

[4+2]-Cycloaddition to 5-Methylidene-Hydantoins and 5-Methylidene-2-Thiohydantoins in the Synthesis of Spiro-2-Chalcogenimidazolones
 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
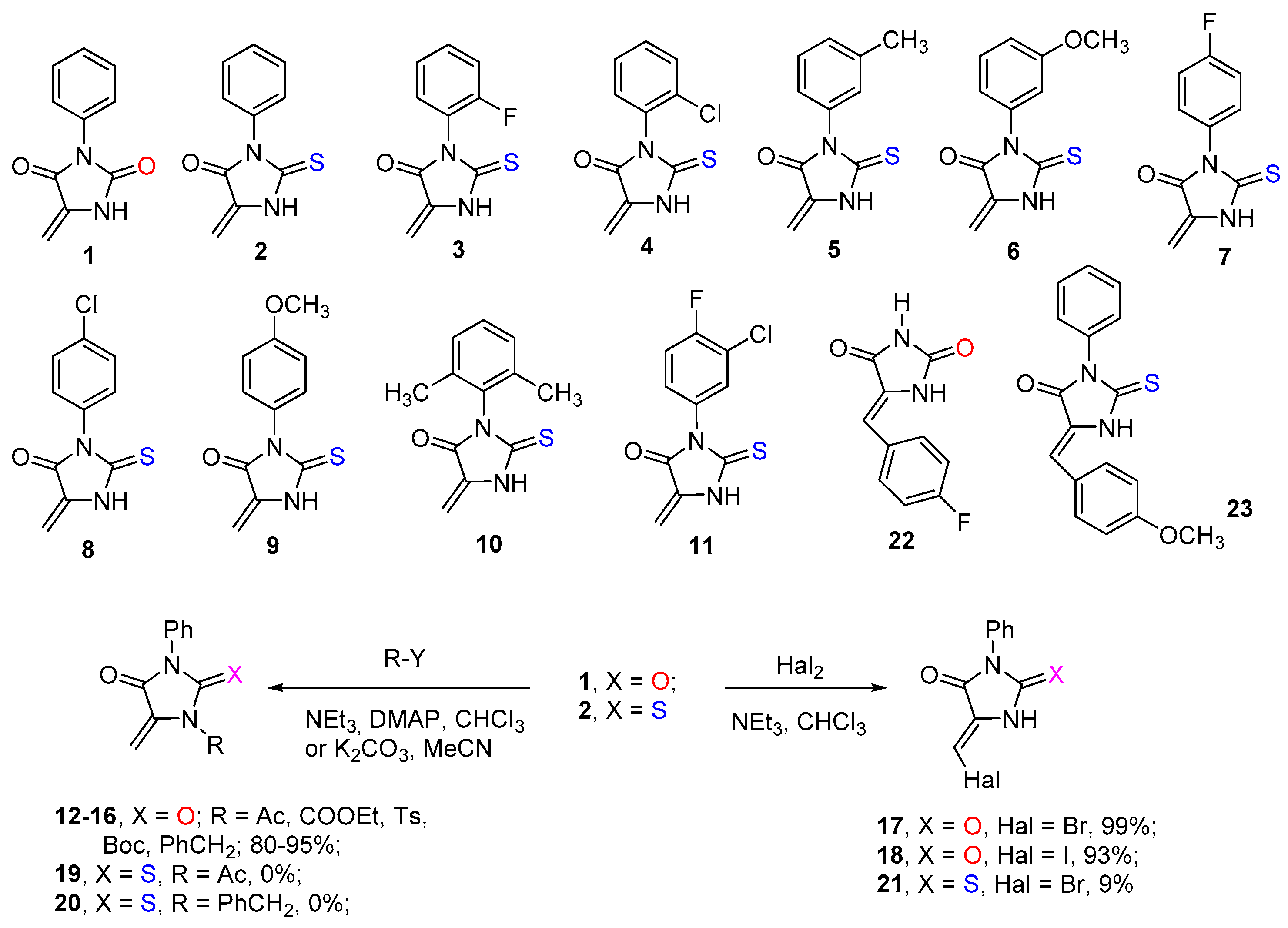
2.1. Synthesis of Dienophiles
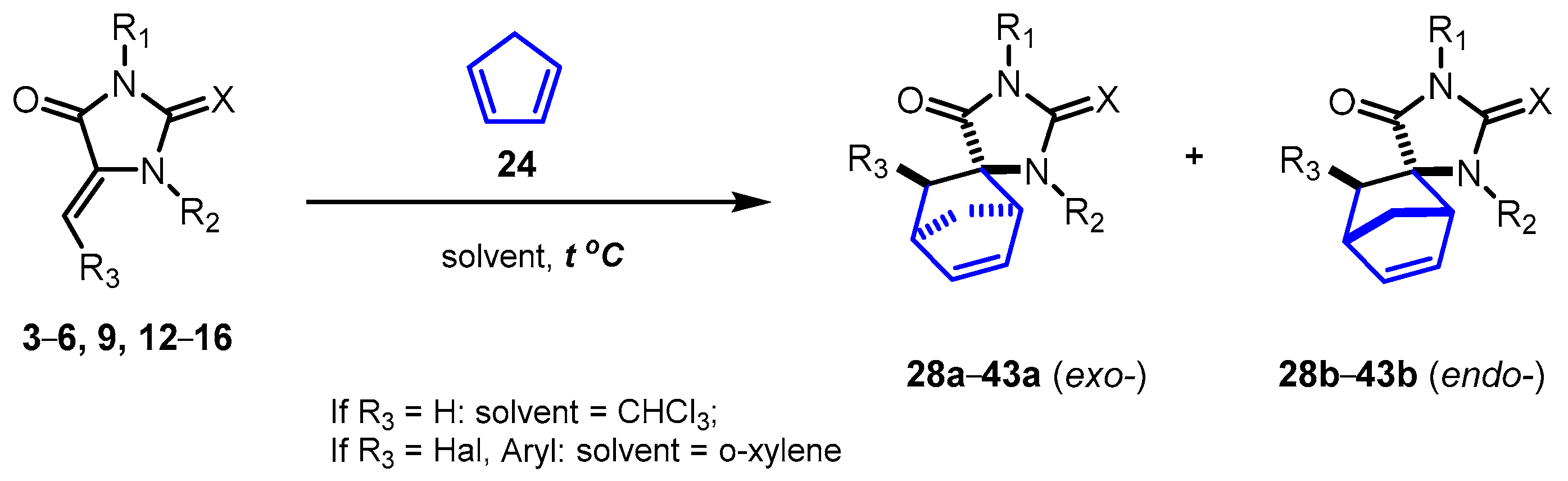
2.2. Reactions of 5-Methylideneimidazolones 1-6, 9, 12-18, 22, and 23 with Cyclopentadiene
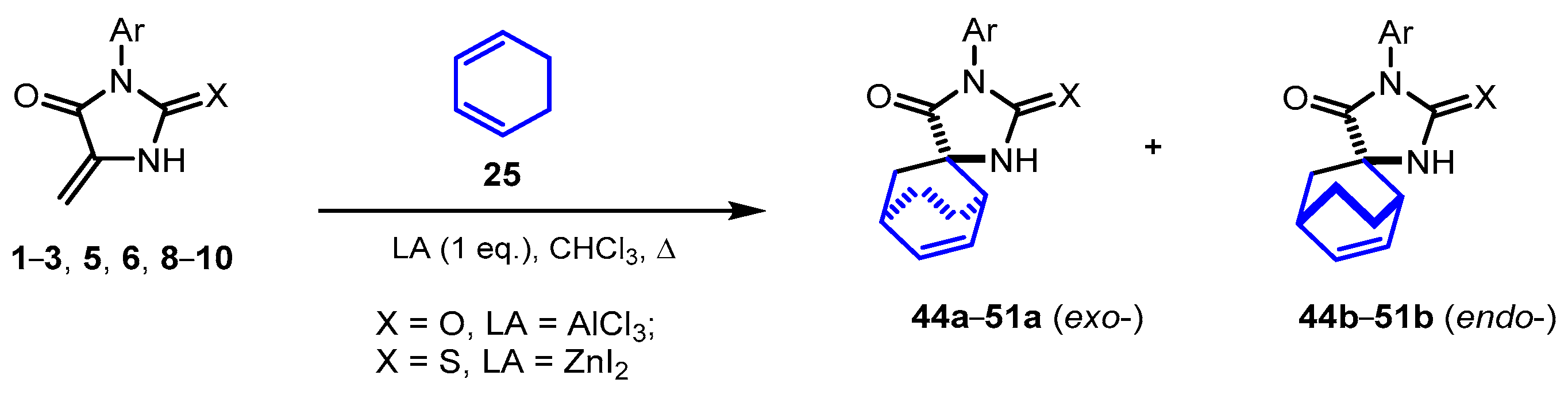
2.3. Reactions of 5-Methylideneimidazolones 1-3, 5, 6, 8-10 with Cyclohexadiene
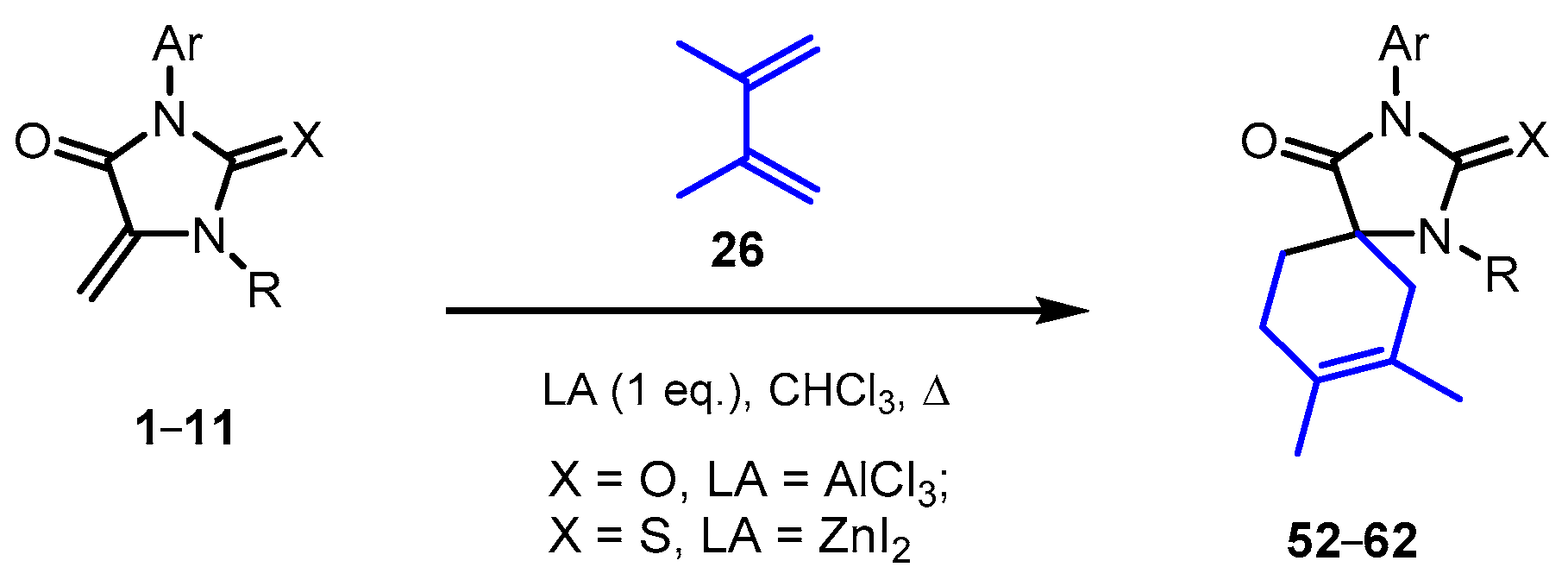
2.4. Reactions of 5-Methylideimidazolones 1-12 with 2,3-Dimethylbutadiene
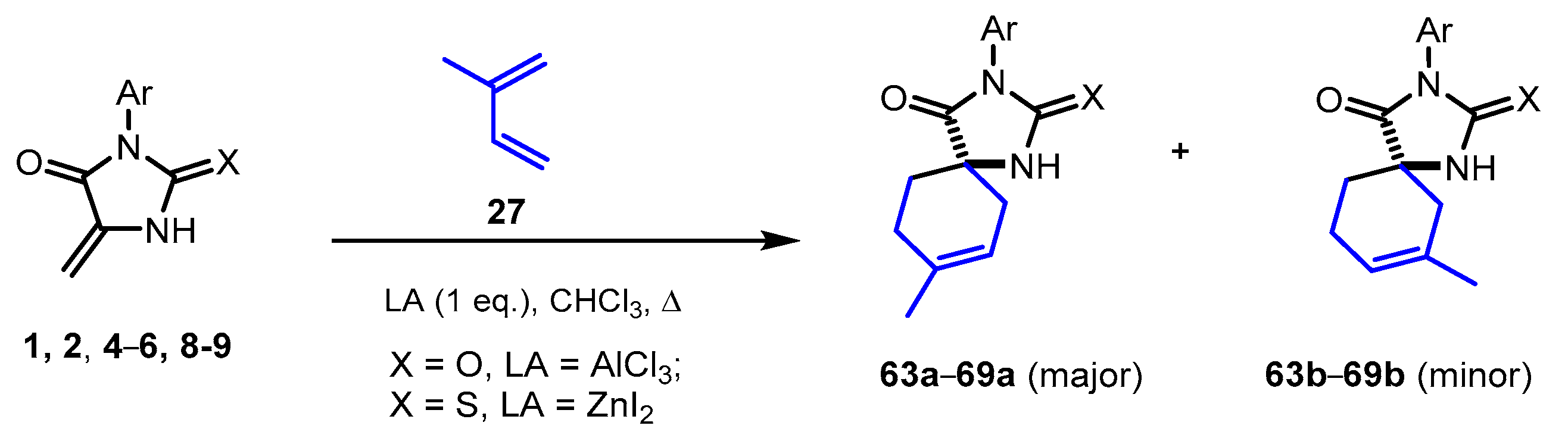
2.5. Reactions of 5-Methylideneimidazolones 1, 2, 4-6, 8, and 9 with Isoprene
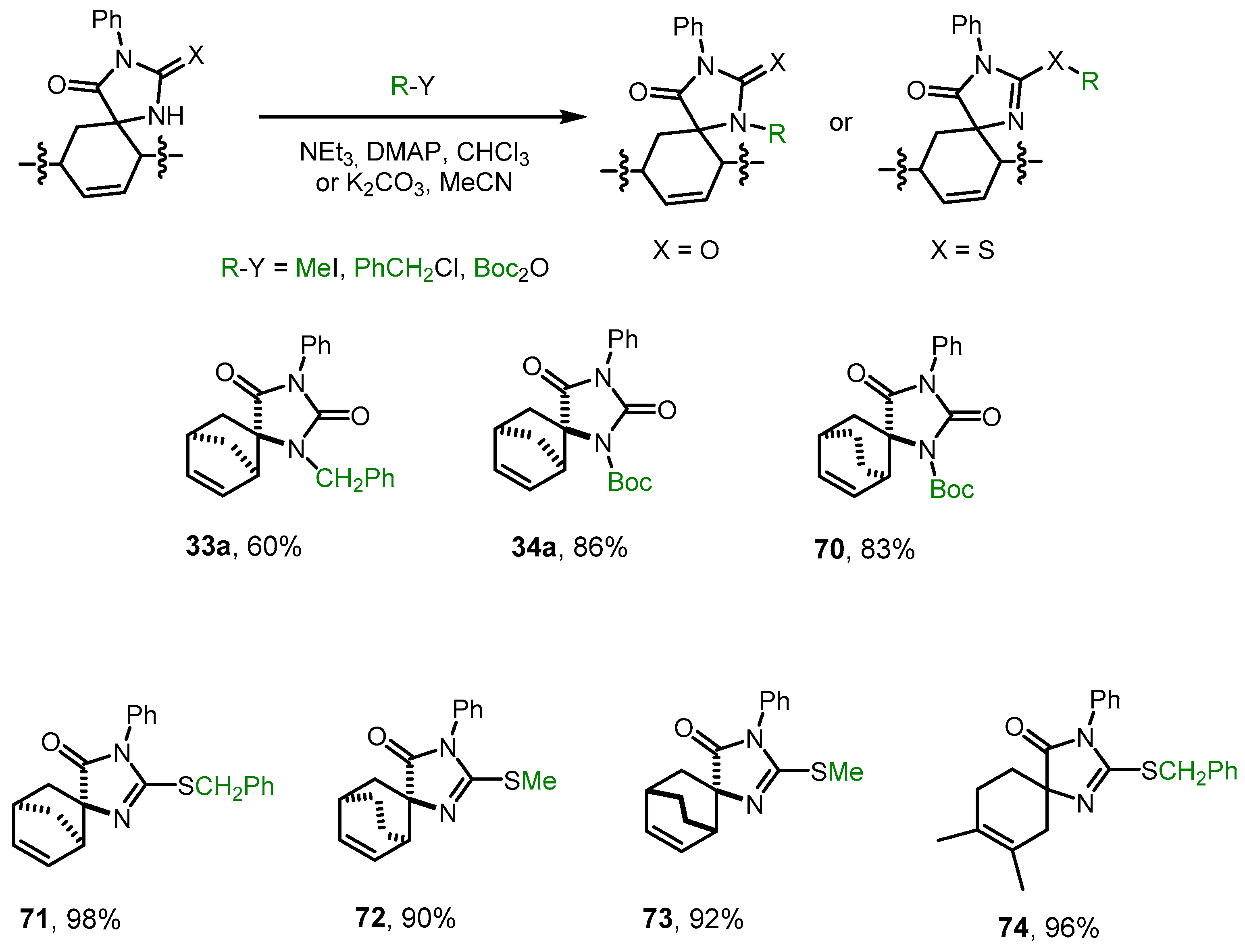
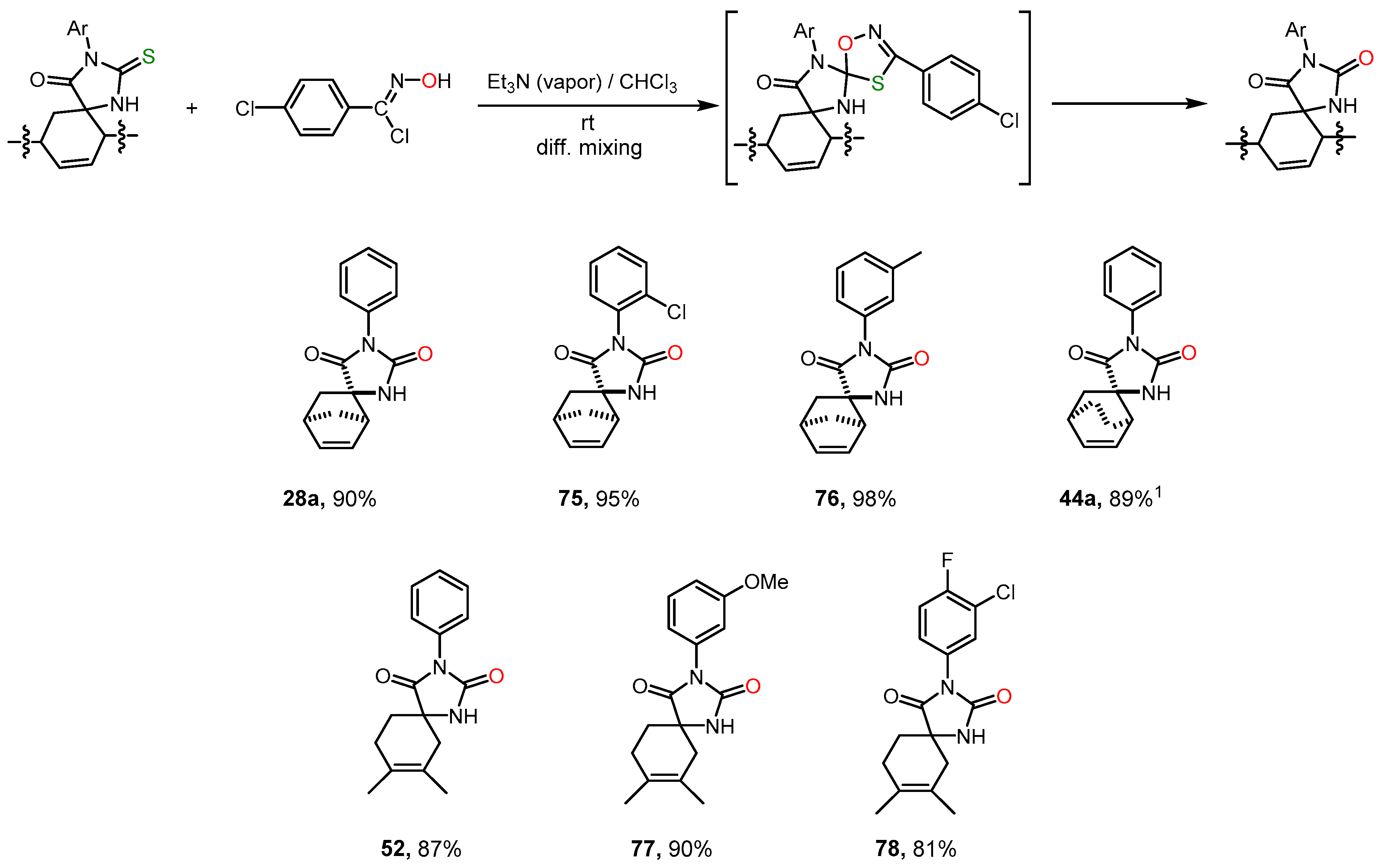
2.6. Alkylation and Acylation of [4+2]-Cycloaddition Reaction Products
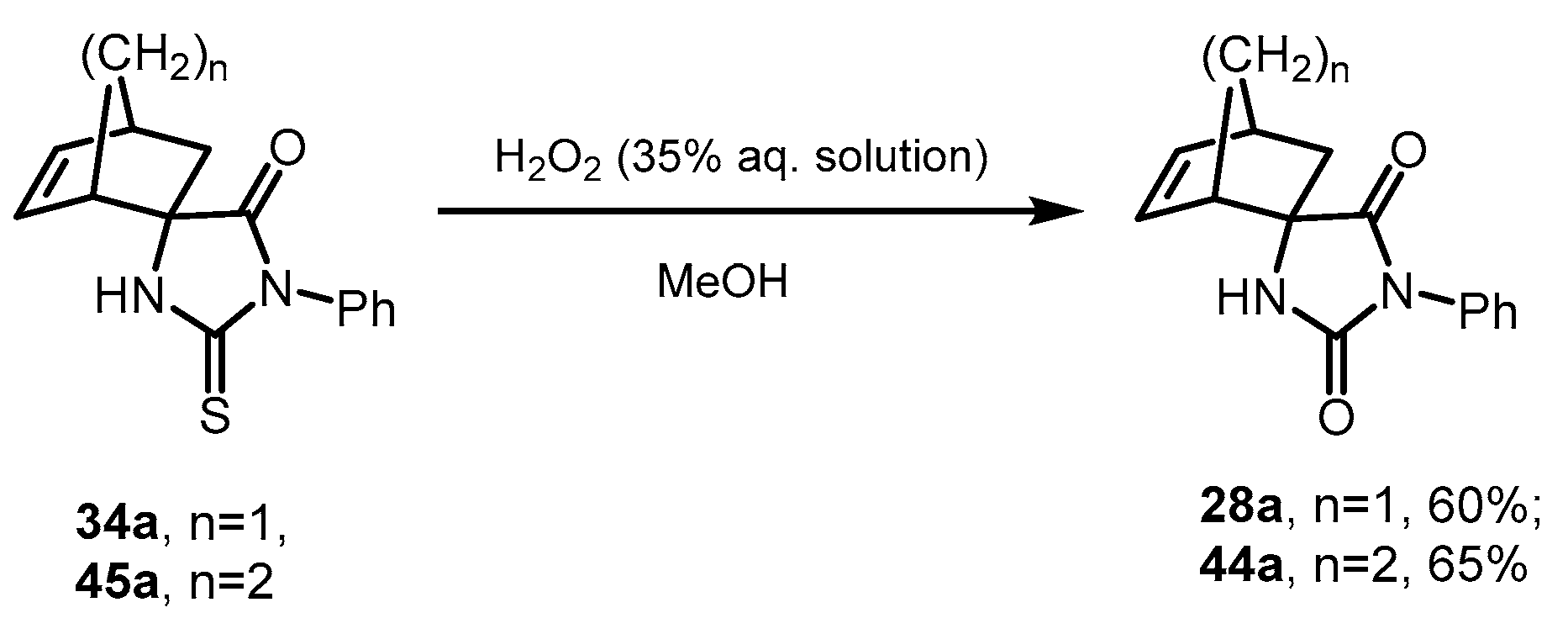
2.7. Desulfurization of Spiro-Thiohydantoins
2.8. Cytotoxicity against Human Cell Lines
2.9. Cytotoxicity against E. coli
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scammells, P.J.; Baker, S.P.; Luiz Bellardinelli, R.A.O.; Russell, R.A.; Wright, D.M. The Synthesis of the Novel Adenosine Agonists, Exo- and Endo. N6-(5,6-Epoxynorborn-2-Yl)Adenosine. Tetrahedron 1996, 52, 4735–4744. [Google Scholar] [CrossRef]
- Lee, W.G.; Lee, S.D.; Cho, J.H.; Jung, Y.; Kim, J.H.; Hien, T.T.; Kang, K.W.; Ko, H.; Kim, Y.C. Structure-Activity Relationships and Optimization of 3,5-Dichloropyridine Derivatives as Novel P2X 7 Receptor Antagonists. J. Med. Chem. 2012, 55, 3687–3698. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Sakashita, H.; Murakami, K.; Sugiura, M.; Kondo, T.; Fukaya, C. Novel Histamine H3 Receptor Antagonists: Synthesis and Evaluation of Formamidine and S-Methylisothiourea Derivatives. Chem. Pharm. Bull. 1997, 45, 305–311. [Google Scholar] [CrossRef] [Green Version]
- Meguro, K.; Tawada, H.; Sugiyama, Y.; Fujita, T.; Kawamatsu, Y. Studies on Antidiabetic Agents. VII Synthesis and Hypoglycemic Activity of 4-Oxazoleacetic Acid Derivatives. Chem. Pharm. Bull. 1986, 34, 2840–2851. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.L.; Roche, V.F.; Pendola, K.; Kearley, M.; Lei, L.; Romstedt, K.J.; Herdman, M.; Shams, G.; Kaisare, V.; Feller, D.R. Synthesis and in Vitro Platelet Aggregation and TP Receptor Binding Studies on Bicyclic 5,8-Ethanooctahydroisoquinolines and 5,8-Ethanotetrahydroisoquinolines. Bioorg. Med. Chem. 2002, 10, 2779–2793. [Google Scholar] [CrossRef] [PubMed]
