
Transcriptome and Metabolome Profiling Unveil Pigment Formation Variations in Brown Cotton Lines (Gossypium hirsutum L.)


Abstract
:1. Introduction
2. Results
2.1. Overview of Metabolomics Profiling
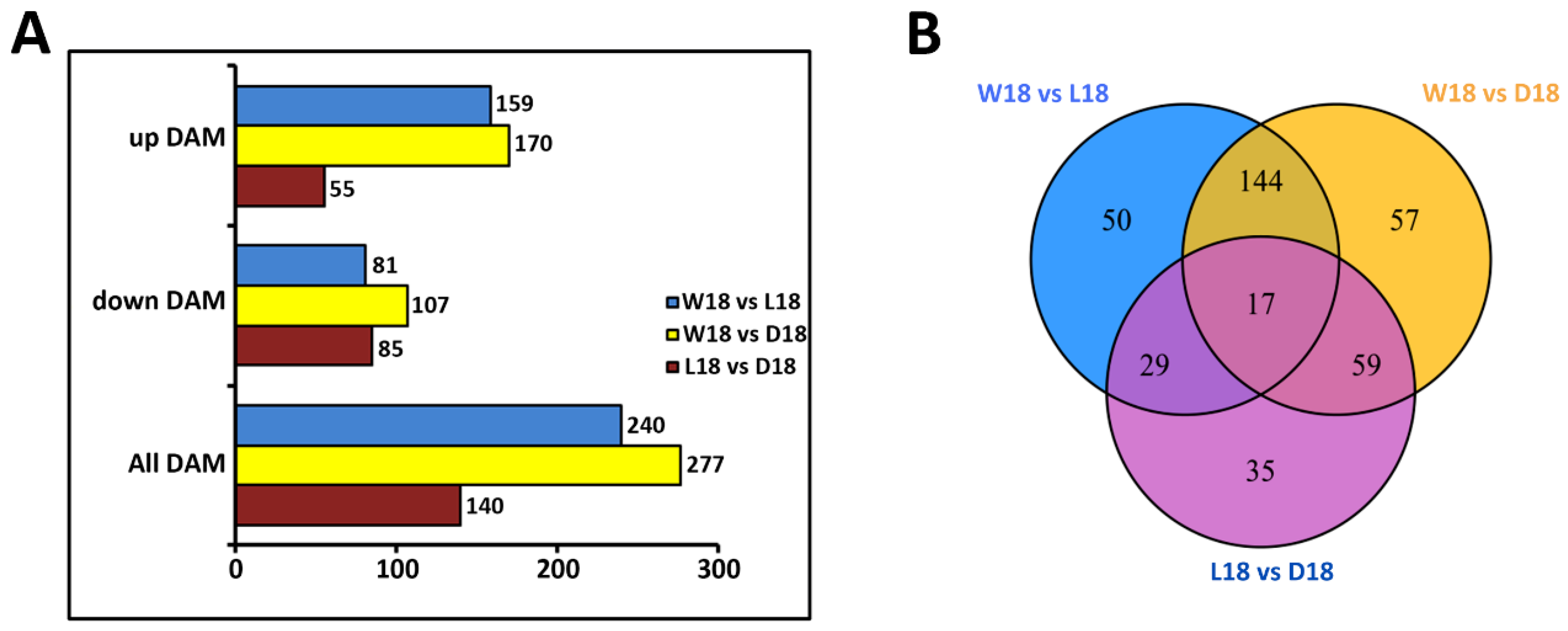
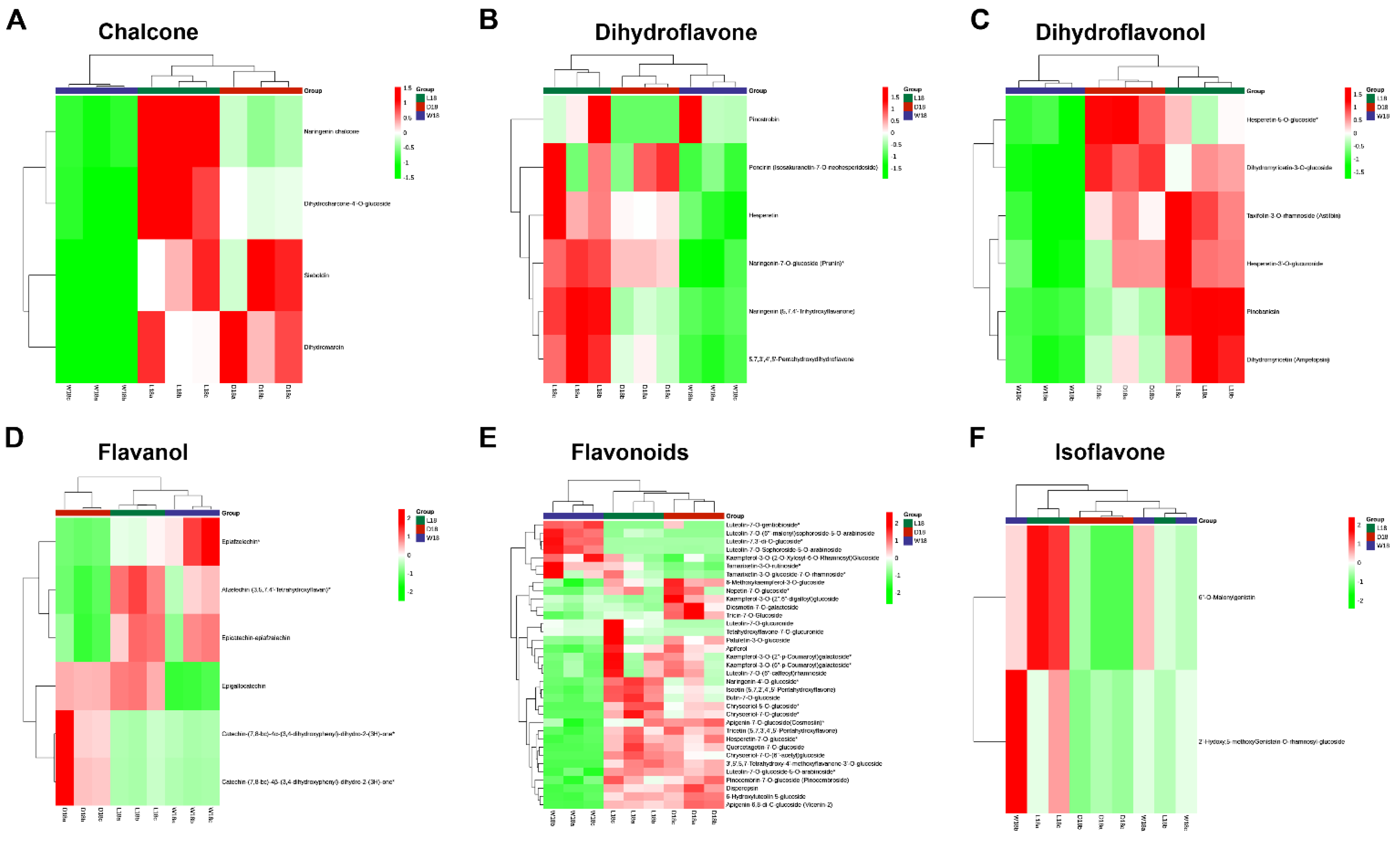
2.2. Untargeted Metabolome Analysis of Fiber Coloration in Cotton at 18 DPA
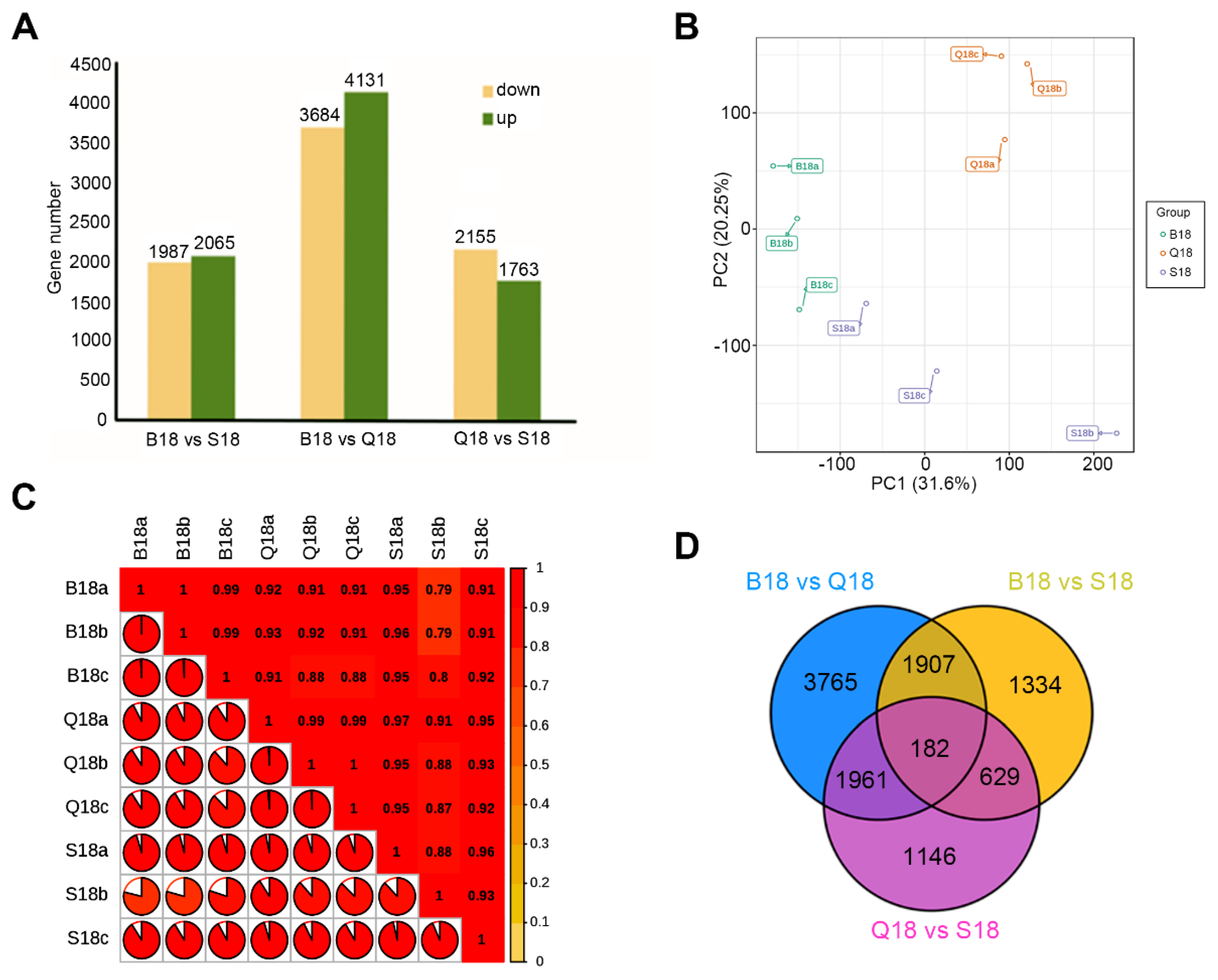
2.3. De Novo Transcriptome Assembly and Gene Expression Profiles in WCF, LCF and DCF at 18 DPA
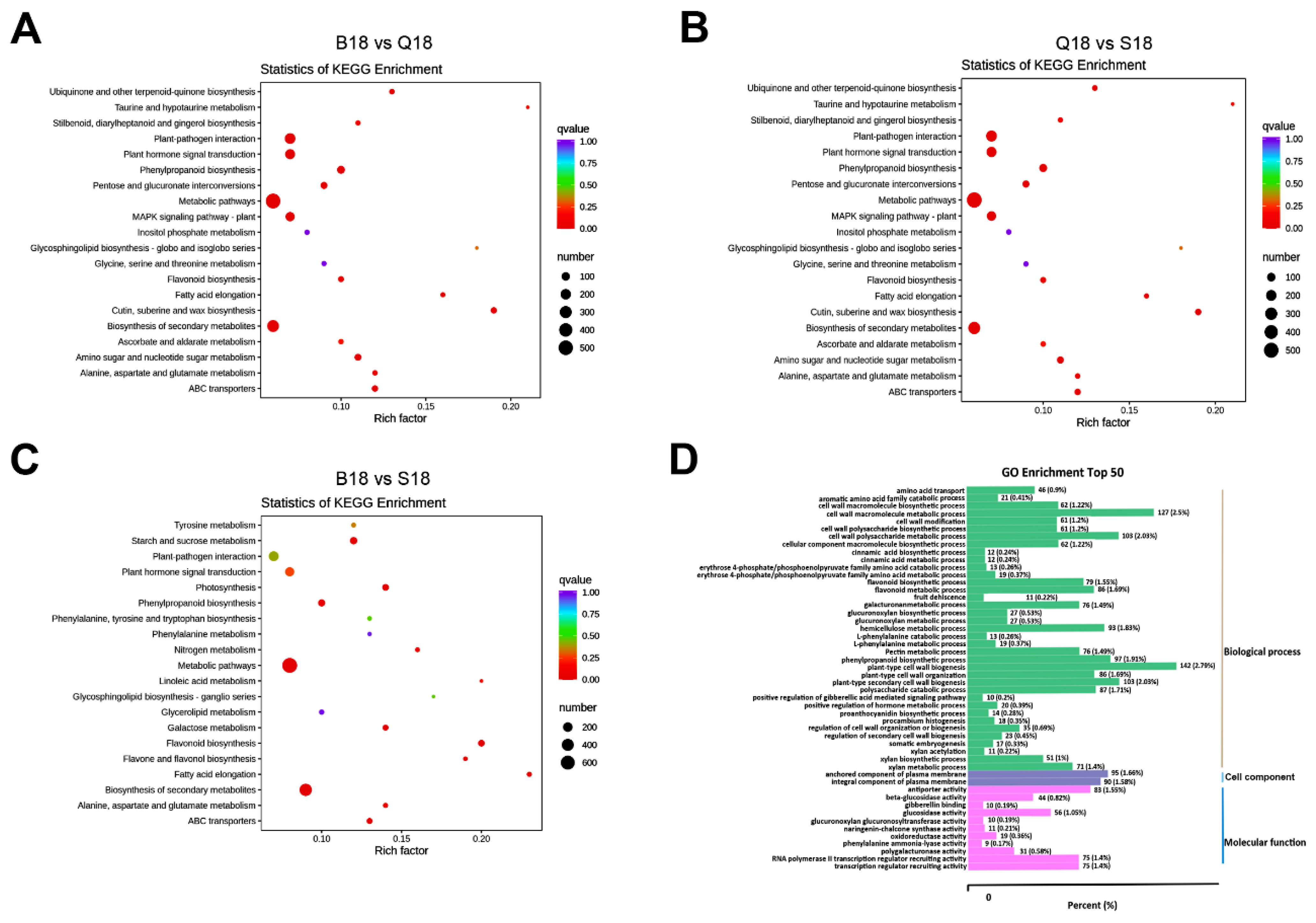
2.4. KEGG Pathway Enrichment and GO Functional Classification Analysis of DEGs
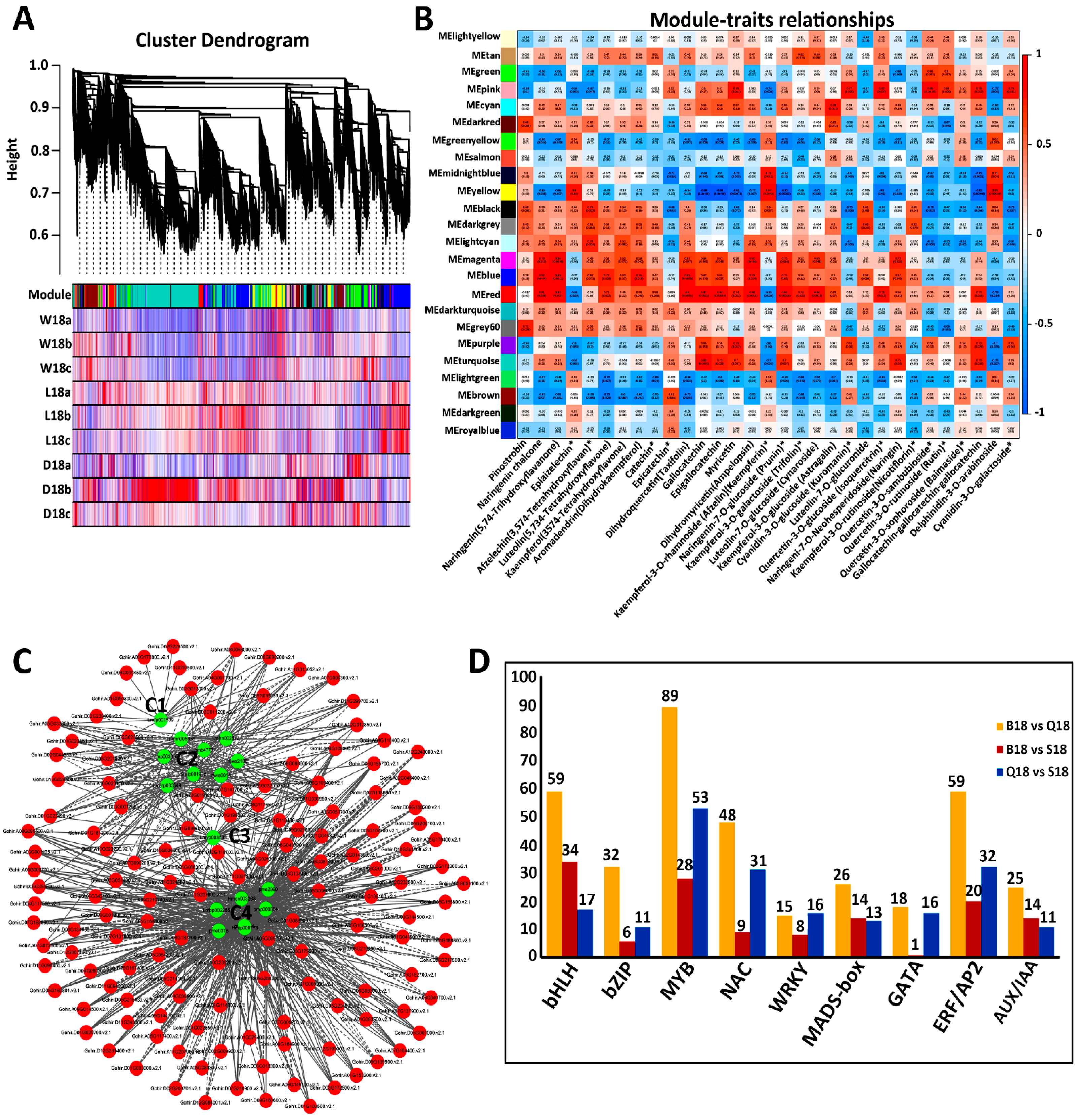
2.5. Co-Expression Network Analysis of Color-Related DEGs
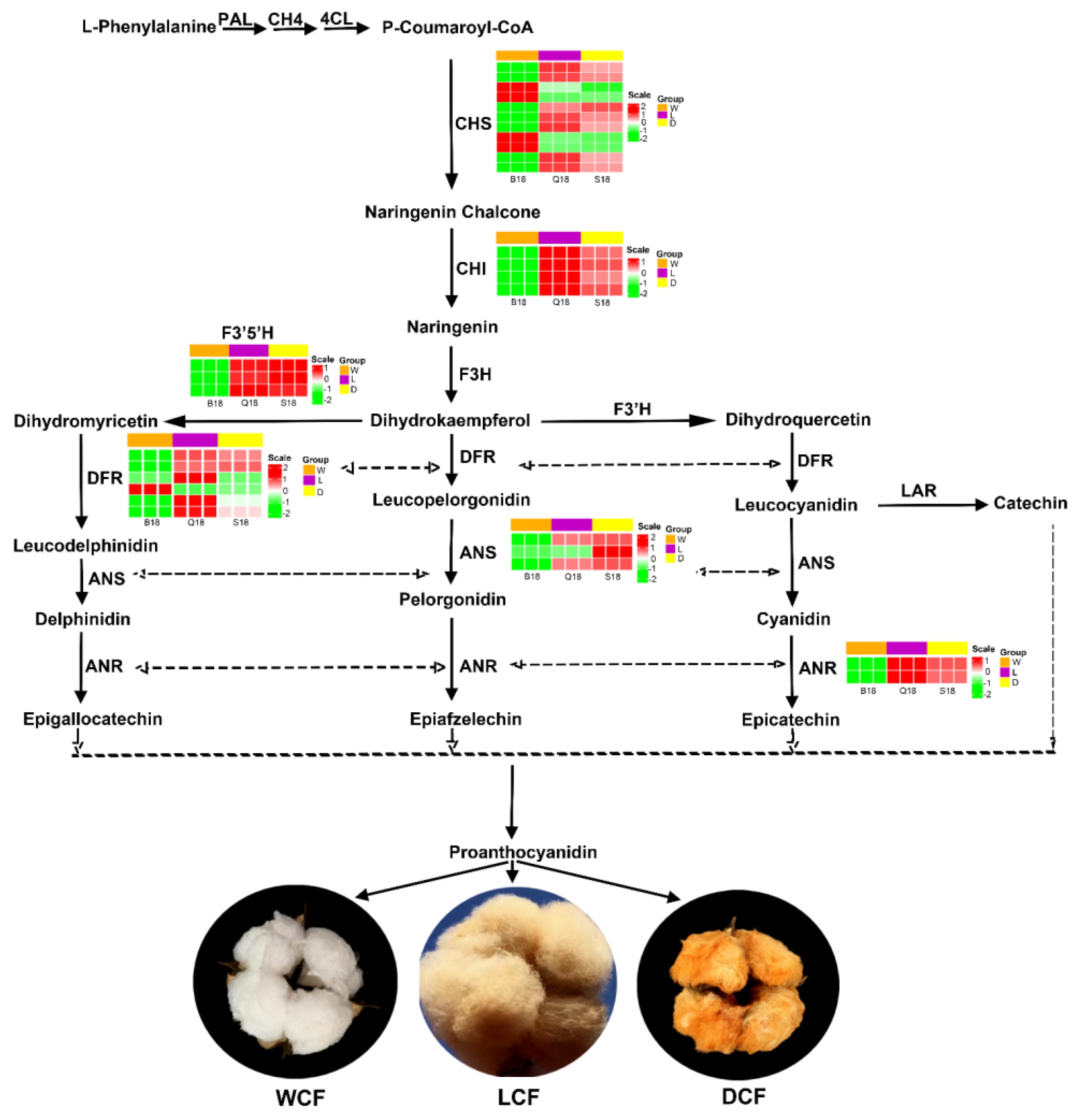
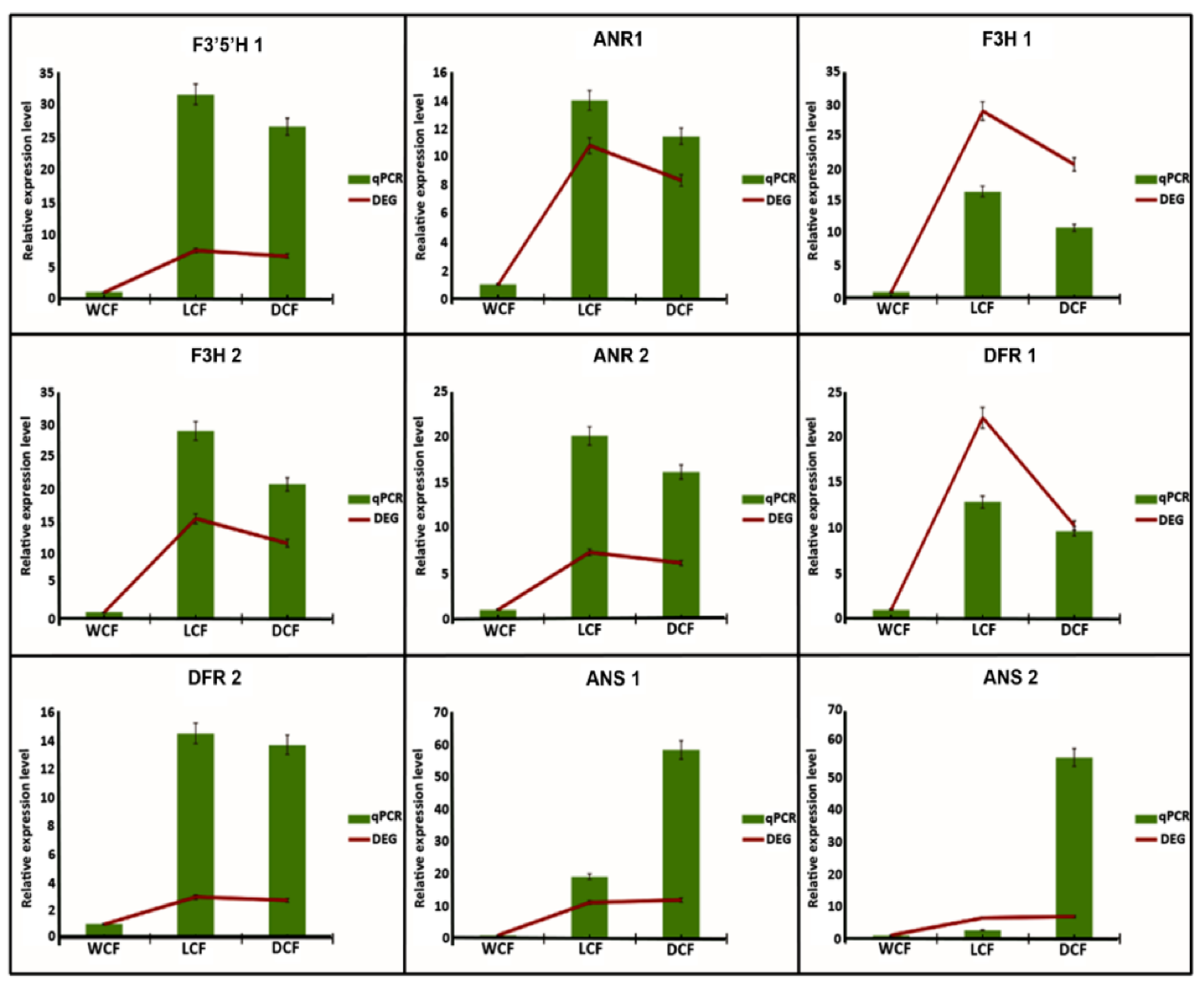
2.6. Analysis and Verification of Structural Genes Involved Anthocyanin Biosynthesis Pathway
3. Discussion
3.1. Classifying Metabolites Differences of Pigment-Related in DCF, WCF and LCF
3.2. Genes Involved in Anthocyanidin Pathway Influence Fiber Pigment Development in NBBC
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction and RNA-Seq Analysis
4.3. Functional Annotation and Expression Level Analysis
4.4. Weighted Gene Co-Expression Network Analysis (WGCNA)
4.5. Sample Preparation, Metabolite Extraction, and UPLC-ESI-MS/MS Analysis
4.6. ESI-Q TRAP-MS/MS
4.7. Selection of Differential Metabolites
4.8. qRT-PCR Validation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| NBCC | Naturally brown cotton |
| DCF | dark brown cotton fiber |
| LCF | light brown cotton fiber |
| WCF | white cotton fiber |
| DEGs | differentially expressed genes |
| DeMs | differentially expressed metabolites |
| PA | Proanthocyanidin |
| DPA | days post-anthesis |
| CHS | chalcone isomerase |
| F3H | flavanone 3-hydroxylase |
| F3′H | flavanone 3-hydroxylase |
| DFR | dihydroflavonol 4-reductase |
| F3′5′H | flavanone-3′5′-hydroxylase |
