
Novel Aspects of the Immune Response Involved in the Peritoneal Damage in Chronic Kidney Disease Patients under Dialysis

 , , , , , , ,
, , , , , , ,  and
and 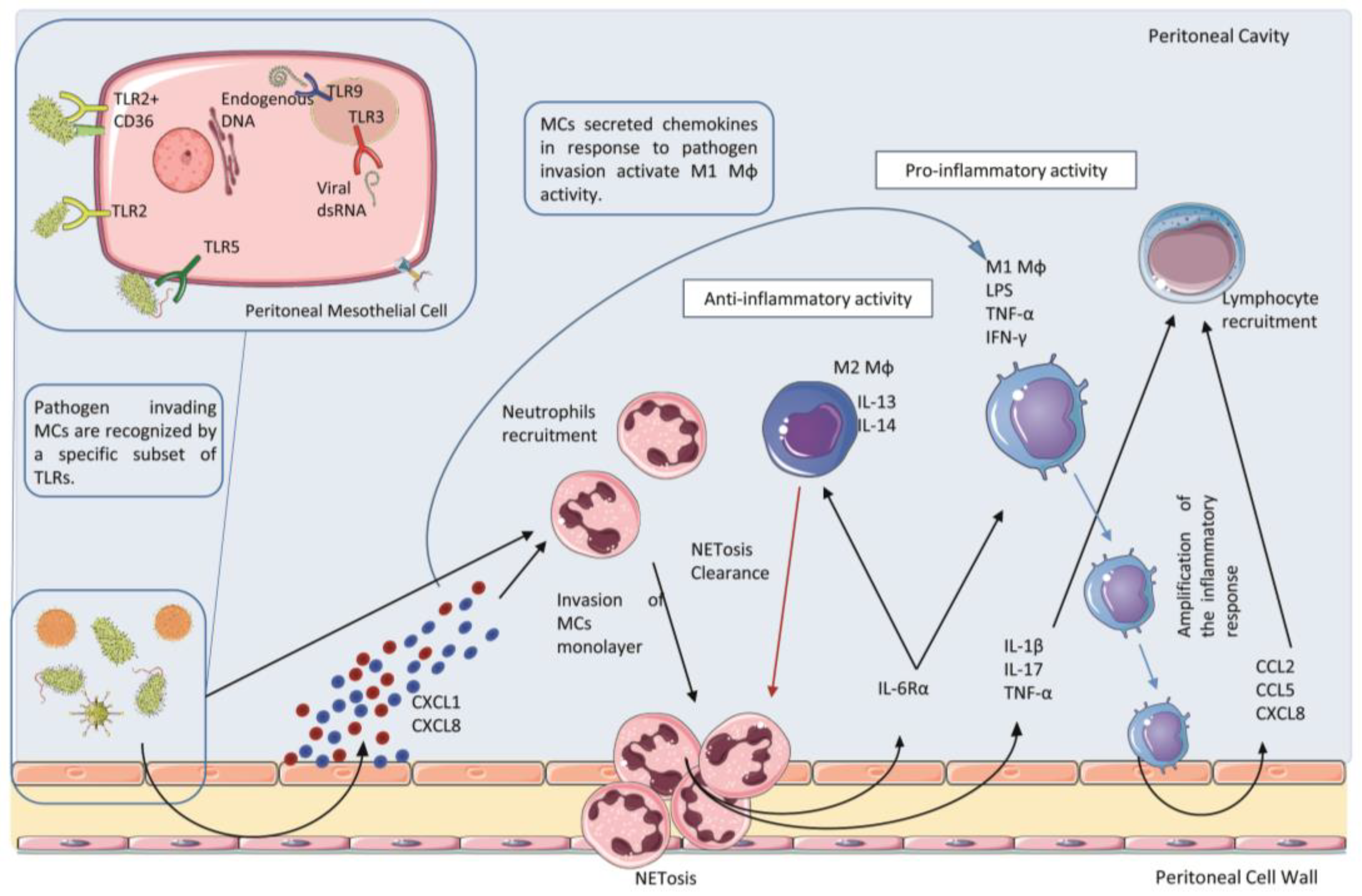{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cellular and Molecular Mechanisms Implicated in the Damage of the Peritoneum
2.1. Microorganisms Involved in Peritonitis Episodes during PD
2.2. Receptors and Ligands Implicated in the Infection of the Peritoneum
2.3. Role of Polymorphonucleate Neutrophils in Acute Infection and Early Innate Response
2.4. Role of Polymorphonucleate Macrophages and Dendritic Cells in Acute Infection and Chronic Inflammation
2.5. Role of Th17 Lymphocytes and Its Effector Cytokine IL-17A in Acute Infection and Chronic Inflammation
2.6. Other Cells Implicated in the Peritoneal Response to Infections: Mastocytes and Natural Killer Cells
3. Resident Peritoneal Cells: Activation of Proinflammatory and Immunomodulatory Signals
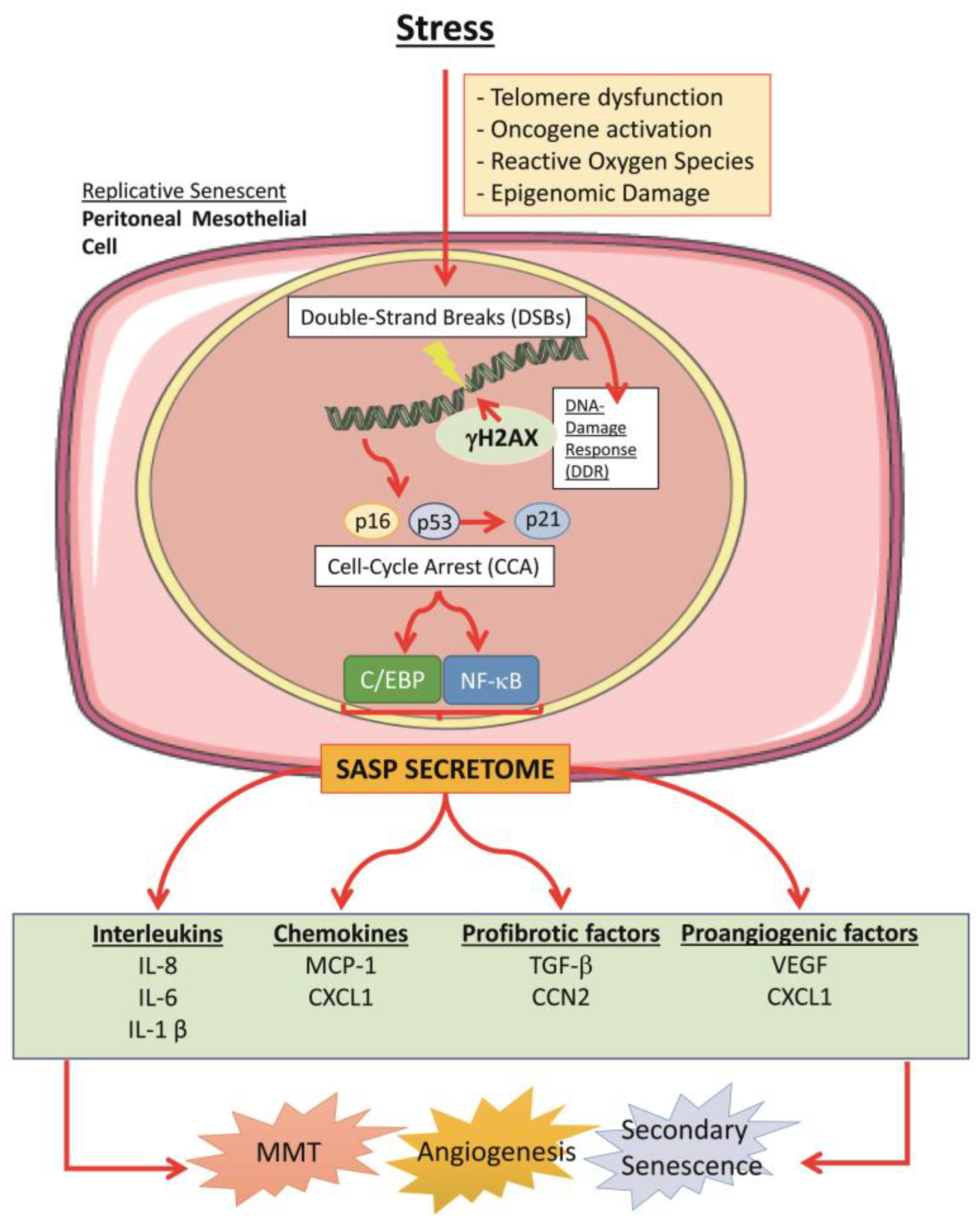
4. Cellular Senescence in the Peritoneum
5. Potential Anti-Inflammatory Treatments in the Preservation of the PM Integrity
5.1. Current Clinical Treatments in CKD Patients That Exert Anti-Inflammatory Actions
5.2. Anti-Inflammatory Actions of Current Clinical Treatments for CKD in the Preservation of the PM Integrity
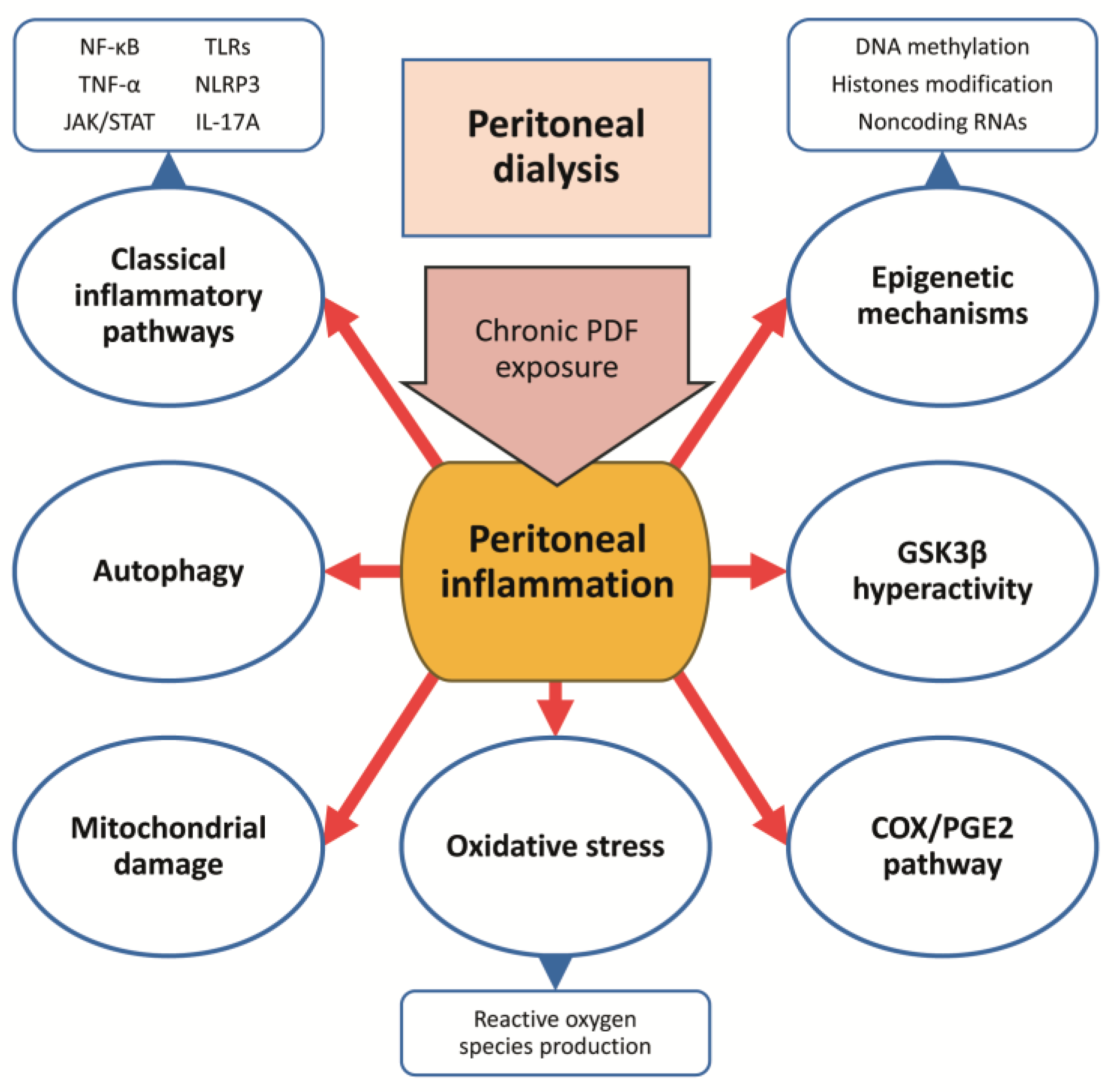
5.3. Novel Anti-Inflammatory Treatments in Experimental PD
5.3.1. Inhibition of Classical Proinflammatory Cytokines and Chemokines
5.3.2. Glycogen Synthase Kinase 3 Beta Inhibition
5.3.3. Cyclooxygenase-2/ Prostaglandin-E2 Pathway Blockade
5.3.4. Targeting Autophagy
5.3.5. Targeting Mitochondrial Dysfunction and Oxidative Stress
5.3.6. Modulation of Epigenetic Mechanisms
6. COVID-19 in CKD Patients under KRT and in Mesothelium
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ortiz, A.; Asociacion Informacion Enfermedades Renales Geneticas (AIRG-E); European Kidney Patients’ Federation (EKPF); Federación Nacional de Asociaciones para la Lucha Contra las Enfermedades del Riñón (ALCER); Fundación Renal Íñigo Álvarez de Toledo (FRIAT); Red de Investigación Renal (REDINREN); Resultados en Salud 2040 (RICORS2040); Sociedad Española de Nefrología (SENEFRO) Council; Sociedad Española de Trasplante (SET) Council; Organización Nacional de Trasplantes (ONT). RICORS2040: The need for collaborative research in chronic kidney disease. Clin. Kidney J. 2022, 15, 372–387. [Google Scholar] [CrossRef] [PubMed]
- Stevens, P.E.; Levin, A.; Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group, M. Evaluation and management of chronic kidney disease: Synopsis of the kidney disease: Improving global outcomes 2012 clinical practice guideline. Ann. Intern. Med. 2013, 158, 825–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, A.; Covic, A.; Fliser, D.; Fouque, D.; Goldsmith, D.; Kanbay, M.; Mallamaci, F.; Massy, Z.A.; Rossignol, P.; Vanholder, R.; et al. Epidemiology, contributors to, and clinical trials of mortality risk in chronic kidney failure. Lancet 2014, 383, 1831–1843. [Google Scholar] [CrossRef] [PubMed]
- Council, E.-E.; Group, E.W. Chronic kidney disease is a key risk factor for severe COVID-19: A call to action by the ERA-EDTA. Nephrol. Dial. Transplant. 2021, 36, 87–94. [Google Scholar] [CrossRef]
- Chu, K.H.; Tsang, W.K.; Tang, C.S.; Lam, M.F.; Lai, F.M.; To, K.F.; Fung, K.S.; Tang, H.L.; Yan, W.W.; Chan, H.W.; et al. Acute renal impairment in coronavirus-associated severe acute respiratory syndrome. Kidney Int. 2005, 67, 698–705. [Google Scholar] [CrossRef] [Green Version]
- del Peso, G.; Jimenez-Heffernan, J.A.; Selgas, R.; Remon, C.; Ossorio, M.; Fernandez-Perpen, A.; Sanchez-Tomero, J.A.; Cirugeda, A.; de Sousa, E.; Sandoval, P.; et al. Biocompatible Dialysis Solutions Preserve Peritoneal Mesothelial Cell and Vessel Wall Integrity. A Case-Control Study on Human Biopsies. Perit. Dial. Int. 2016, 36, 129–134. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Bajo, M.A.; Del Peso, G.; Yu, X.; Selgas, R. Preventing peritoneal membrane fibrosis in peritoneal dialysis patients. Kidney Int. 2016, 90, 515–524. [Google Scholar] [CrossRef]
- Devuyst, O.; Margetts, P.J.; Topley, N. The pathophysiology of the peritoneal membrane. J. Am. Soc. Nephrol. 2010, 21, 1077–1085. [Google Scholar] [CrossRef] [Green Version]
- Bonomini, M.; Masola, V.; Procino, G.; Zammit, V.; Divino-Filho, J.C.; Arduini, A.; Gambaro, G. How to Improve the Biocompatibility of Peritoneal Dialysis Solutions (without Jeopardizing the Patient’s Health). Int. J. Mol. Sci. 2021, 22, 7955. [Google Scholar] [CrossRef]
- Grantham, C.E.; Hull, K.L.; Graham-Brown, M.P.M.; March, D.S.; Burton, J.O. The Potential Cardiovascular Benefits of Low-Glucose Degradation Product, Biocompatible Peritoneal Dialysis Fluids: A Review of the Literature. Perit. Dial. Int. 2017, 37, 375–383. [Google Scholar] [CrossRef]
- Kopytina, V.; Pascual-Antón, L.; Toggweiler, N.; Arriero-País, E.M.; Strahl, L.; Albar-Vizcaíno, P.; Sucunza, D.; Vaquero, J.J.; Steppan, S.; Piecha, D.; et al. Steviol glycosides as an alternative osmotic agent for peritoneal dialysis fluid. Front. Pharmacol. 2022, 13, 868374. [Google Scholar] [CrossRef] [PubMed]
- Bazzato, G.; Coli, U.; Landini, S.; Fracasso, A.; Morachiello, P.; Righetto, F.; Scanferla, F.; Onesti, G. Xylitol as osmotic agent in CAPD: An alternative to glucose for uremic diabetic patients? Trans.–Am. Soc. Artif. Intern. Organs 1982, 28, 280–286. [Google Scholar] [PubMed]
- Rago, C.; Lombardi, T.; Di Fulvio, G.; Di Liberato, L.; Arduini, A.; Divino-Filho, J.C.; Bonomini, M. A New Peritoneal Dialysis Solution Containing L-Carnitine and Xylitol for Patients on Continuous Ambulatory Peritoneal Dialysis: First Clinical Experience. Toxins 2021, 13, 174. [Google Scholar] [CrossRef]
- Wang, A.Y.; Brimble, K.S.; Brunier, G.; Holt, S.G.; Jha, V.; Johnson, D.W.; Kang, S.W.; Kooman, J.P.; Lambie, M.; McIntyre, C.; et al. ISPD Cardiovascular and Metabolic Guidelines in Adult Peritoneal Dialysis Patients Part II–Management of Various Cardiovascular Complications. Perit. Dial. Int. 2015, 35, 388–396. [Google Scholar] [CrossRef] [Green Version]