- Burmistrov, V.; Morisseau, C.; Karlov, D.; Pitushkin, D.; Vernigora, A.; Rasskazova, E.; Butov, G.M.; Hammock, B.D. Bioisosteric Substitution of Adamantane with Bicyclic Lipophilic Groups Improves Water Solubility of Human Soluble Epoxide Hydrolase Inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127430. [Google Scholar] [CrossRef] [PubMed]
- Saccomano, N.A.; Vinick, F.J.; Koe, B.K.; Nielsen, J.A.; Whalen, W.M.; Meltz, M.; Phillips, D.; Thadieo, P.F.; Jung, S.; Chapin, D.S.; et al. Calcium-Independent Phosphodiesterase Inhibitors as Putative Antidepressants: [3-(Bicycloalkyloxy)-4-Methoxyphenyl]-2-Imidazolidinones. J. Med. Chem. 1991, 34, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Wang, Y.; Meng, X.; Yang, Y. Design, Synthesis and Biological Evaluation of Novel Thiohydantoin Derivatives as Potent Androgen Receptor Antagonists for the Treatment of Prostate Cancer. Bioorg. Med. Chem. 2021, 31, 115953. [Google Scholar] [CrossRef]
- Isizumi, K.; Kojima, A.; Antoku, F.; Saji, I.; Yoshigi, M. Succinimide Derivatives. II. Synthesis and Antipsychotic Activity of N-[4-[4-(1,2-Benzisothiazol-3-Yl)-1-Piperazinyl] Butyl]-1,2-Cis-Cyclohexanedicarboximide (SM-9018) and Related Compounds. Chem. Pharm. Bull. 1995, 43, 2139–2151. [Google Scholar] [CrossRef] [Green Version]
- Valderrama, J.A.; Zamorano, C.; González, M.F.; Prina, E.; Fournet, A. Studies on Quinones. Part 39: Synthesis and Leishmanicidal Activity of Acylchloroquinones and Hydroquinones. Bioorg. Med. Chem. 2005, 13, 4153–4159. [Google Scholar] [CrossRef]
- Bertilsson, S.K.; Ekegren, J.K.; Modin, S.A.; Andersson, P.G. The Aza-Diels-Alder Reaction Protocol—A Useful Approach to Chiral, Sterically Constrained α-Amino Acid Derivatives. Tetrahedron 2001, 57, 6399–6406. [Google Scholar] [CrossRef]