| ANS | anthocyanindin synthase |
| CHI | chalcone isomerase |
References
- Zahra, R.; Id, N.; Id, S.S.Z.; Mukhtar, M.S.; Amin, I.; Mishra, B.; Strickler, S.; Mueller, L.A.; Asif, M.; Mansoor, S. Transcriptomic Analysis of Cultivated Cotton Gossypium Hirsutum Provides Insights into Host Responses upon Whitefly-Mediated Transmission of Cotton Leaf Curl Disease. PLoS ONE 2019, 14, e0210011. [Google Scholar] [CrossRef]
- Ma, M.; Hussain, M.; Memon, H.; Zhou, W. Structure of Pigment Compositions and Radical Scavenging Activity of Naturally Green-Colored Cotton Fiber. Cellulose 2016, 23, 955–963. [Google Scholar] [CrossRef]
- Sundram, V.; Narayanan, S.S. Naturally Coloured Cotton. Asian Text. J. 2001, 6, 43–46. [Google Scholar]
- Liu, H.F.; Luo, C.; Song, W.; Shen, H.; Li, G.; He, Z.G.; Chen, W.G.; Cao, Y.Y.; Huang, F.; Tang, S.W.; et al. Flavonoid Biosynthesis Controls Fiber Color in Naturally Colored Cotton. PeerJ 2018, 2018, e4537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.J.; Zhang, X.Y.; Wang, F.X.; Yang, C.L.; Liu, F.; Xia, G.X.; Sun, J. A Comparative Proteomic Analysis Provides Insights into Pigment Biosynthesis in Brown Color Fiber. J. Proteom. 2013, 78, 374–388. [Google Scholar] [CrossRef]
- Xiang, Z.; Pan, L.P.; Wei, D.M.; Chen, Z.; Hua, W.C.; Yuan, C.; Di, D.Z.; Hua, C.D. Physiological Characteristics Associated with Fiber Development in Two Naturally Colored Cotton Cultivars. Agron. J. 2019, 111, 1190–1197. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Luo, S.; Hu, Z.; Tang, Z.; Zhou, W. Antioxidant Properties of Naturally Brown-Colored Cotton Fibers. Text. Res. J. 2016, 86, 256–263. [Google Scholar] [CrossRef]
- Sun, S.; Xiong, X.P.; Zhu, Q.; Li, Y.J.; Sun, J. Transcriptome Sequencing and Metabolome Analysis Reveal Genes Involved in Pigmentation of Green-Colored Cotton Fibers. Int. J. Mol. Sci. 2019, 20, 4838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmutz, A.; Jenny, T.; Ryser, U. A Caffeoyl-Fatty Acid-Glycerol Ester from Wax Associated with Green Cotton Fibre Suberin. Phytochemistry 1994, 36, 1343–1346. [Google Scholar] [CrossRef]