- De Angelis, R.; Sant, M.; Coleman, M.P.; Francisci, S.; Baili, P.; Pierannunzio, D.; Trama, A.; Visser, O.; Brenner, H.; Ardanaz, E.; et al. Cancer survival in Europe 1999-2007 by country and age: Results of EUROCARE--5-a population-based study. Lancet Oncol. 2014, 15, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Terri, M.; Trionfetti, F.; Montaldo, C.; Cordani, M.; Tripodi, M.; Lopez-Cabrera, M.; Strippoli, R. Mechanisms of Peritoneal Fibrosis: Focus on Immune Cells-Peritoneal Stroma Interactions. Front. Immunol. 2021, 12, 607204. [Google Scholar] [CrossRef]
- Luo, Q.; Hu, Q.; Zheng, Q.; Gong, L.; Su, L.; Ren, B.; Ju, Y.; Jia, Z.; Dou, X. Enhanced mPGES-1 Contributes to PD-Related Peritoneal Fibrosis via Activation of the NLRP3 Inflammasome. Front. Med. 2021, 8, 675363. [Google Scholar] [CrossRef]
- Ludemann, W.M.; Heide, D.; Kihm, L.; Zeier, M.; Scheurich, P.; Schwenger, V.; Ranzinger, J. TNF Signaling in Peritoneal Mesothelial Cells: Pivotal Role of cFLIP(L). Perit. Dial. Int. 2017, 37, 250–258. [Google Scholar] [CrossRef]
- Mutsaers, S.E.; Birnie, K.; Lansley, S.; Herrick, S.E.; Lim, C.B.; Prele, C.M. Mesothelial cells in tissue repair and fibrosis. Front. Pharmacol. 2015, 6, 113. [Google Scholar] [CrossRef] [Green Version]
- Raby, A.C.; Gonzalez-Mateo, G.T.; Williams, A.; Topley, N.; Fraser, D.; Lopez-Cabrera, M.; Labeta, M.O. Targeting Toll-like receptors with soluble Toll-like receptor 2 prevents peritoneal dialysis solution-induced fibrosis. Kidney Int. 2018, 94, 346–362. [Google Scholar] [CrossRef]
- Karki, R.; Kanneganti, T.D. The ’cytokine storm’: Molecular mechanisms and therapeutic prospects. Trends Immunol. 2021, 42, 681–705. [Google Scholar] [CrossRef] [PubMed]
- Thodis, E.; Passadakis, P.; Lyrantzopooulos, N.; Panagoutsos, S.; Vargemezis, V.; Oreopoulos, D. Peritoneal catheters and related infections. Int. Urol. Nephrol. 2005, 37, 379–393. [Google Scholar] [CrossRef] [PubMed]
- De Waele, J.; Lipman, J.; Sakr, Y.; Marshall, J.C.; Vanhems, P.; Barrera Groba, C.; Leone, M.; Vincent, J.L.; Investigators, E.I. Abdominal infections in the intensive care unit: Characteristics, treatment and determinants of outcome. BMC Infect. Dis. 2014, 14, 420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartelli, M.; Catena, F.; Ansaloni, L.; Coccolini, F.; Corbella, D.; Moore, E.E.; Malangoni, M.; Velmahos, G.; Coimbra, R.; Koike, K.; et al. Complicated intra-abdominal infections worldwide: The definitive data of the CIAOW Study. World J. Emerg. Surg. 2014, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Goodlad, C.; George, S.; Sandoval, S.; Mepham, S.; Parekh, G.; Eberl, M.; Topley, N.; Davenport, A. Measurement of innate immune response biomarkers in peritoneal dialysis effluent using a rapid diagnostic point-of-care device as a diagnostic indicator of peritonitis. Kidney Int. 2020, 97, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Aufricht, C.; Beelen, R.; Eberl, M.; Fischbach, M.; Fraser, D.; Jorres, A.; Kratochwill, K.; LopezCabrera, M.; Rutherford, P.; Schmitt, C.P.; et al. Biomarker research to improve clinical outcomes of peritoneal dialysis: Consensus of the European Training and Research in Peritoneal Dialysis (EuTRiPD) network. Kidney Int. 2017, 92, 824–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauwels, S.; De Moor, B.; Stas, K.; Magerman, K.; Gyssens, I.C.; Van Ranst, M.; Cartuyvels, R. Coxsackievirus B1 peritonitis in a patient treated with continuous ambulatory peritoneal dialysis: A case report and brief review of the literature. Clin. Microbiol. Infect. 2012, 18, E431–E434. [Google Scholar] [CrossRef]
- Castro, R.; Abos, B.; Gonzalez, L.; Granja, A.G.; Tafalla, C. Expansion and differentiation of IgM(+) B cells in the rainbow trout peritoneal cavity in response to different antigens. Dev. Comp. Immunol. 2017, 70, 119–127. [Google Scholar] [CrossRef]
- Miles, R.; Hawley, C.M.; McDonald, S.P.; Brown, F.G.; Rosman, J.B.; Wiggins, K.J.; Bannister, K.M.; Johnson, D.W. Predictors and outcomes of fungal peritonitis in peritoneal dialysis patients. Kidney Int. 2009, 76, 622–628. [Google Scholar] [CrossRef]
- Tomita, T.; Arai, S.; Kitada, K.; Mizuno, M.; Suzuki, Y.; Sakata, F.; Nakano, D.; Hiramoto, E.; Takei, Y.; Maruyama, S.; et al. Apoptosis inhibitor of macrophage ameliorates fungus-induced peritoneal injury model in mice. Sci. Rep. 2017, 7, 6450. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune. Netw. 2018, 18, e27. [Google Scholar] [CrossRef] [PubMed]
- Colmont, C.S.; Raby, A.C.; Dioszeghy, V.; Lebouder, E.; Foster, T.L.; Jones, S.A.; Labeta, M.O.; Fielding, C.A.; Topley, N. Human peritoneal mesothelial cells respond to bacterial ligands through a specific subset of Toll-like receptors. Nephrol. Dial. Transplant. 2011, 26, 4079–4090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Aubel, R.A.; Keestra, A.M.; Krooshoop, D.J.; van Eden, W.; van Putten, J.P. Ligand-induced differential cross-regulation of Toll-like receptors 2, 4 and 5 in intestinal epithelial cells. Mol. Immunol. 2007, 44, 3702–3714. [Google Scholar] [CrossRef]
- Gewirtz, A.T.; Navas, T.A.; Lyons, S.; Godowski, P.J.; Madara, J.L. Cutting edge: Bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J. Immunol. 2001, 167, 1882–1885. [Google Scholar] [CrossRef] [Green Version]
- Wornle, M.; Sauter, M.; Kastenmuller, K.; Ribeiro, A.; Roeder, M.; Schmid, H.; Krotz, F.; Mussack, T.; Ladurner, R.; Sitter, T. Novel role of toll-like receptor 3, RIG-I and MDA5 in poly (I:C) RNA-induced mesothelial inflammation. Mol. Cell. Biochem. 2009, 322, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Merkle, M.; Ribeiro, A.; Sauter, M.; Ladurner, R.; Mussack, T.; Sitter, T.; Wornle, M. Effect of activation of viral receptors on the gelatinases MMP-2 and MMP-9 in human mesothelial cells. Matrix. Biol. 2010, 29, 202–208. [Google Scholar] [CrossRef]
- Zarember, K.A.; Godowski, P.J. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 2002, 168, 554–561. [Google Scholar] [CrossRef] [Green Version]
- Hurst, S.M.; Wilkinson, T.S.; McLoughlin, R.M.; Jones, S.; Horiuchi, S.; Yamamoto, N.; Rose-John, S.; Fuller, G.M.; Topley, N.; Jones, S.A. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 2001, 14, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Jackson-Jones, L.H.; Smith, P.; Portman, J.R.; Magalhaes, M.S.; Mylonas, K.J.; Vermeren, M.M.; Nixon, M.; Henderson, B.E.P.; Dobie, R.; Vermeren, S.; et al. Stromal Cells Covering Omental Fat-Associated Lymphoid Clusters Trigger Formation of Neutrophil Aggregates to Capture Peritoneal Contaminants. Immunity 2020, 52, 700–715.e6. [Google Scholar] [CrossRef]