- Kukushkin, M.E.; Skvortsov, D.A.; Kalinina, M.A.; Tafeenko, V.A.; Burmistrov, V.V.; Butov, G.M.; Zyk, N.V.; Majouga, A.G.; Beloglazkina, E.K. Synthesis and Cytotoxicity of Oxindoles Dispiro Derivatives with Thiohydantoin and Adamantane Fragments. Phosphorus Sulfur Silicon Relat. Elem. 2020, 195, 544–555. [Google Scholar] [CrossRef]
- Beloglazkina, A.; Barashkin, A.; Polyakov, V.; Kotovsky, G.; Karpov, N.; Mefedova, S.; Zagribelny, B.; Ivanenkov, Y.; Kalinina, M.; Skvortsov, D.; et al. Synthesis and Biological Evaluation of Novel Dispiro Compounds Based on 5-Arylidenehydantoins and Isatins as Inhibitors of P53–MDM2 Protein–Protein Interaction. Chem. Heterocycl. Compd. 2020, 56, 747–755. [Google Scholar] [CrossRef]
- Novotortsev, V.K.; Kukushkin, M.E.; Tafeenko, V.A.; Skvortsov, D.A.; Kalinina, M.A.; Timoshenko, R.V.; Chmelyuk, N.S.; Vasilyeva, L.A.; Tarasevich, B.N.; Gorelkin, P.V.; et al. Dispirooxindoles Based on 2-Selenoxo-Imidazolidin-4-Ones: Synthesis, Cytotoxicity and Ros Generation Ability. Int. J. Mol. Sci. 2021, 22, 2613. [Google Scholar] [CrossRef] [PubMed]
- Cernak, T.A.; Gleason, J.L. Density Functional Theory Guided Design of Exo-Selective Dehydroalanine Dienophiles for Application toward the Synthesis of Palau’amine. J. Org. Chem. 2008, 73, 102–110. [Google Scholar] [CrossRef]
- Sankhavasi, W.; Kohmoto, S.; Yamamoto, M.; Nishio, T.; Iida, I.; Yamada, K. Chiral Hydantoin; A New Dienophile for Diels–Alder Reaction. Bull. Chem. Soc. Jpn. 1992, 65, 935–937. [Google Scholar] [CrossRef]
- Fraile, J.M.; Lafuente, G.; Mayoral, J.A.; Pallarés, A. Synthesis and Reactivity of 5-Methylenehydantoins. Tetrahedron 2011, 67, 8639–8647. [Google Scholar] [CrossRef] [Green Version]
- Soengas, R.G.; Da Silva, G.; Estévez, J.C. Synthesis of Spironucleosides: Past and Future Perspectives. Molecules 2017, 22, 2028. [Google Scholar] [CrossRef] [Green Version]
- Wadghane, S.; Bhor, R.; Shinde, G.; Kolhe, M.; Rathod, P. A Review on the Some Biological Activities of the Hydantoin Derivatives. J. Drug Deliv. Ther. 2023, 13, 171–178. [Google Scholar] [CrossRef]
- Fujisaki, F.; Shoji, K.; Sumoto, K. A Synthetic Application of β-Aminoalanines to Some New 5-Dialkylaminomethyl-3-Phenylhydantoin Derivatives. Heterocycles 2009, 78, 213–220. [Google Scholar] [CrossRef]
- Kukushkin, M.E.; Karpov, N.A.; Shybanov, D.E.; Zyk, N.V.; Beloglazkina, E.K. A Convenient Synthesis of 3-Aryl-5-Methylidene-2-Thiohydantoins. Mendeleev Commun. 2022, 32, 126–128. [Google Scholar] [CrossRef]