- Li, Y.J.; Wang, F.X.; Wang, Y.Q.; Liu, Y.C.; Zhang, X.Y.; Sun, Y.Q.; Sun, J. Identification of the Proteins in Green Cotton Fiber Using a Proteomics-Based Approach. Biotechnol. Lett. 2013, 35, 1519–1523. [Google Scholar] [CrossRef]
- Xiao, Y.H.; Yan, Q.; Ding, H.; Luo, M.; Hou, L.; Zhang, M.; Yao, D.; Liu, H.S.; Li, X.; Zhao, J.; et al. Transcriptome and Biochemical Analyses Revealed a Detailed Proanthocyanidin Biosynthesis Pathway in Brown Cotton Fiber. PLoS ONE 2014, 9, e86344. [Google Scholar] [CrossRef]
- Feng, H.; Li, Y.; Wang, S.; Zhang, L.; Liu, Y.; Xue, F.; Sun, Y.; Wang, Y.; Sun, J. Molecular Analysis of Proanthocyanidins Related to Pigmentation in Brown Cotton Fibre (Gossypium Hirsutum L.). J. Exp. Bot. 2014, 65, 5759–5769. [Google Scholar] [CrossRef]
- Mikhailova, A.; Strygina, K.; Khlestkina, E. The Genes Determining Synthesis of Pigments in Cotton. Biol. Commun. 2019, 64, 133–145. [Google Scholar] [CrossRef]
- Li, T.; Fan, H.; Li, Z.; Wei, J.; Lin, Y.; Cai, Y. The Accumulation of Pigment in Fiber Related to Proanthocyanidins Synthesis for Brown Cotton. Acta Physiol. Plant. 2012, 34, 813–818. [Google Scholar] [CrossRef]
- Gao, J.; Shen, L.; Yuan, J.; Zheng, H.; Su, Q.; Yang, W.; Zhang, L.; Nnaemeka, V.E.; Sun, J.; Ke, L.; et al. Functional Analysis of GhCHS, GhANR and GhLAR in Colored Fiber Formation of Gossypium Hirsutum L. BMC Plant Biol. 2019, 19, 455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Tian, X.; Liu, Y.; Li, Y.; Zhang, X.; Jones, B.J.; Sun, Y.; Sun, J. Analysis of Flavonoids and the Flavonoid Structural Genes in Brown Fiber of Upland Cotton. PLoS ONE 2013, 8, e58820. [Google Scholar] [CrossRef] [Green Version]