- Buechler, M.B.; Turley, S.J. Neutrophils Follow Stromal Omens to Limit Peritoneal Inflammation. Immunity 2020, 52, 578–580. [Google Scholar] [CrossRef]
- Fielding, C.A.; McLoughlin, R.M.; McLeod, L.; Colmont, C.S.; Najdovska, M.; Grail, D.; Ernst, M.; Jones, S.A.; Topley, N.; Jenkins, B.J. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J. Immunol. 2008, 181, 2189–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.A. Directing transition from innate to acquired immunity: Defining a role for IL-6. J. Immunol. 2005, 175, 3463–3468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirado, M.; Koss, W. Differentiation of mesothelial cells into macrophage phagocytic cells in a patient with clinical sepsis. Blood 2018, 132, 1460. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kubes, P. A Reservoir of Mature Cavity Macrophages that Can Rapidly Invade Visceral Organs to Affect Tissue Repair. Cell 2016, 165, 668–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catar, R.A.; Chen, L.; Cuff, S.M.; Kift-Morgan, A.; Eberl, M.; Kettritz, R.; Kamhieh-Milz, J.; Moll, G.; Li, Q.; Zhao, H.; et al. Control of neutrophil influx during peritonitis by transcriptional cross-regulation of chemokine CXCL1 by IL-17 and IFN-gamma. J. Pathol. 2020, 251, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Erreni, M.; Allavena, P.; Porta, C. Macrophage polarization in pathology. Cell. Mol. Life Sci. 2015, 72, 4111–4126. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Cassado Ados, A.; D’Imperio Lima, M.R.; Bortoluci, K.R. Revisiting mouse peritoneal macrophages: Heterogeneity, development, and function. Front. Immunol. 2015, 6, 225. [Google Scholar] [CrossRef] [Green Version]
- Liao, C.T.; Andrews, R.; Wallace, L.E.; Khan, M.W.; Kift-Morgan, A.; Topley, N.; Fraser, D.J.; Taylor, P.R. Peritoneal macrophage heterogeneity is associated with different peritoneal dialysis outcomes. Kidney Int. 2017, 91, 1088–1103. [Google Scholar] [CrossRef]
- Kubicka, U.; Olszewski, W.L.; Tarnowski, W.; Bielecki, K.; Ziolkowska, A.; Wierzbicki, Z. Normal human immune peritoneal cells: Subpopulations and functional characteristics. Scand. J. Immunol. 1996, 44, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Accarias, S.; Genthon, C.; Rengel, D.; Boullier, S.; Foucras, G.; Tabouret, G. Single-cell analysis reveals new subset markers of murine peritoneal macrophages and highlights macrophage dynamics upon Staphylococcus aureus peritonitis. Innate Immun. 2016, 22, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Barth, M.W.; Hendrzak, J.A.; Melnicoff, M.J.; Morahan, P.S. Review of the macrophage disappearance reaction. J. Leukoc. Biol. 1995, 57, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, G.J.; Barter, P.J. Apolipoprotein A-I inhibits transformation of high density lipoprotein subpopulations during incubation of human plasma. Atherosclerosis 1989, 75, 73–82. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, S.; Li, L.; Li, L.; Zhou, X.; Wan, M.; Lou, P.; Zhao, M.; Lv, K.; Yuan, Y.; et al. Peritoneal M2 macrophage-derived extracellular vesicles as natural multitarget nanotherapeutics to attenuate cytokine storms after severe infections. J. Control. Release 2022, 349, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, T.E.; Shaw, T.N.; Lennon, R.; Herrick, S.E.; Ruckerl, D. Ongoing Exposure to Peritoneal Dialysis Fluid Alters Resident Peritoneal Macrophage Phenotype and Activation Propensity. Front. Immunol. 2021, 12, 715209. [Google Scholar] [CrossRef]
- Li, Q.; Zheng, M.; Liu, Y.; Sun, W.; Shi, J.; Ni, J.; Wang, Q. A pathogenetic role for M1 macrophages in peritoneal dialysis-associated fibrosis. Mol. Immunol. 2018, 94, 131–139. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, Z.P.; Su, N.; Fan, J.J.; Ruan, Y.P.; Peng, W.X.; Li, Y.F.; Yu, X.Q. The role of peritoneal alternatively activated macrophages in the process of peritoneal fibrosis related to peritoneal dialysis. Int. J. Mol. Sci. 2013, 14, 10369–10382. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.T.; Hsu, H.; Lin, C.C.; Pan, S.Y.; Liu, S.Y.; Wu, C.F.; Tsai, P.Z.; Liao, C.T.; Cheng, H.T.; Chiang, W.C.; et al. Inflammatory macrophages switch to CCL17-expressing phenotype and promote peritoneal fibrosis. J. Pathol. 2020, 250, 55–66. [Google Scholar] [CrossRef] [Green Version]
- de Fijter, C.W.; Verbrugh, H.A.; Peters, E.D.; Oe, P.L.; van der Meulen, J.; Verhoef, J.; Donker, A.J. In vivo exposure to the currently available peritoneal dialysis fluids decreases the function of peritoneal macrophages in CAPD. Clin. Nephrol. 1993, 39, 75–80. [Google Scholar] [PubMed]
- Fieren, M.W. Mechanisms regulating cytokine release from peritoneal macrophages during continuous ambulatory peritoneal dialysis. Blood Purif. 1996, 14, 179–187. [Google Scholar] [CrossRef]
- Bellon, T.; Martinez, V.; Lucendo, B.; del Peso, G.; Castro, M.J.; Aroeira, L.S.; Rodriguez-Sanz, A.; Ossorio, M.; Sanchez-Villanueva, R.; Selgas, R.; et al. Alternative activation of macrophages in human peritoneum: Implications for peritoneal fibrosis. Nephrol. Dial. Transplant. 2011, 26, 2995–3005. [Google Scholar] [CrossRef] [Green Version]
- Kono, M. New insights into the metabolism of Th17 cells. Immunol. Med. 2023, 46, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Takeuchi, Y.; Hirota, K. The pathogenicity of Th17 cells in autoimmune diseases. Semin. Immunopathol. 2019, 41, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Diez, R.; Aroeira, L.S.; Orejudo, M.; Bajo, M.A.; Heffernan, J.J.; Rodrigues-Diez, R.R.; Rayego-Mateos, S.; Ortiz, A.; Gonzalez-Mateo, G.; Lopez-Cabrera, M.; et al. IL-17A is a novel player in dialysis-induced peritoneal damage. Kidney Int. 2014, 86, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Chung, D.R.; Kasper, D.L.; Panzo, R.J.; Chitnis, T.; Grusby, M.J.; Sayegh, M.H.; Tzianabos, A.O. CD4+ T cells mediate abscess formation in intra-abdominal sepsis by an IL-17-dependent mechanism. J. Immunol. 2003, 170, 1958–1963. [Google Scholar] [CrossRef] [Green Version]