- Jia, H.H.; Ni, F.; Wang, J.; Wei, P.; Ouyang, P.K. 5-(4-Fluorobenzylidene)Imidazolidine-2,4-Dione. Acta Crystallogr. Sect. E Struct. Rep. Online 2005, 61, 2784–2786. [Google Scholar] [CrossRef]
- Maccari, R.; Ettari, R.; Adornato, I.; Naß, A.; Wolber, G.; Bitto, A.; Mannino, F.; Aliquò, F.; Bruno, G.; Nicolò, F.; et al. Identification of 2-Thioxoimidazolidin-4-One Derivatives as Novel Noncovalent Proteasome and Immunoproteasome Inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 278–283. [Google Scholar] [CrossRef]
- Shybanov, D.E.; Kukushkin, M.E.; Tafeenko, V.A.; Zyk, N.V.; Grishin, Y.K.; Roznyatovsky, V.A.; Beloglazkina, E.K. Different Addition Modes of Cyclopentadiene and Furan at Methylidene(Thio)Hydantoins. Mendeleev Commun. 2021, 31, 246–247. [Google Scholar] [CrossRef]
- Sarigul, S.; Dogan, I. Atroposelective Synthesis of Axially Chiral Thiohydantoin Derivatives. J. Org. Chem. 2016, 81, 5895–5902. [Google Scholar] [CrossRef] [PubMed]
- Kukushkin, M.E.; Kondratieva, A.A.; Karpov, A.; Shybanov, D.E.; Tafeenko, V.A.; Roznyatovsky, V.A.; Grishin, Y.K.; Moiseeva, A.; Zyk, N.V.; Beloglazkina, E.K. Access to Dispiro Indolinone-Pyrrolidine-Imidazolones. R. Soc. Open Sci. 2022, 9. [Google Scholar] [CrossRef]
- Sarigul Ozbek, S.; Bacak Erdik, M.; Dogan, I. Aldol Reactions of Conformationally Stable Axially Chiral Thiohydantoin Derivatives. ACS Omega 2021, 6, 27823–27832. [Google Scholar] [CrossRef]
- Kukushkin, M.; Novotortsev, V.; Filatov, V.; Ivanenkov, Y.; Skvortsov, D.; Veselov, M.; Shafikov, R.; Moiseeva, A.; Zyk, N.; Majouga, A.; et al. Synthesis and Biological Evaluation of S-, O- and Se-Containing Dispirooxindoles. Molecules 2021, 26, 7645. [Google Scholar] [CrossRef]
- Vilková, M.; Prokaiová, M.; Imrich, J. Spontaneous Cyclization of (Acridin-9-Ylmethyl)Thioureas to Spiro [Dihydroacridine-9′(10′H),5-Imidazolidine]-2-Thiones, a Novel Type of Acridine Spirocycles. Tetrahedron 2014, 70, 944–961. [Google Scholar] [CrossRef]
- Hewitt, R.J.; Ong, M.J.H.; Lim, Y.W.; Burkett, B.A. Investigations of the Thermal Responsiveness of 1,4,2-Oxathiazoles. Eur. J. Org. Chem. 2015, 2015, 6687–6700. [Google Scholar] [CrossRef]