- Yan, Q.; Wang, Y.; Li, Q.; Zhang, Z.; Ding, H.; Zhang, Y.; Liu, H.; Luo, M.; Liu, D.; Song, W.; et al. Up-Regulation of GhTT2-3A in Cotton Fibres during Secondary Wall Thickening Results in Brown Fibres with Improved Quality. Plant Biotechnol. J. 2018, 16, 1735–1747. [Google Scholar] [CrossRef] [Green Version]
- Bogs, J.; Jaffé, F.W.; Takos, A.M.; Walker, A.R.; Robinson, S.P. The Grapevine Transcription Factor VvMYBPA1 Regulates Proanthocyanidin Synthesis during Fruit Development. Plant Physiol. 2007, 143, 1347–1361. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Peng, H.; Chen, F.; Luo, M.; Li, W. Genome-Wide Analysis of Transcription Factors Related to Anthocyanin Biosynthesis in Carmine Radish (Raphanus Sativus L.) Fleshy Roots. PeerJ 2019, 2019, e8041. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Thrimawithana, A.H.; Cooney, J.M.; Jensen, D.J.; Espley, R.V.; Allan, A.C. The Proanthocyanin-Related Transcription Factors MYBC1 and WRKY44 Regulate Branch Points in the Kiwifruit Anthocyanin Pathway. Sci. Rep. 2020, 10, 14161. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.Z.; Hu, F.C.; Hu, G.B.; Li, X.J.; Huang, X.M.; Wang, H.C. Differential Expression of Anthocyanin Biosynthetic Genes in Relation to Anthocyanin Accumulation in the Pericarp of Litchi Chinensis Sonn. PLoS ONE 2011, 6, e19455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, R.A.; Strack, D. Phytochemistry Meets Genome Analysis, and Beyond. Phytochemistry 2003, 62, 815–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiehn, O. Metabolomics—The Link between Genotypes and Phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Fan, Y.; Zhang, L.; Zheng, C.; Chen, A.; Sun, Y.; Guo, H.; Wu, J.; Li, T.; Fan, Y.; et al. Quantitative Metabolome and Transcriptome Analysis Reveals Complex Regulatory Pathway Underlying Photoinduced Fiber Color Formation in Cotton. Gene 2021, 767, 145180. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Kelm, M.A.; Hammerstone, J.F.; Beecher, G.; Holden, J.; Haytowitz, D.; Gebhardt, S.; Prior, R.L. Concentrations of Proanthocyanidins in Common Foods and Estimations of Normal Consumption. J. Nutr. 2004, 134, 613–617. [Google Scholar] [CrossRef] [Green Version]