- Murphy, A.G.; O’Keeffe, K.M.; Lalor, S.J.; Maher, B.M.; Mills, K.H.; McLoughlin, R.M. Staphylococcus aureus infection of mice expands a population of memory gammadelta T cells that are protective against subsequent infection. J. Immunol. 2014, 192, 3697–3708. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, Y.; Lou, J.; Zhu, J.; He, M.; Deng, X.; Cai, Z. Neutralisation of peritoneal IL-17A markedly improves the prognosis of severe septic mice by decreasing neutrophil infiltration and proinflammatory cytokines. PLoS ONE 2012, 7, e46506. [Google Scholar] [CrossRef]
- Witowski, J.; Ksiazek, K.; Warnecke, C.; Kuzlan, M.; Korybalska, K.; Tayama, H.; Wisniewska-Elnur, J.; Pawlaczyk, K.; Trominska, J.; Breborowicz, A.; et al. Role of mesothelial cell-derived granulocyte colony-stimulating factor in interleukin-17-induced neutrophil accumulation in the peritoneum. Kidney Int. 2007, 71, 514–525. [Google Scholar] [CrossRef] [Green Version]
- Witowski, J.; Pawlaczyk, K.; Breborowicz, A.; Scheuren, A.; Kuzlan-Pawlaczyk, M.; Wisniewska, J.; Polubinska, A.; Friess, H.; Gahl, G.M.; Frei, U.; et al. IL-17 stimulates intraperitoneal neutrophil infiltration through the release of GRO alpha chemokine from mesothelial cells. J. Immunol. 2000, 165, 5814–5821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liappas, G.; Gonzalez-Mateo, G.T.; Majano, P.; Sanchez-Tomero, J.A.; Ruiz-Ortega, M.; Rodrigues Diez, R.; Martin, P.; Sanchez-Diaz, R.; Selgas, R.; Lopez-Cabrera, M.; et al. T Helper 17/Regulatory T Cell Balance and Experimental Models of Peritoneal Dialysis-Induced Damage. BioMed Res. Int. 2015, 2015, 416480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.; Gomez, M.; Lamana, A.; Cruz-Adalia, A.; Ramirez-Huesca, M.; Ursa, M.A.; Yanez-Mo, M.; Sanchez-Madrid, F. CD69 association with Jak3/Stat5 proteins regulates Th17 cell differentiation. Mol. Cell. Biol. 2010, 30, 4877–4889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liappas, G.; Gonzalez-Mateo, G.T.; Sanchez-Diaz, R.; Lazcano, J.J.; Lasarte, S.; Matesanz-Marin, A.; Zur, R.; Ferrantelli, E.; Ramirez, L.G.; Aguilera, A.; et al. Immune-Regulatory Molecule CD69 Controls Peritoneal Fibrosis. J. Am. Soc. Nephrol. 2016, 27, 3561–3576. [Google Scholar] [CrossRef] [Green Version]
- Vila Cuenca, M.; Keuning, E.D.; Talhout, W.; Paauw, N.J.; van Ittersum, F.J.; Ter Wee, P.M.; Beelen, R.H.J.; Vervloet, M.G.; Ferrantelli, E. Differences in peritoneal response after exposure to low-GDP bicarbonate/lactate-buffered dialysis solution compared to conventional dialysis solution in a uremic mouse model. Int. Urol. Nephrol. 2018, 50, 1151–1161. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.T.; Ng, H.Y.; Hsu, C.Y.; Tsai, Y.C.; Yang, Y.K.; Chen, T.C.; Chiou, T.T.; Kuo, C.C.; Lee, W.C.; Hsu, K.T. Proinflammatory cytokines, hepatocyte growth factor and adipokines in peritoneal dialysis patients. Artif. Organs. 2010, 34, E222–E229. [Google Scholar] [CrossRef]
- Parikova, A.; Zweers, M.M.; Struijk, D.G.; Krediet, R.T. Peritoneal effluent markers of inflammation in patients treated with icodextrin-based and glucose-based dialysis solutions. Adv. Perit. Dial. 2003, 19, 186–190. [Google Scholar]
- Sanz, A.B.; Aroeira, L.S.; Bellon, T.; del Peso, G.; Jimenez-Heffernan, J.; Santamaria, B.; Sanchez-Nino, M.D.; Blanco-Colio, L.M.; Lopez-Cabrera, M.; Ruiz-Ortega, M.; et al. TWEAK promotes peritoneal inflammation. PLoS ONE 2014, 9, e90399. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Roberts, G.W.; Kift-Morgan, A.; Donovan, K.L.; Topley, N.; Eberl, M. Pathogen-specific local immune fingerprints diagnose bacterial infection in peritoneal dialysis patients. J Am Soc Nephrol 2013, 24, 2002–2009. [Google Scholar] [CrossRef] [Green Version]
- Zareie, M.; Fabbrini, P.; Hekking, L.H.; Keuning, E.D.; Ter Wee, P.M.; Beelen, R.H.; van den Born, J. Novel role for mast cells in omental tissue remodeling and cell recruitment in experimental peritoneal dialysis. J. Am. Soc. Nephrol. 2006, 17, 3447–3457. [Google Scholar] [CrossRef] [Green Version]
- Alscher, D.M.; Braun, N.; Biegger, D.; Fritz, P. Peritoneal mast cells in peritoneal dialysis patients, particularly in encapsulating peritoneal sclerosis patients. Am. J. Kidney Dis. 2007, 49, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Anuforo, O.U.U.; Bjarnarson, S.P.; Jonasdottir, H.S.; Giera, M.; Hardardottir, I.; Freysdottir, J. Natural killer cells play an essential role in resolution of antigen-induced inflammation in mice. Mol. Immunol. 2018, 93, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Thoren, F.B.; Riise, R.E.; Ousback, J.; Della Chiesa, M.; Alsterholm, M.; Marcenaro, E.; Pesce, S.; Prato, C.; Cantoni, C.; Bylund, J.; et al. Human NK Cells induce neutrophil apoptosis via an NKp46- and Fas-dependent mechanism. J. Immunol. 2012, 188, 1668–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, B.M.P.; Wilkinson, R.; Wang, X.; Kildey, K.; Lindner, M.; Rist, M.J.; Beagley, K.; Healy, H.; Kassianos, A.J. Interferon-gamma production by tubulointerstitial human CD56(bright) natural killer cells contributes to renal fibrosis and chronic kidney disease progression. Kidney Int. 2017, 92, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Steis, R.G.; Urba, W.J.; VanderMolen, L.A.; Bookman, M.A.; Smith, J.W., 2nd; Clark, J.W.; Miller, R.L.; Crum, E.D.; Beckner, S.K.; McKnight, J.E.; et al. Intraperitoneal lymphokine-activated killer-cell and interleukin-2 therapy for malignancies limited to the peritoneal cavity. J. Clin. Oncol. 1990, 8, 1618–1629. [Google Scholar] [CrossRef]
- Gulyas, M.; Hjerpe, A. Proteoglycans and WT1 as markers for distinguishing adenocarcinoma, epithelioid mesothelioma, and benign mesothelium. J. Pathol. 2003, 199, 479–487. [Google Scholar] [CrossRef]
- Wilm, B.; Munoz-Chapuli, R. The Role of WT1 in Embryonic Development and Normal Organ Homeostasis. Methods Mol. Biol. 2016, 1467, 23–39. [Google Scholar] [CrossRef] [Green Version]
- Strippoli, R.; Sandoval, P.; Moreno-Vicente, R.; Rossi, L.; Battistelli, C.; Terri, M.; Pascual-Anton, L.; Loureiro, M.; Matteini, F.; Calvo, E.; et al. Caveolin1 and YAP drive mechanically induced mesothelial to mesenchymal transition and fibrosis. Cell Death Dis. 2020, 11, 647. [Google Scholar] [CrossRef]