- Shybanov, D.E.; Filkina, M.E.; Kukushkin, M.E.; Grishin, Y.K.; Roznyatovsky, V.A.; Zyk, N.V.; Beloglazkina, E.K. Diffusion Mixing with a Volatile Tertiary Amine as a Very Efficient Technique for 1,3-Dipolar Cycloaddition Reactions Proceeding via Dehydrohalogenation of Stable Precursors of Reactive Dipoles. New J. Chem. 2022, 46, 18575–18586. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Lmmunological Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Ghoncheh, M.; Mohammadian, M.; Mohammadian-Hafshejani, A.; Salehiniya, H. The Incidence and Mortality of Colorectal Cancer and Its Relationship with the Human Development Index in Asia. Ann. Glob. Health 2016, 82, 726–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osterman, I.A.; Komarova, E.S.; Shiryaev, D.I.; Korniltsev, I.A.; Khven, I.M.; Lukyanov, D.A.; Tashlitsky, V.N.; Serebryakova, M.V.; Efremenkova, O.V.; Ivanenkov, Y.A.; et al. Sorting out Antibiotics’ Mechanisms of Action: A Double Fluorescent Protein Reporter for High-Throughput Screening of Ribosome and DNA Biosynthesis Inhibitors. Antimicrob. Agents Chemother. 2016, 60, 7481–7489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tietze, L.F.; Eicher, T. Reaktionen Und Synthesen Im Organisch-Chemischen Praktikum Und Forschungslaboratorium (2. Auflage), 117th ed.; Wiley: Thieme Verlag, Stuttgart, Germany; New York, NY, USA, 1991; ISBN 3527308741. [Google Scholar]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandenburg, K. DIAMOND, Release 2.1d; Crystal Impact GbR: Bonn, Germany, 2000. [Google Scholar]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia Coli K-12 in-Frame, Single-Gene Knockout Mutants: The Keio Collection. Mol. Syst. Biol. 2006, 2, 2006.0008. [Google Scholar] [CrossRef] [Green Version]
- Sulavik, M.C.; Houseweart, C.; Cramer, C.; Jiwani, N.; Murgolo, N.; Greene, J.; Didomenico, B.; Shaw, K.J.; Miller, G.H.; Hare, R.; et al. Antibiotic Susceptibility Profiles of Escherichia Coli Strains Lacking Multidrug Efflux Pump Genes. Antimicrob. Agents Chemother. 2001, 45, 1126–1136. [Google Scholar] [CrossRef] [Green Version]