- Pisitsak, P.; Hutakamol, J.; Thongcharoen, R.; Phokaew, P.; Kanjanawan, K.; Saksaeng, N. Improving the Dyeability of Cotton with Tannin-Rich Natural Dye through Pretreatment with Whey Protein Isolate. Ind. Crops Prod. 2016, 79, 47–56. [Google Scholar] [CrossRef]
- Nix, A.; Paull, C.; Colgrave, M. Flavonoid Profile of the Cotton Plant, Gossypium Hirsutum: A Review. Plants 2017, 43, 43. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Du, X. Transcriptome Co-Expression Network and Metabolome Analysis Identifies Key Genes and Regulators of Proanthocyanidins Biosynthesis in Brown Cotton. Front. Plant Sci. 2022, 12, 3429. [Google Scholar] [CrossRef]
- Gunawardena, D.; Govindaraghavan, S.; Münch, G. Anti-Inflammatory Properties of Cinnamon Polyphenols and Their Monomeric Precursors. Polyphen. Hum. Heal. Dis. 2013, 1, 409–425. [Google Scholar] [CrossRef]
- Lin, Y.; Shi, R.; Wang, X.; Shen, H.M. Luteolin, a Flavonoid with Potentials for Cancer Prevention and Therapy. Curr Cancer Drug Targets 2009, 8, 634–646. [Google Scholar] [CrossRef]
- Božić, D.D.; Milenković, M.; Ivković, B.; Ćirković, I. Antibacterial Activity of Three Newly-Synthesized Chalcones & Synergism with Antibiotics against Clinical Isolates of Methicillin-Resistant Staphylococcus Aureus. Indian J. Med. Res. 2014, 140, 130–137. [Google Scholar] [PubMed]
- Wang, X.; Luo, S.; Li, Q.; Song, L.; Zhang, W.; Yu, P.; Xuan, S.; Wang, Y.; Zhao, J.; Chen, X.; et al. Delphinidins and Naringenin Chalcone Underlying the Fruit Color Changes during Maturity Stages in Eggplant. Agronomy 2022, 12, 1036. [Google Scholar] [CrossRef]