- Namvar, S.; Woolf, A.S.; Zeef, L.A.; Wilm, T.; Wilm, B.; Herrick, S.E. Functional molecules in mesothelial-to-mesenchymal transition revealed by transcriptome analyses. J. Pathol. 2018, 245, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Carpio, V.; Sandoval, P.; Aguilera, A.; Albar-Vizcaino, P.; Perez-Lozano, M.L.; Gonzalez-Mateo, G.T.; Acuna-Ruiz, A.; Garcia-Cantalejo, J.; Botias, P.; Bajo, M.A.; et al. Genomic reprograming analysis of the Mesothelial to Mesenchymal Transition identifies biomarkers in peritoneal dialysis patients. Sci. Rep. 2017, 7, 44941. [Google Scholar] [CrossRef] [Green Version]
- Lambie, M.; Chess, J.; Donovan, K.L.; Kim, Y.L.; Do, J.Y.; Lee, H.B.; Noh, H.; Williams, P.F.; Williams, A.J.; Davison, S.; et al. Independent effects of systemic and peritoneal inflammation on peritoneal dialysis survival. J. Am. Soc. Nephrol. 2013, 24, 2071–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, G.W.; Baird, D.; Gallagher, K.; Jones, R.E.; Pepper, C.J.; Williams, J.D.; Topley, N. Functional effector memory T cells enrich the peritoneal cavity of patients treated with peritoneal dialysis. J. Am. Soc. Nephrol. 2009, 20, 1895–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cailhier, J.F.; Partolina, M.; Vuthoori, S.; Wu, S.; Ko, K.; Watson, S.; Savill, J.; Hughes, J.; Lang, R.A. Conditional macrophage ablation demonstrates that resident macrophages initiate acute peritoneal inflammation. J. Immunol. 2005, 174, 2336–2342. [Google Scholar] [CrossRef] [Green Version]
- Yung, S.; Chan, T.M. Intrinsic cells: Mesothelial cells -- central players in regulating inflammation and resolution. Perit. Dial. Int. 2009, 29, S21–S27. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, Y.G.; Shaw, M.; Kanneganti, T.D.; Fujimoto, Y.; Fukase, K.; Inohara, N.; Nunez, G. Nod1/RICK and TLR signaling regulate chemokine and antimicrobial innate immune responses in mesothelial cells. J. Immunol. 2007, 179, 514–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S.; Yuzawa, Y.; Tsuboi, N.; Maruyama, S.; Morita, Y.; Matsuguchi, T.; Matsuo, S. Endotoxin-induced chemokine expression in murine peritoneal mesothelial cells: The role of toll-like receptor 4. J. Am. Soc. Nephrol. 2004, 15, 1289–1299. [Google Scholar]
- Beelen, R.H.; Oosterling, S.J.; van Egmond, M.; van den Born, J.; Zareie, M. Omental milky spots in peritoneal pathophysiology (spots before your eyes). Perit. Dial. Int. 2005, 25, 30–32. [Google Scholar] [CrossRef]
- Rangel-Moreno, J.; Moyron-Quiroz, J.E.; Carragher, D.M.; Kusser, K.; Hartson, L.; Moquin, A.; Randall, T.D. Omental milky spots develop in the absence of lymphoid tissue-inducer cells and support B and T cell responses to peritoneal antigens. Immunity 2009, 30, 731–743. [Google Scholar] [CrossRef] [Green Version]
- Mebius, R.E. Lymphoid organs for peritoneal cavity immune response: Milky spots. Immunity 2009, 30, 670–672. [Google Scholar] [CrossRef] [Green Version]
- Perez-Shibayama, C.; Gil-Cruz, C.; Cheng, H.W.; Onder, L.; Printz, A.; Morbe, U.; Novkovic, M.; Li, C.; Lopez-Macias, C.; Buechler, M.B.; et al. Fibroblastic reticular cells initiate immune responses in visceral adipose tissues and secure peritoneal immunity. Sci. Immunol. 2018, 3, 26. [Google Scholar] [CrossRef] [Green Version]
- Witowski, J.; Tayama, H.; Ksiazek, K.; Wanic-Kossowska, M.; Bender, T.O.; Jorres, A. Human peritoneal fibroblasts are a potent source of neutrophil-targeting cytokines: A key role of IL-1beta stimulation. Lab. Investig. 2009, 89, 414–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riese, J.; Denzel, C.; Zowe, M.; Mehler, C.; Hohenberger, W.; Haupt, W. Secretion of IL-6, monocyte chemoattractant protein-1, macrophage inflammatory protein-1alpha, and TNFalpha by cultured intact human peritoneum. Eur. Surg. Res. 1999, 31, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Catar, R.A.; Bartosova, M.; Kawka, E.; Chen, L.; Marinovic, I.; Zhang, C.; Zhao, H.; Wu, D.; Zickler, D.; Stadnik, H.; et al. Angiogenic Role of Mesothelium-Derived Chemokine CXCL1 During Unfavorable Peritoneal Tissue Remodeling in Patients Receiving Peritoneal Dialysis as Renal Replacement Therapy. Front. Immunol. 2022, 13, 821681. [Google Scholar] [CrossRef] [PubMed]
- Margetts, P.J.; Bonniaud, P. Basic mechanisms and clinical implications of peritoneal fibrosis. Perit. Dial. Int. 2003, 23, 530–541. [Google Scholar] [CrossRef]
- Jorres, A.; Ludat, K.; Lang, J.; Sander, K.; Gahl, G.M.; Frei, U.; DeJonge, K.; Williams, J.D.; Topley, N. Establishment and functional characterization of human peritoneal fibroblasts in culture: Regulation of interleukin-6 production by proinflammatory cytokines. J. Am. Soc. Nephrol. 1996, 7, 2192–2201. [Google Scholar] [CrossRef]
- Witowski, J.; Thiel, A.; Dechend, R.; Dunkel, K.; Fouquet, N.; Bender, T.O.; Langrehr, J.M.; Gahl, G.M.; Frei, U.; Jorres, A. Synthesis of C-X-C and C-C chemokines by human peritoneal fibroblasts: Induction by macrophage-derived cytokines. Am. J. Pathol. 2001, 158, 1441–1450. [Google Scholar] [CrossRef]
- Witowski, J.; Kawka, E.; Rudolf, A.; Jorres, A. New developments in peritoneal fibroblast biology: Implications for inflammation and fibrosis in peritoneal dialysis. BioMed Res. Int. 2015, 2015, 134708. [Google Scholar] [CrossRef] [Green Version]
- Kawka, E.; Witowski, J.; Fouqet, N.; Tayama, H.; Bender, T.O.; Catar, R.; Dragun, D.; Jorres, A. Regulation of chemokine CCL5 synthesis in human peritoneal fibroblasts: A key role of IFN-gamma. Mediators Inflamm. 2014, 2014, 590654. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Burton, D.G.; Krizhanovsky, V. Physiological and pathological consequences of cellular senescence. Cell. Mol. Life Sci. 2014, 71, 4373–4386. [Google Scholar] [CrossRef] [Green Version]
- Bernadotte, A.; Mikhelson, V.M.; Spivak, I.M. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging 2016, 8, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastian, T.; Malik, R.; Thomas, S.; Sage, J.; Johnson, P.F. C/EBPbeta cooperates with RB:E2F to implement Ras(V12)-induced cellular senescence. EMBO J. 2005, 24, 3301–3312. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, K. Mesothelial cell: A multifaceted model of aging. Ageing Res. Rev. 2013, 12, 595–604. [Google Scholar] [CrossRef]