- Orelle, C.; Carlson, S.; Kaushal, B.; Almutairi, M.M.; Liu, H.; Ochabowicz, A.; Quan, S.; Pham, V.C.; Squires, C.L.; Murphy, B.T.; et al. Tools for Characterizing Bacterial Protein Synthesis Inhibitors. Antimicrob. Agents Chemother. 2013, 57, 5994–6004. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; McCandlish, A.C.; Gronenberg, L.S.; Chng, S.S.; Silhavy, T.J.; Kahne, D. Identification of a Protein Complex That Assembles Lipopolysaccharide in the Outer Membrane of Escherichia Coli. Proc. Natl. Acad. Sci. USA 2006, 103, 11754–11759. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
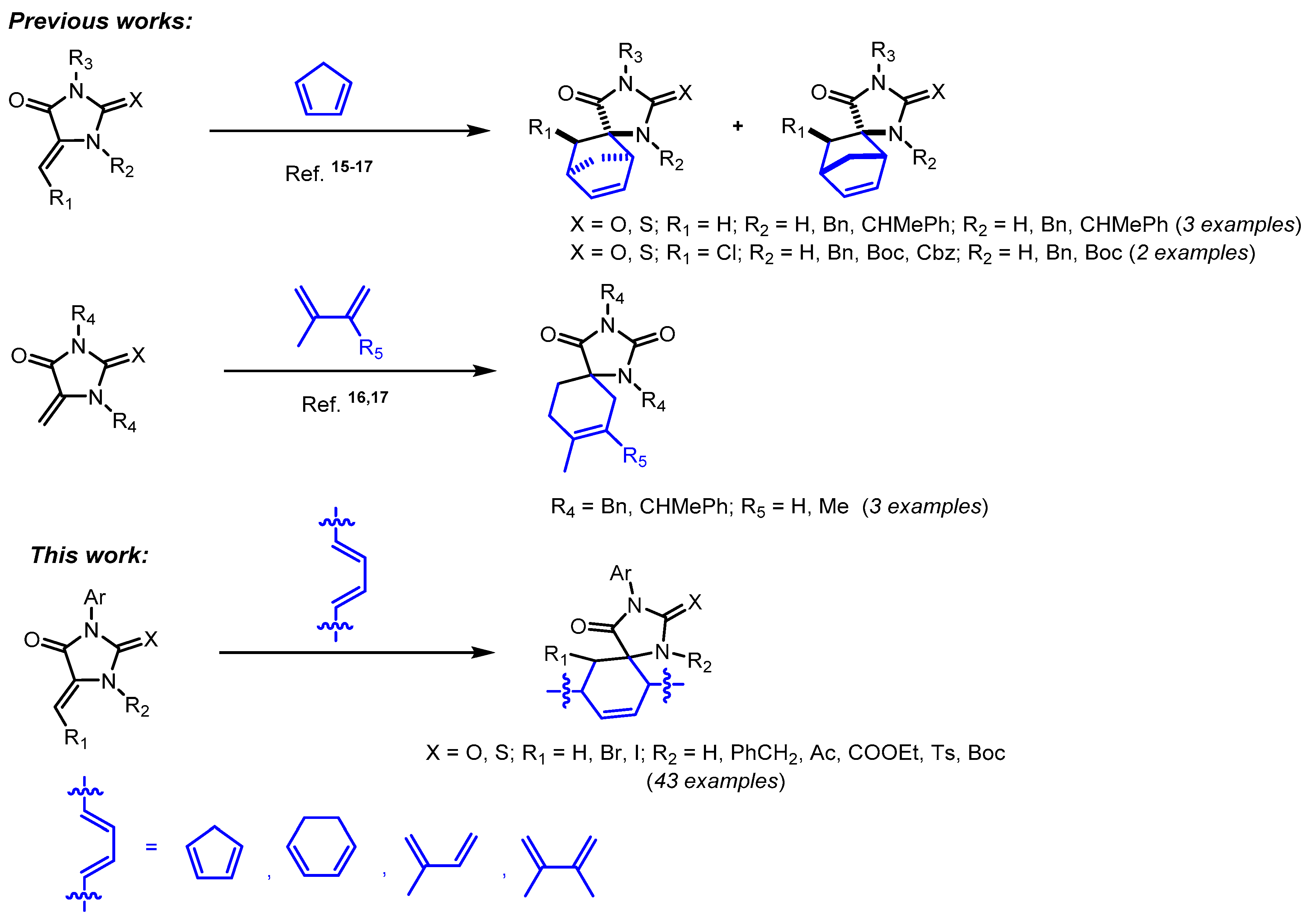{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Products | X | R1 | R2 | R3 | exo:endo1 | Yield, % |
|---|---|---|---|---|---|---|
| 28a + 28b | O | Ph | H | H | 91:9 | 81 + 9 2,3 [24] |
| 29a + 29b | O | Ph | Ac | H | 77:23 | 73 (78:22) 3,4 |
| 30a +30b | O | Ph | COOEt | H | 79:21 | 83 (83:17) 3,4 |
| 31a +31b | O | Ph | Ts | H | 87:13 | 91 (89:11) 3,4 |
| 32a + 32b | O | Ph | Boc | H | 85:15 | 73 (85:15) 3,4 |
| 33a | O | Ph | CH2Ph | H | 100:0 | 10 3,5 |
| 34a + 34b | S | Ph | H | H | 91:9 | 79 + 8 2,3 [24] |
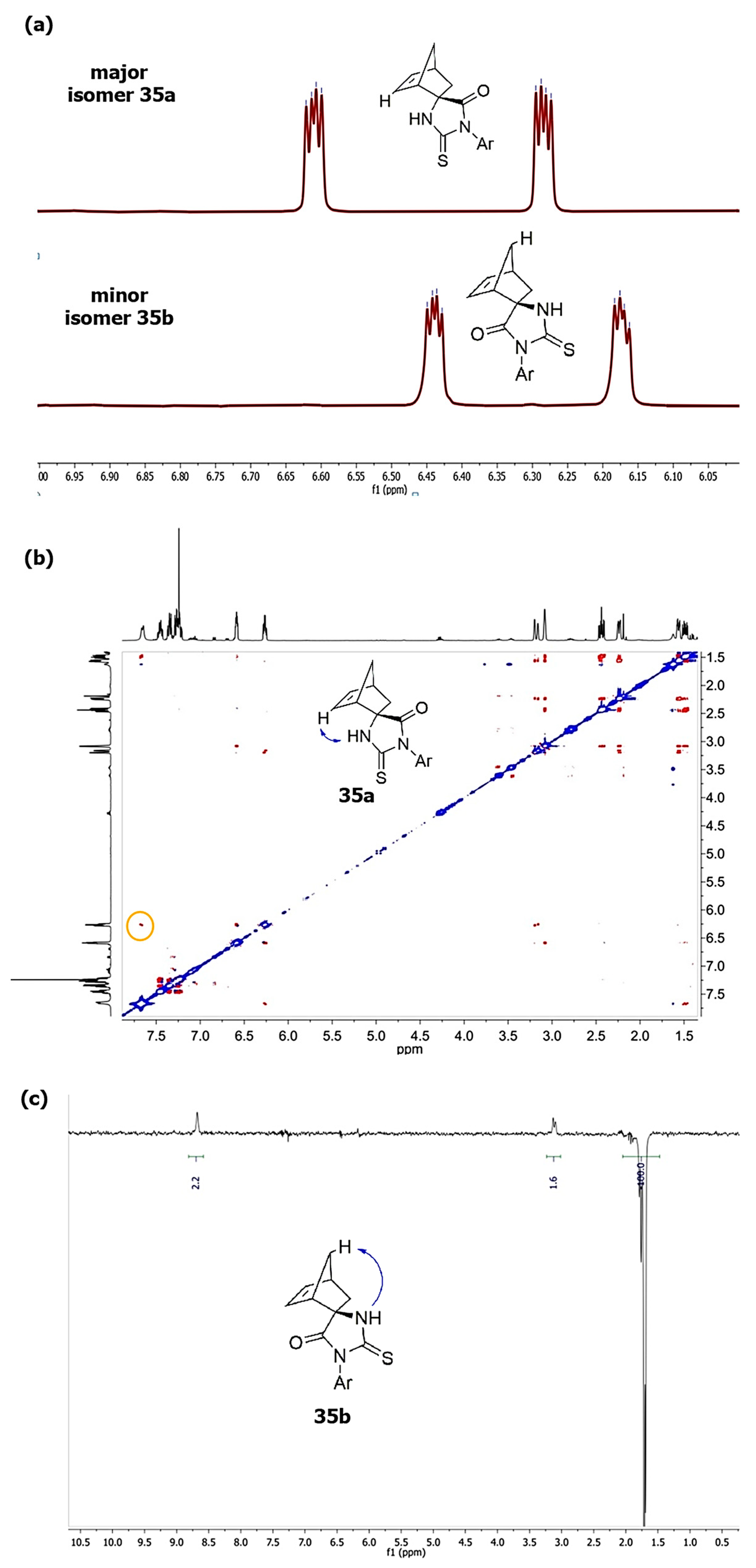
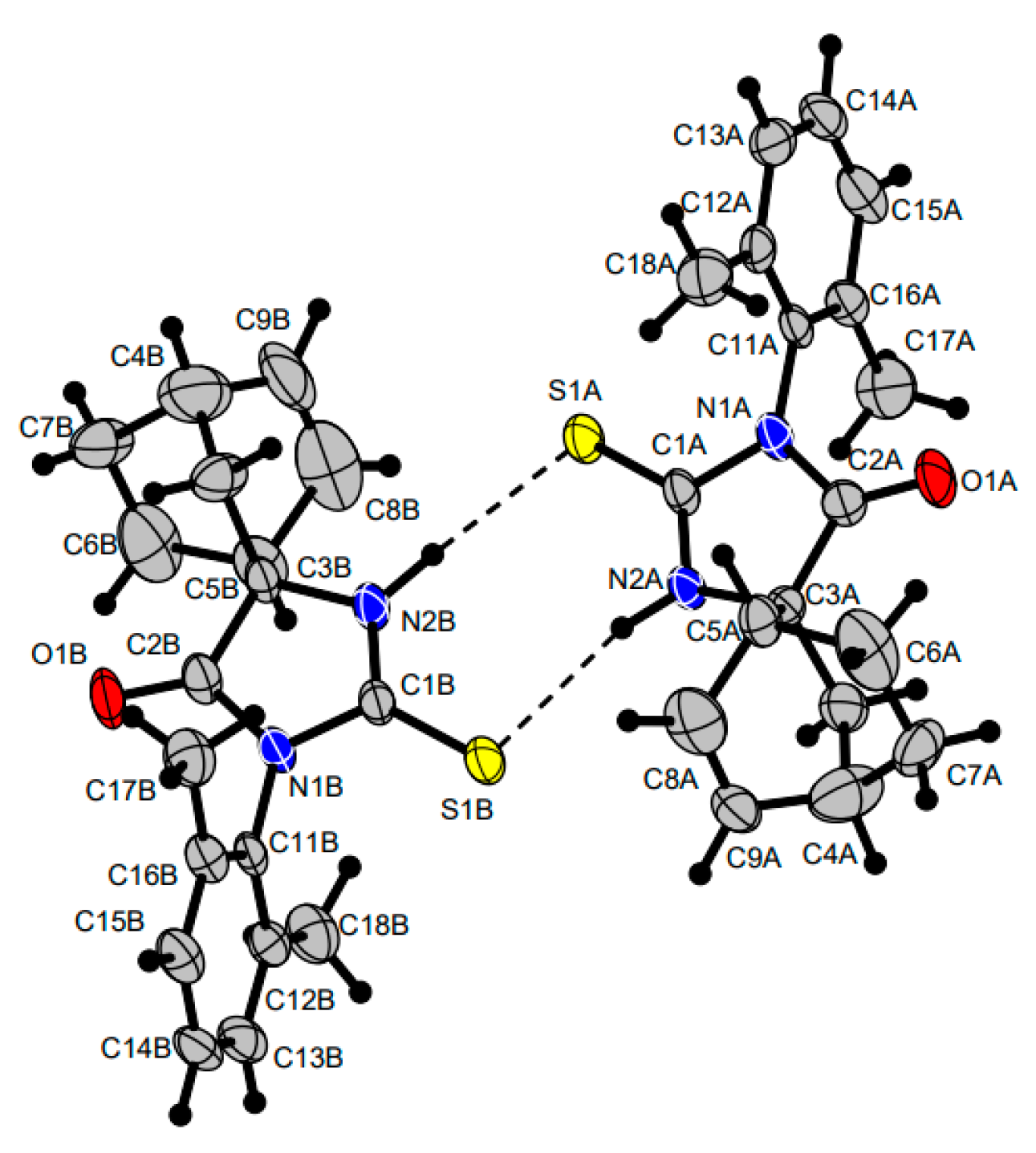
| 35a + 35b | S | 2-FC6H4 | H | H | 91:9 | 71 + 7 2,3 |
| 36a + 36b | S | 2-ClC6H4 | H | H | 91:9 | 68 + 0 2,3 |
| 37a + 37b | S | 3-MeC6H4 | H | H | 91:9 | 80 + 8 2,3 |
| 38a + 38b | S | 3-MeOC6H4 | H | H | 91:9 | 81 + 9 2,3 |
| 39a + 39b | S | 4-MeOC6H4 | H | H | 91:9 | 78 + 8 2,3 |
| 40a | O | Ph | H | Br | 100:0 | 64 6,7 |
| 41a | O | Ph | H | I | 100:0 | 37 6,7 |
| 42a | O | H | H | 4-FC6H4 | - | 07 |
| 43a | S | Ph | H | 4-MeOC6H4 | - | 07 |
| Products | X | LA | Ar | Yield a 1, % | Yield b 1, % |
|---|---|---|---|---|---|
| 44a + 44b | O | AlCl3 | Ph | 16 | 0 |
| ZnI2 | 0 | 0 | |||
| 45a + 45b | S | ZnI2 | Ph | 54 | 19 |
| 46a + 46b | S | ZnI2 | 2-FC6H4 | 44 | 15 |
| 47a + 47b | S | ZnI2 | 3-MeC6H4 | 60 | 25 |
| 48a + 48b | S | ZnI2 | 3-MeOC6H4 | 51 | 21 |
| 49a + 49b | S | ZnI2 | 4-ClC6H4 | 55 | 20 |
| 50a + 50b | S | ZnI2 | 4-MeOC6H4 | 50 | 19 |
| 51a + 51b | S | ZnI2 | 2,6-Me2C6H3 | 44 | 15 |
| Product | X | LA | Ar | R | Yield 1, % |
|---|---|---|---|---|---|
| 52 | O | AlCl3 | Ph | H | 47 |
| ZnI2 | 0 | ||||
| 53 | S | AlCl3 | Ph | H | 45 |
| ZnI2 | 92 | ||||
| 54 | S | ZnI2 | 2-FC6H4 | H | 84 |
| 55 | S | ZnI2 | 2-ClC6H4 | H | 71 |