- Canavar, Ö.; Rausher, M.D. Molecular Analysis of Structural Genes Involved in Flavonoids Biosynthesis in Naturally Colored Cotton. Crop Sci. 2020, 61, 1117–1126. [Google Scholar] [CrossRef]
- Wang, C.; Han, P.; Zhao, Y.; Ji, X.; Yu, J.; You, C.; Hu, D.; Hao, Y. Auxin Regulates Anthocyanin Biosynthesis through the Auxin Repressor Protein MdIAA26. Biochem. Biophys. Res. Commun. 2020, 533, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Han, P.; Zhao, Y.; Yu, J.; Hu, D.; Hao, Y. Genome-Wide Analysis of Auxin Response Factor (ARF) Genes and Functional Identification of MdARF2 Reveals the Involvement in the Regulation of Anthocyanin Accumulation in Apple. New Zeal. J. Crop Hortic. Sci. 2021, 49, 78–91. [Google Scholar] [CrossRef]
- An, J.P.; Zhang, X.W.; You, C.X.; Bi, S.Q.; Wang, X.F.; Hao, Y.J. MdWRKY40 Promotes Wounding-Induced Anthocyanin Biosynthesis in Association with MdMYB1 and Undergoes MdBT2-Mediated Degradation. New Phytol. 2019, 224, 380–395. [Google Scholar] [CrossRef]
- Zhou, L.; Ye, Y. A Genetic Pathway for Naturally Colored Cotton Production. Plant Physiol. 2022, 189, 1190–1191. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.R.; Davies, K.M.; Mp, M.; Petunia, P. MYB and BHLH Transcription Factor Transgenes Increase Anthocyanin Pigmentation in Petunia and Lisianthus Plants, and the Petunia Phenotypes Are Strongly Enhanced under Field Conditions. Front. Plant Sci. 2014, 5, 603. [Google Scholar] [CrossRef]
- Gallego, A.M.; Rojas, L.F.; Parra, O.; Rodriguez, H.A.; Rivas, J.C.M.; Urrea, A.I.; Atehortúa, L.; Fister, A.S.; Guiltinan, M.J.; Maximova, S.N. Transcriptomic Analyses of Cacao Cell Suspensions in Light and Dark Provide Target Genes for Controlled Flavonoid Production. Sci. Rep. 2018, 8, 13575. [Google Scholar] [CrossRef] [Green Version]
- Cavallini, E.; Zenoni, S.; Finezzo, L.; Guzzo, F.; Zamboni, A.; Avesani, L.; Tornielli, G.B. Functional Diversification of Grapevine MYB5a and MYB5b in the Control of Flavonoid Biosynthesis in a Petunia Anthocyanin Regulatory Mutant. Plant Cell Physiol. 2014, 55, 517–534. [Google Scholar] [CrossRef]