- Lopez-Cabrera, M.; Aguilera, A.; Aroeira, L.S.; Ramirez-Huesca, M.; Perez-Lozano, M.L.; Jimenez-Heffernan, J.A.; Bajo, M.A.; del Peso, G.; Sanchez-Tomero, J.A.; Selgas, R. Ex vivo analysis of dialysis effluent-derived mesothelial cells as an approach to unveiling the mechanism of peritoneal membrane failure. Perit. Dial. Int. 2006, 26, 26–34. [Google Scholar] [CrossRef]
- Yanez-Mo, M.; Lara-Pezzi, E.; Selgas, R.; Ramirez-Huesca, M.; Dominguez-Jimenez, C.; Jimenez-Heffernan, J.A.; Aguilera, A.; Sanchez-Tomero, J.A.; Bajo, M.A.; Alvarez, V.; et al. Peritoneal dialysis and epithelial-to-mesenchymal transition of mesothelial cells. N. Engl. J. Med. 2003, 348, 403–413. [Google Scholar] [CrossRef]
- Kawka, E.; Witowski, J.; Sandoval, P.; Rudolf, A.; Vidal, A.R.; Cabrera, M.L.; Jorres, A. Epithelial-to-Mesenchymal Transition and Migration of Human Peritoneal Mesothelial Cells Undergoing Senescence. Perit. Dial. Int. 2019, 39, 35–41. [Google Scholar] [CrossRef]
- Witowski, J.; Bender, T.O.; Wisniewska-Elnur, J.; Ksiazek, K.; Passlick-Deetjen, J.; Breborowicz, A.; Jorres, A. Mesothelial toxicity of peritoneal dialysis fluids is related primarily to glucose degradation products, not to glucose per se. Perit. Dial. Int. 2003, 23, 381–390. [Google Scholar] [CrossRef]
- Yang, L.; Wu, L.; Zhang, X.; Hu, Y.; Fan, Y.; Ma, J. 1,25(OH)2D3/VDR attenuates high glucose-induced epithelial-mesenchymal transition in human peritoneal mesothelial cells via the TGFbeta/Smad3 pathway. Mol. Med. Rep. 2017, 15, 2273–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotloib, L.; Shostak, A.; Wajsbrot, V.; Kushnier, R. High glucose induces a hypertrophic, senescent mesothelial cell phenotype after long in vivo exposure. Nephron 1999, 82, 164–173. [Google Scholar] [CrossRef]
- Ksiazek, K.; Mikula-Pietrasik, J.; Jorres, A.; Witowski, J. Oxidative stress-mediated early senescence contributes to the short replicative life span of human peritoneal mesothelial cells. Free Radic. Biol. Med. 2008, 45, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Gotloib, L.; Wajsbrot, V.; Shostak, A. Icodextrin-induced lipid peroxidation disrupts the mesothelial cell cycle engine. Free Radic. Biol. Med. 2003, 34, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Gotloib, L.; Gotloib, L.C.; Khrizman, V. The use of peritoneal mesothelium as a potential source of adult stem cells. Int. J. Artif. Organs 2007, 30, 501–512. [Google Scholar] [CrossRef]
- Bartosova, M.; Zhang, C.; Schaefer, B.; Herzog, R.; Ridinger, D.; Damgov, I.; Levai, E.; Marinovic, I.; Eckert, C.; Romero, P.; et al. Glucose Derivative Induced Vasculopathy in Children on Chronic Peritoneal Dialysis. Circ. Res. 2021, 129, e102–e118. [Google Scholar] [CrossRef]
- Perez-Gomez, M.V.; Sanchez-Nino, M.D.; Sanz, A.B.; Zheng, B.; Martin-Cleary, C.; Ruiz-Ortega, M.; Ortiz, A.; Fernandez-Fernandez, B. Targeting inflammation in diabetic kidney disease: Early clinical trials. Expert Opin. Investig. Drugs 2016, 25, 1045–1058. [Google Scholar] [CrossRef]
- Shao, Y. Bibliometric Study of Trends in the Diabetic Nephropathy Research Space from 2016 to 2020. Oxid. Med. Cell. Longev. 2022, 2022, 8050137. [Google Scholar] [CrossRef]
- Rayego-Mateos, S.; Rodrigues-Diez, R.R.; Fernandez-Fernandez, B.; Mora-Fernandez, C.; Marchant, V.; Donate-Correa, J.; Navarro-Gonzalez, J.F.; Ortiz, A.; Ruiz-Ortega, M. Targeting inflammation to treat diabetic kidney disease: The road to 2030. Kidney Int. 2022, 103, 282–296. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, B.; Li, Y.; Zhang, L.; Wang, Y.; Yang, S.; Xiao, X.; Qin, Q. The use of renin-angiotensin-aldosterone system (RAAS) inhibitors is associated with a lower risk of mortality in hypertensive COVID-19 patients: A systematic review and meta-analysis. J. Med. Virol. 2021, 93, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; Adler, S.G.; Barratt, J.; Bridoux, F.; Burdge, K.A.; Chan, T.M.; Cook, H.T.; Fervenza, F.C.; Gibson, K.L.; Glassock, R.J.; et al. Executive summary of the KDIGO 2021 Guideline for the Management of Glomerular Diseases. Kidney Int. 2021, 100, 753–779. [Google Scholar] [CrossRef] [PubMed]
- de Boer, I.H.; Khunti, K.; Sadusky, T.; Tuttle, K.R.; Neumiller, J.J.; Rhee, C.M.; Rosas, S.E.; Rossing, P.; Bakris, G. Diabetes management in chronic kidney disease: A consensus report by the American Diabetes Association (ADA) and Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2022, 102, 974–989. [Google Scholar] [CrossRef] [PubMed]
- Li, P.K.; Chow, K.M.; Wong, T.Y.; Leung, C.B.; Szeto, C.C. Effects of an angiotensin-converting enzyme inhibitor on residual renal function in patients receiving peritoneal dialysis. A randomized, controlled study. Ann. Intern. Med. 2003, 139, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Stefansson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Group, E.-K.C.; Herrington, W.G.; Staplin, N.; Wanner, C.; Green, J.B.; Hauske, S.J.; Emberson, J.R.; Preiss, D.; Judge, P.; Mayne, K.J.; et al. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2022, 388, 117–127. [Google Scholar] [CrossRef]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Back, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. J. Prev. Cardiol. 2022, 29, 5–115. [Google Scholar] [CrossRef]
- Bakris, G.L.; Ruilope, L.M.; Anker, S.D.; Filippatos, G.; Pitt, B.; Rossing, P.; Fried, L.; Roy-Chaudhury, P.; Sarafidis, P.; Ahlers, C.; et al. A prespecified exploratory analysis from FIDELITY examined finerenone use and kidney outcomes in patients with chronic kidney disease and type 2 diabetes. Kidney Int. 2023, 103, 196–206. [Google Scholar] [CrossRef]
- Agarwal, R.; Filippatos, G.; Pitt, B.; Anker, S.D.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Gebel, M.; Ruilope, L.M.; et al. Cardiovascular and kidney outcomes with finerenone in patients with type 2 diabetes and chronic kidney disease: The FIDELITY pooled analysis. Eur. Heart J. 2022, 43, 474–484. [Google Scholar] [CrossRef]
- Frimodt-Moller, M.; Persson, F.; Rossing, P. Mitigating risk of aldosterone in diabetic kidney disease. Curr. Opin. Nephrol. Hypertens. 2020, 29, 145–151. [Google Scholar] [CrossRef]