| 56 | S | ZnI2 | 3-MeC6H4 | H | 85 |
| 57 | S | ZnI2 | 3-MeOC6H4 | H | 79 |
| 58 | S | ZnI2 | 4-FC6H4 | H | 74 |
| 69 | S | ZnI2 | 4-ClC6H4 | H | 82 |
| 60 | S | ZnI2 | 4-MeOC6H4 | H | 88 |
| 61 | S | ZnI2 | 2,6-Me2C6H3 | H | 68 |
| 62 | S | ZnI2 | 3-Cl,4-FC6H3 | H | 81 |
| Product | X | LA | Ar | Yield a + b 1, % |
|---|---|---|---|---|
| 63a + 63b | O | AlCl3 | Ph | 70 |
| 64a + 64b | S | ZnI2 | Ph | 82 |
| 65a + 65b | S | ZnI2 | 2-ClC6H4 | 61 |
| 66a + 66b | S | ZnI2 | 3-MeC6H4 | 64 |
| 67a + 67b | S | ZnI2 | 3-MeOC6H4 | 76 |
| 68a + 68b | S | ZnI2 | 4-ClC6H4 | 68 |
| 69a + 69b | S | ZnI2 | 4-MeOC6H4 | 71 |
| Cell line | ||||||
|---|---|---|---|---|---|---|
| Compound | HEK293T | MCF7 | VA13 | A549 | Cmax | Units |
| 28a | >>100 | >>100 | >>100 | >>100 | 100 | µM |
| 28b | >>100 | >>100 | >>100 | >>100 | 100 | µM |
| 34a | 83.0 ± 3.7 | >>100 | >>100 | >>100 | 100 | µM |
| 35b | 87.4 ± 9.9 | >>100 | >>100 | >>100 | 100 | µM |
| 37a | >>100 | >>100 | >>100 | >>100 | 100 | µM |
| 37b | 115 ± 17 ** | >>100 | >>100 | >>100 | 100 | µM |
| 38a | 111 ± 9 ** | >>100 | >>100 | >>100 | 100 | µM |
| 38b | 112 ± 8 ** | >>100 | >>100 | >>100 | 100 | µM |
| 39b | 120 ± 13 ** | >>100 | >>100 | >>100 | 100 | µM |
| 40a | 56.4 ± 2.5 | >>100 | >>100 | >>100 | 100 | µM |
| 41a | 69.1 ± 4.8 | >>100 | >>100 | >>100 | 100 | µM |
| 45a | 21.3 ± 2.3 | 141 ± 28 ** | >>100 | 85.2 ± 10.1 | 100 | µM |
| 48a | 98.0 ± 9.7 | >>100 | >>100 | >>100 | 100 | µM |
| 48b | 108 ± 12 ** | >>100 | >>100 | >>100 | 100 | µM |
| 49a | 6.7 ± 0.7 | 37.8 ± 6.1 | 30.9 ± 3.4 | 16.9 ± 1.9 | 100 | µM |
| 50a | 41.9 ± 3.8 | >>100 | >>100 | >>100 | 100 | µM |
| 50b | 109 ± 12 ** | >>100 | >>100 | >>100 | 100 | µM |
| 51a | >>100 | >>100 | >>100 | >>100 | 100 | µM |
| 51b | 87.5 ± 7.2 | 104 ± 11 ** | >>100 | 140 ± 21 ** | 100 | µM |
| 54 | 29.6 ± 3 | 122 ± 29 ** | >>100 | 49.5 ± 5 | 100 | µM |
| 55 * | ~5 | ~100 | >>100 | ~10 | 100 | µM |
| 56 | 22.0 ± 8.0 | 102 ± 17 ** | 98.7 ± 17.6 | >>100 | 100 | µM |
| 57 | 18.3 ± 1.7 | 50.6 ± 5.2 | 80.8 ± 13.7 | 35.4 ± 4.8 | 100 | µM |
| 58 | 13.7 ± 2.3 | 59.0 ± 13.3 | 45.0 ± 7.5 | 23.2 ± 3.5 | 100 | µM |
| 59 | 4.4 ± 0.6 | 76.6 ± 16 | 52.6 ± 7.8 | 26.2 ± 3.9 | 100 | µM |
| 60 | 10.2 ± 1.3 | 98.3 ± 19.8 | 75.3 ± 17 | 52.2 ± 5.7 | 100 | µM |