- Zhou, C.; Mei, X.; Rothenberg, D.O.; Yang, Z.; Zhang, W.; Wan, S.; Yang, H.; Zhang, L. Metabolome and Transcriptome Analysis Reveals Putative Genes Involved in Anthocyanin Accumulation and Coloration in White and Pink Tea (Camellia sinensis). Molecules 2020, 25, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Zhang, D.; Zheng, H.; Wu, Y.; Mei, J.; Ke, L.; Yu, D.; Sun, Y. Biochemical and Expression Analyses Revealed the Involvement of Proanthocyanidins and/or Their Derivatives in Fiber Pigmentation of Gossypium Stocksii. Int. J. Mol. Sci. 2022, 23, 1008. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Symbol | DCF vs. LCF | DCF vs. WCF | LCF vs. WCF | Description |
|---|---|---|---|---|---|
| Log2FC | Log2FC | Log2FC | |||
| Gohir.A05G167300.v2.1 | ANR | 2.88 | 2.62 | −0.24 | Anthocyanidin reductase |
| Gohir.D05G170300.v2.1 | ANR | 3.44 | 3.07 | −0.36 | |
| Gohir.A08G176400.v2.1 | LDOX/ANS | 3.48 | 3.57 | 0.11 | Leucoanthocyanidin dioxygenase |
| Gohir.D04G067900.v2.1 | LDOX/ANS | 0.12 | 1.63 | 1.51 | |
| Gohir.D08G195100.v2.1 | LDOX/ANS | 2.67 | 2.77 | 0.11 | |
| Gohir.A05G403200.v2.1 | CHI | 2.15 | 1.86 | −0.28 | Chalcone isomerase |
| Gohir.A13G020400.v2.1 | CHI | 2.42 | 2.21 | −0.19 | |
| Gohir.D04G012300.v2.1 | CHI | 2.29 | 1.87 | −0.41 | |
| Gohir.D13G021000.v2.1 | CHI | 1.67 | 1.45 | −0.21 | |
| Gohir.A02G024200.v2.1 | CHS | 3.16 | 2.80 | −0.36 | Chalcone synthase |
| Gohir.A05G318401.v2.1 | CHS | 2.72 | 2.52 | −0.20 | |
| Gohir.A09G000148.v2.1 | CHS | −1.00 | −1.55 | −0.53 | |
| Gohir.A09G000500.v2.1 | CHS | −1.79 | −1.61 | 0.18 | |
| Gohir.A10G121700.v2.1 | CHS | 4.75 | 4.92 | 0.17 | |
| Gohir.A10G121800.v2.1 | CHS | 3.61 | 3.37 | −0.22 | |
| Gohir.D02G031600.v2.1 | CHS | 2.96 | 2.60 | −0.35 | |
| Gohir.D09G000301.v2.1 | CHS | −1.30 | −1.40 | −0.09 | |
| Gohir.D09G000401.v2.1 | CHS | −1.58 | −1.61 | −0.02 | |