- Terami, N.; Ogawa, D.; Tachibana, H.; Hatanaka, T.; Wada, J.; Nakatsuka, A.; Eguchi, J.; Horiguchi, C.S.; Nishii, N.; Yamada, H.; et al. Long-term treatment with the sodium glucose cotransporter 2 inhibitor, dapagliflozin, ameliorates glucose homeostasis and diabetic nephropathy in db/db mice. PLoS ONE 2014, 9, e100777. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, Y.; Matsui, T.; Yamagishi, S.; Tofogliflozin, A. Highly Selective Inhibitor of SGLT2 Blocks Proinflammatory and Proapoptotic Effects of Glucose Overload on Proximal Tubular Cells Partly by Suppressing Oxidative Stress Generation. Horm. Metab. Res. 2016, 48, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Chimal, J.; Estrela, G.R.; Lechner, S.M.; Giraud, S.; El Moghrabi, S.; Kaaki, S.; Kolkhof, P.; Hauet, T.; Jaisser, F. The myeloid mineralocorticoid receptor controls inflammatory and fibrotic responses after renal injury via macrophage interleukin-4 receptor signaling. Kidney Int. 2018, 93, 1344–1355. [Google Scholar] [CrossRef] [PubMed]
- Egido, J.; Rojas-Rivera, J.; Mas, S.; Ruiz-Ortega, M.; Sanz, A.B.; Gonzalez Parra, E.; Gomez-Guerrero, C. Atrasentan for the treatment of diabetic nephropathy. Expert Opin. Investig. Drugs 2017, 26, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Fernandez-Fernandez, B. Atrasentan: The Difficult Task of Integrating Endothelin A Receptor Antagonists into Current Treatment Paradigm for Diabetic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2021, 16, 1775–1778. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, B.; Fernandez-Prado, R.; Gorriz, J.L.; Martinez-Castelao, A.; Navarro-Gonzalez, J.F.; Porrini, E.; Soler, M.J.; Ortiz, A. Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation and Study of Diabetic Nephropathy with Atrasentan: What was learned about the treatment of diabetic kidney disease with canagliflozin and atrasentan? Clin. Kidney J. 2019, 12, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Heerspink, H.J.L.; Parving, H.H.; Andress, D.L.; Bakris, G.; Correa-Rotter, R.; Hou, F.F.; Kitzman, D.W.; Kohan, D.; Makino, H.; McMurray, J.J.V.; et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): A double-blind, randomised, placebo-controlled trial. Lancet 2019, 393, 1937–1947. [Google Scholar] [CrossRef]
- Trachtman, H.; Nelson, P.; Adler, S.; Campbell, K.N.; Chaudhuri, A.; Derebail, V.K.; Gambaro, G.; Gesualdo, L.; Gipson, D.S.; Hogan, J.; et al. DUET: A Phase 2 Study Evaluating the Efficacy and Safety of Sparsentan in Patients with FSGS. J. Am. Soc. Nephrol. 2018, 29, 2745–2754. [Google Scholar] [CrossRef] [Green Version]
- Vergara, A.; Jacobs-Cacha, C.; Llorens-Cebria, C.; Ortiz, A.; Martinez-Diaz, I.; Martos, N.; Dominguez-Baez, P.; Van den Bosch, M.M.; Bermejo, S.; Pieper, M.P.; et al. Enhanced Cardiorenal Protective Effects of Combining SGLT2 Inhibition, Endothelin Receptor Antagonism and RAS Blockade in Type 2 Diabetic Mice. Int. J. Mol. Sci. 2022, 23, 12823. [Google Scholar] [CrossRef]
- Ortiz, A. Complement and protection from tissue injury in COVID-19. Clin. Kidney J. 2020, 13, 734–738. [Google Scholar] [CrossRef]
- Perez-Gomez, M.V.; Ortiz, A. Aliskiren and the dual complement inhibition concept. Clin. Kidney J. 2020, 13, 35–38. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Cui, Z.; Zhao, M.H. Complement C3a and C3a Receptor Activation Mediates Podocyte Injuries in the Mechanism of Primary Membranous Nephropathy. J. Am. Soc. Nephrol. 2022, 33, 1742–1756. [Google Scholar] [CrossRef]
- Anwar, I.J.; DeLaura, I.; Ladowski, J.; Gao, Q.; Knechtle, S.J.; Kwun, J. Complement-targeted therapies in kidney transplantation-insights from preclinical studies. Front. Immunol. 2022, 13, 984090. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.M.; Snelson, M.; Ostergaard, J.A.; Coughlan, M.T. The Complement Pathway: New Insights into Immunometabolic Signaling in Diabetic Kidney Disease. Antioxid. Redox Signal. 2022, 37, 781–801. [Google Scholar] [CrossRef]
- Jayne, D.R.W.; Merkel, P.A.; Schall, T.J.; Bekker, P.; Group, A.S. Avacopan for the Treatment of ANCA-Associated Vasculitis. N. Engl. J. Med. 2021, 384, 599–609. [Google Scholar] [CrossRef]
- Lavoz, C.; Rayego-Mateos, S.; Orejudo, M.; Opazo-Rios, L.; Marchant, V.; Marquez-Exposito, L.; Tejera-Munoz, A.; Navarro-Gonzalez, J.F.; Droguett, A.; Ortiz, A.; et al. Could IL-17A Be a Novel Therapeutic Target in Diabetic Nephropathy? J. Clin. Med. 2020, 9, 272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Martinez, J.; Perez-Martinez, F.C.; Carrion, B.; Masia, J.; Ortega, A.; Simarro, E.; Nam-Cha, S.H.; Cena, V. Aliskiren prevents the toxic effects of peritoneal dialysis fluids during chronic dialysis in rats. PLoS ONE 2012, 7, e36268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocak, G.; Azak, A.; Astarci, H.M.; Huddam, B.; Karaca, G.; Ceri, M.; Can, M.; Sert, M.; Duranay, M. Effects of renin-angiotensin-aldosterone system blockade on chlorhexidine gluconate-induced sclerosing encapsulated peritonitis in rats. Ther. Apher. Dial. 2012, 16, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Duman, S.; Wieczorowska-Tobis, K.; Styszynski, A.; Kwiatkowska, B.; Breborowicz, A.; Oreopoulos, D.G. Intraperitoneal enalapril ameliorates morphologic changes induced by hypertonic peritoneal dialysis solutions in rat peritoneum. Adv. Perit. Dial. 2004, 20, 31–36. [Google Scholar]
- Duman, S.; Sen, S.; Duman, C.; Oreopoulos, D.G. Effect of valsartan versus lisinopril on peritoneal sclerosis in rats. Int. J. Artif. Organs 2005, 28, 156–163. [Google Scholar] [CrossRef]
- Nessim, S.J.; Perl, J.; Bargman, J.M. The renin-angiotensin-aldosterone system in peritoneal dialysis: Is what is good for the kidney also good for the peritoneum? Kidney Int. 2010, 78, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, F.; Scheen, A.J. Effects of SGLT2 inhibitors on systemic and tissue low-grade inflammation: The potential contribution to diabetes complications and cardiovascular disease. Diabetes Metab. 2018, 44, 457–464. [Google Scholar] [CrossRef]
- Scisciola, L.; Cataldo, V.; Taktaz, F.; Fontanella, R.A.; Pesapane, A.; Ghosh, P.; Franzese, M.; Puocci, A.; De Angelis, A.; Sportiello, L.; et al. Anti-inflammatory role of SGLT2 inhibitors as part of their anti-atherosclerotic activity: Data from basic science and clinical trials. Front. Cardiovasc. Med. 2022, 9, 1008922. [Google Scholar] [CrossRef] [PubMed]