| 61 * | ~4 | ~100 | N/A | ~10 | 100 | µM |
| 62 | 6.8 ± 0.3 | 11.1 ± 0.6 | 9.8 ± 0.4 | 7.9 ± 0.4 | 100 | µM |
| 66a + 66b | 44.6 ± 4.9 | 146 ± 22 ** | 110 ± 13 ** | 144 ± 36 ** | 100 | µM |
| 72 | 60.1 ± 3.4 | >>100 | >>100 | >>100 | 100 | µM |
| 75 | 99.0 ± 11.1 | >>100 | >>100 | >>100 | 100 | µM |
| 76 | 48.6 ± 2.4 | 86.4 ± 7.3 | 132 ± 17 ** | 135 ± 14 ** | 100 | µM |
| Strain | 28a | 28b | 35b | 40a | 41a | 51a | 55 | 60 | 62 | 72 | 75 | 76 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E. coli DTC | >1000 | >1000 | 1000 | 1000 | 1000 | >1000 | >1000 | >1000 | 1000 | 500 | 500 | 125 |
| E. coli K12 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shybanov, D.E.; Kukushkin, M.E.; Hrytseniuk, Y.S.; Grishin, Y.K.; Roznyatovsky, V.A.; Tafeenko, V.A.; Skvortsov, D.A.; Zyk, N.V.; Beloglazkina, E.K. [4+2]-Cycloaddition to 5-Methylidene-Hydantoins and 5-Methylidene-2-Thiohydantoins in the Synthesis of Spiro-2-Chalcogenimidazolones. Int. J. Mol. Sci. 2023, 24, 5037. https://doi.org/10.3390/ijms24055037
Shybanov DE, Kukushkin ME, Hrytseniuk YS, Grishin YK, Roznyatovsky VA, Tafeenko VA, Skvortsov DA, Zyk NV, Beloglazkina EK. [4+2]-Cycloaddition to 5-Methylidene-Hydantoins and 5-Methylidene-2-Thiohydantoins in the Synthesis of Spiro-2-Chalcogenimidazolones. International Journal of Molecular Sciences. 2023; 24(5):5037. https://doi.org/10.3390/ijms24055037
Chicago/Turabian StyleShybanov, Dmitry E., Maxim E. Kukushkin, Yanislav S. Hrytseniuk, Yuri K. Grishin, Vitaly A. Roznyatovsky, Viktor A. Tafeenko, Dmitry A. Skvortsov, Nikolai V. Zyk, and Elena K. Beloglazkina. 2023. "[4+2]-Cycloaddition to 5-Methylidene-Hydantoins and 5-Methylidene-2-Thiohydantoins in the Synthesis of Spiro-2-Chalcogenimidazolones" International Journal of Molecular Sciences 24, no. 5: 5037. https://doi.org/10.3390/ijms24055037
APA StyleShybanov, D. E., Kukushkin, M. E., Hrytseniuk, Y. S., Grishin, Y. K., Roznyatovsky, V. A., Tafeenko, V. A., Skvortsov, D. A., Zyk, N. V., & Beloglazkina, E. K. (2023). [4+2]-Cycloaddition to 5-Methylidene-Hydantoins and 5-Methylidene-2-Thiohydantoins in the Synthesis of Spiro-2-Chalcogenimidazolones. International Journal of Molecular Sciences, 24(5), 5037. https://doi.org/10.3390/ijms24055037