| Gohir.D10G144300.v2.1 | CHS | 3.07 | 2.70 | −0.36 | |
| Gohir.D10G144400.v2.1 | CHS | 3.17 | 2.87 | −0.29 | |
| Gohir.A05G192500.v2.1 | DFR | 1.55 | 1.43 | −0.11 | Dihydroflavonol 4-reductase |
| Gohir.A06G008700.v2.1 | DFR | 2.83 | 2.81 | −0.02 | |
| Gohir.A07G138900.v2.1 | DFR | 4.20 | 0.27 | −3.86 | |
| Gohir.D01G059900.v2.1 | DFR | −1.46 | −1.21 | 0.27 | |
| Gohir.D05G195700.v2.1 | DFR | 4.47 | 3.36 | −1.10 | |
| Gohir.D06G004300.v2.1 | DFR | 2.98 | 2.36 | −0.61 | |
| Gohir.A07G120800.v2.1 | F3′5′H | 3.14 | 3.24 | 0.11 | Flavonoid 3′,5′-hydroxylase |
| Gohir.A07G120900.v2.1 | F3′5′H | 2.78 | 2.98 | 0.21 | |
| Gohir.D07G125100.v2.1 | F3′5′H | 2.90 | 2.71 | −0.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, Y.-P.; Zhao, G.; Xie, Y.-F.; Owusu, A.G.; Wu, Y.; Gao, J.-S. Transcriptome and Metabolome Profiling Unveil Pigment Formation Variations in Brown Cotton Lines (Gossypium hirsutum L.). Int. J. Mol. Sci. 2023, 24, 5249. https://doi.org/10.3390/ijms24065249
Lv Y-P, Zhao G, Xie Y-F, Owusu AG, Wu Y, Gao J-S. Transcriptome and Metabolome Profiling Unveil Pigment Formation Variations in Brown Cotton Lines (Gossypium hirsutum L.). International Journal of Molecular Sciences. 2023; 24(6):5249. https://doi.org/10.3390/ijms24065249
Chicago/Turabian StyleLv, Yin-Ping, Gang Zhao, Yong-Fei Xie, Anane Gideon Owusu, Yong Wu, and Jun-Shan Gao. 2023. "Transcriptome and Metabolome Profiling Unveil Pigment Formation Variations in Brown Cotton Lines (Gossypium hirsutum L.)" International Journal of Molecular Sciences 24, no. 6: 5249. https://doi.org/10.3390/ijms24065249
APA StyleLv, Y. -P., Zhao, G., Xie, Y. -F., Owusu, A. G., Wu, Y., & Gao, J. -S. (2023). Transcriptome and Metabolome Profiling Unveil Pigment Formation Variations in Brown Cotton Lines (Gossypium hirsutum L.). International Journal of Molecular Sciences, 24(6), 5249. https://doi.org/10.3390/ijms24065249