature 1988, 331, 622–624. [Google Scholar] [CrossRef]
- Al-Awqati, Q. Terminal Differentiation in Epithelia: The Role of Integrins in Hensin Polymerization. Annu. Rev. Physiol. 2011, 73, 401–412. [Google Scholar] [CrossRef]
- Purkerson, J.M.; Tsuruoka, S.; Suter, D.Z.; Nakamori, A.; Schwartz, G.J. Adaptation to Metabolic Acidosis and Its Recovery Are Associated with Changes in Anion Exchanger Distribution and Expression in the Cortical Collecting Duct. Kidney Int. 2010, 78, 993–1005. [Google Scholar] [CrossRef] [Green Version]
- Takito, J.; Hikita, C.; Al-Awqati, Q. Hensin, a New Collecting Duct Protein Involved in the in Vitro Plasticity of Intercalated Cell Polarity. J. Clin. Investig. 1996, 98, 2324–2331. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Eladari, D.; Leviel, F.; Tew, B.Y.; Miró-Julià, C.; Cheema, F.H.; Miller, L.; Nelson, R.; Paunescu, T.G.; McKee, M.; et al. Deletion of Hensin/DMBT1 Blocks Conversion of Beta- to Alpha-Intercalated Cells and Induces Distal Renal Tubular Acidosis. Proc. Natl. Acad. Sci. USA 2010, 107, 21872–21877. [Google Scholar] [CrossRef] [Green Version]
- Bastani, B.; Purcell, H.; Hemken, P.; Trigg, D.; Gluck, S. Expression and Distribution of Renal Vacuolar Proton-Translocating Adenosine Triphosphatase in Response to Chronic Acid and Alkali Loads in the Rat. J. Clin. Investig. 1991, 88, 126–136. [Google Scholar] [CrossRef]
- Sabolić, I.; Brown, D.; Gluck, S.L.; Alper, S.L. Regulation of AE1 Anion Exchanger and H(+)-ATPase in Rat Cortex by Acute Metabolic Acidosis and Alkalosis. Kidney Int. 1997, 51, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Verlander, J.W.; Lee, H.-W.; Wall, S.M.; Harris, A.N.; Weiner, I.D. The Proximal Tubule through an NBCe1-Dependent Mechanism Regulates Collecting Duct Phenotypic and Remodeling Responses to Acidosis. Am. J. Physiol. Ren. Physiol. 2023, 324, F12–F29. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Tisher, C.C.; Linser, P.J.; Madsen, K.M. Ultrastructural Localization of Carbonic Anhydrase II in Subpopulations of Intercalated Cells of the Rat Kidney. J. Am. Soc. Nephrol. JASN 1990, 1, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Nagami, G.T.; Hamm, L.L. Regulation of Acid-Base Balance in Chronic Kidney Disease. Adv. Chronic Kidney Dis. 2017, 24, 274–279. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene (Protein) Name | mRNA Expression | Protein Expression | ||||||
|---|---|---|---|---|---|---|---|---|
| Very Low | Low | Medium | High | Low | Medium | High | N/A * | |
| SLC4A1 (AE1) | x | x | ||||||
| SLC4A2 (AE2) | x | x | ||||||
| SLC4A3 (AE3) | x | x | ||||||
| SLC4A4 (NBCe1) | x | x | ||||||
| SLC4A5 (NBCe2) | x | x | ||||||
| SLC4A7 (NBCn1) | x | x | ||||||
| SLC4A8 (NDCBE) | x | x | ||||||
| SLC4A9 (AE4) | x | x | ||||||
| SLC4A10 (NBCn2) | x | x | ||||||
| Gene (Protein) Name | mRNA Expression | Protein Expression | ||||||
|---|---|---|---|---|---|---|---|---|
| Very Low | Low | Medium | High | Low | Medium | High | N/A * | |
| CA1 (CA I) | x | x | ||||||
| CA2 (CA II) | x | x | ||||||
| CA3 (CA III) | x | x | ||||||
| CA4 (CA IV) | x | x | ||||||
| CA5A (CA VA) | x | x | ||||||
| CA5B (CA VB) | x | x | ||||||
| CA6 (CA VI) | x | x | ||||||
| CA7 (CA VII) | x | x | ||||||
| CA8 (CA VIII) | x | x | ||||||
| CA9 (CA IX) | x | x | ||||||
| CA10 (CA X) | x | x | ||||||
| CA11 (CA XI) | x | x | ||||||
| CA12 (CA XII) | x | x | ||||||
| CA13 (CA XIII) | x | x | ||||||
| CA14 (CA XIV) | x | x | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.-K.; Boron, W.F.; Occhipinti, R. Potential Novel Role of Membrane-Associated Carbonic Anhydrases in the Kidney. Int. J. Mol. Sci. 2023, 24, 4251. https://doi.org/10.3390/ijms24044251
Lee S-K, Boron WF, Occhipinti R. Potential Novel Role of Membrane-Associated Carbonic Anhydrases in the Kidney. International Journal of Molecular Sciences. 2023; 24(4):4251. https://doi.org/10.3390/ijms24044251
Chicago/Turabian StyleLee, Seong-Ki, Walter F. Boron, and Rossana Occhipinti. 2023. "Potential Novel Role of Membrane-Associated Carbonic Anhydrases in the Kidney" International Journal of Molecular Sciences 24, no. 4: 4251. https://doi.org/10.3390/ijms24044251
APA StyleLee, S. -K., Boron, W. F., & Occhipinti, R. (2023). Potential Novel Role of Membrane-Associated Carbonic Anhydrases in the Kidney. International Journal of Molecular Sciences, 24(4), 4251. https://doi.org/10.3390/ijms24044251