- Balzer, M.S.; Rong, S.; Nordlohne, J.; Zemtsovski, J.D.; Schmidt, S.; Stapel, B.; Bartosova, M.; von Vietinghoff, S.; Haller, H.; Schmitt, C.P.; et al. SGLT2 Inhibition by Intraperitoneal Dapagliflozin Mitigates Peritoneal Fibrosis and Ultrafiltration Failure in a Mouse Model of Chronic Peritoneal Exposure to High-Glucose Dialysate. Biomolecules 2020, 10, 1573. [Google Scholar] [CrossRef] [PubMed]
- Busnadiego, O.; Loureiro-Alvarez, J.; Sandoval, P.; Lagares, D.; Dotor, J.; Perez-Lozano, M.L.; Lopez-Armada, M.J.; Lamas, S.; Lopez-Cabrera, M.; Rodriguez-Pascual, F. A pathogenetic role for endothelin-1 in peritoneal dialysis-associated fibrosis. J. Am. Soc. Nephrol. 2015, 26, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Hao, J.B.; Ren, L.S.; Ding, J.L.; Hao, L.R. The aldosterone receptor antagonist spironolactone prevents peritoneal inflammation and fibrosis. Lab. Investig. 2014, 94, 839–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, H.; Ito, Y.; Mizuno, M.; Tanaka, A.; Morita, Y.; Maruyama, S.; Yuzawa, Y.; Matsuo, S. Mineralocorticoid receptor blockade ameliorates peritoneal fibrosis in new rat peritonitis model. Am. J. Physiol. Renal. Physiol. 2008, 294, F1084–F1093. [Google Scholar] [CrossRef] [Green Version]
- Tayama, Y.; Hasegawa, H.; Takayanagi, K.; Matsuda, A.; Shimizu, T.; Asakura, J.; Iwashita, T.; Ogawa, T.; Katoh, H.; Mitarai, T. Prevention of lipopolysaccharide-induced peritoneal damage by eplerenone in rats undergoing peritoneal dialysis. J. Nephrol. 2013, 26, 1160–1169. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, W.; Peng, X.; Zhu, L.; Wang, Z.; Shen, H.; Chen, C.; Xiao, L.; Li, S.; Zhao, Y.; et al. Parthenolide alleviates peritoneal fibrosis by inhibiting inflammation via the NF-kappaB/ TGF-beta/Smad signaling axis. Lab. Investig. 2022, 102, 1346–1354. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, Q.; Chen, Y.; Peng, X.; Wang, Y.; Li, S.; Wu, J.; Luo, C.; Gong, W.; Yin, B.; et al. Parthenolide, an NF-kappaB inhibitor, alleviates peritoneal fibrosis by suppressing the TGF-beta/Smad pathway. Int. Immunopharmacol. 2020, 78, 106064. [Google Scholar] [CrossRef]
- Dai, T.; Wang, Y.; Nayak, A.; Nast, C.C.; Quang, L.; LaPage, J.; Andalibi, A.; Adler, S.G. Janus kinase signaling activation mediates peritoneal inflammation and injury in vitro and in vivo in response to dialysate. Kidney Int. 2014, 86, 1187–1196. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Gong, Y.; Chen, Y.; Yu, D.; Wang, X.; Zhang, X.; Dou, Y.; Liu, D.; Cheng, G.; Lu, S.; et al. IL-6 promotes epithelial-to-mesenchymal transition of human peritoneal mesothelial cells possibly through the JAK2/STAT3 signaling pathway. Am. J. Physiol. Renal Physiol. 2017, 313, F310–F318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.W.; Wang, L.; Ding, H. Long noncoding RNA AK089579 inhibits epithelial-to-mesenchymal transition of peritoneal mesothelial cells by competitively binding to microRNA-296-3p via DOK2 in peritoneal fibrosis. FASEB J. 2019, 33, 5112–5125. [Google Scholar] [CrossRef] [PubMed]
- Kalble, F.; Damaske, J.; Heide, D.; Arnold, I.; Richter, F.; Maier, O.; Eisel, U.; Scheurich, P.; Pfizenmaier, K.; Zeier, M.; et al. Selective Blocking of TNF Receptor 1 Attenuates Peritoneal Dialysis Fluid Induced Inflammation of the Peritoneum in Mice. PLoS ONE 2016, 11, e0163314. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Lee, J.; Park, M.; Seo, A.; Kim, K.H.; Kim, S.; Kang, M.; Kang, E.; Yoo, K.D.; Lee, S.; et al. Inflammatory chemokine (C-C motif) ligand 8 inhibition ameliorates peritoneal fibrosis. FASEB J 2023, 37, e22632. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Yang, Z.; Xin, Z.; Yang, Y.; Yu, Y.; Cui, J.; Liu, H.; Chen, F. Glycogen synthase kinase-3beta: A promising candidate in the fight against fibrosis. Theranostics 2020, 10, 11737–11753. [Google Scholar] [CrossRef]
- Zhou, F.; Yao, L.; Lu, X.; Li, Y.; Han, X.; Wang, P. Therapeutic Targeting of GSK3beta-Regulated Nrf2 and NFkappaB Signaling Pathways by Salvianolic Acid A Ameliorates Peritoneal Fibrosis. Front. Med. (Lausanne) 2022, 9, 804899. [Google Scholar] [CrossRef]
- Herzog, R.; Sacnun, J.M.; Gonzalez-Mateo, G.; Bartosova, M.; Bialas, K.; Wagner, A.; Unterwurzacher, M.; Sobieszek, I.J.; Daniel-Fischer, L.; Rusai, K.; et al. Lithium preserves peritoneal membrane integrity by suppressing mesothelial cell alphaB-crystallin. Sci. Transl. Med. 2021, 13, 608. [Google Scholar] [CrossRef]
- Aroeira, L.S.; Lara-Pezzi, E.; Loureiro, J.; Aguilera, A.; Ramirez-Huesca, M.; Gonzalez-Mateo, G.; Perez-Lozano, M.L.; Albar-Vizcaino, P.; Bajo, M.A.; del Peso, G.; et al. Cyclooxygenase-2 mediates dialysate-induced alterations of the peritoneal membrane. J. Am. Soc. Nephrol. 2009, 20, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Fabbrini, P.; Schilte, M.N.; Zareie, M.; ter Wee, P.M.; Keuning, E.D.; Beelen, R.H.; van den Born, J. Celecoxib treatment reduces peritoneal fibrosis and angiogenesis and prevents ultrafiltration failure in experimental peritoneal dialysis. Nephrol. Dial. Transplant. 2009, 24, 3669–3676. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.B.; Kim, S.H.; Chang, J.W.; Lee, S.K.; Min, W.K.; Chi, H.S.; Park, J.S. Effects of celecoxib on high-sensitivity C-reactive protein in chronic peritoneal dialysis patients. Ren. Fail. 2004, 26, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Liu, M.; Tan, Y.; Chen, J.; Zhang, W.; Zhong, S.; Pan, J.; Zheng, Q.; Gong, L.; Su, L.; et al. Blockade of prostaglandin E2 receptor 4 ameliorates peritoneal dialysis-associated peritoneal fibrosis. Front. Pharmacol. 2022, 13, 1004619. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. BioMed Pharm. 2018, 104, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xing, C.; Zhang, L.; Mao, H.; Chen, X.; Liang, M.; Wang, F.; Ren, H.; Cui, H.; Jiang, A.; et al. Autophagy promotes fibrosis and apoptosis in the peritoneum during long-term peritoneal dialysis. J. Cell. Mol. Med. 2018, 22, 1190–1201. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Hu, Y.; Wang, Y.; Ma, X.; Tang, L.; Tao, M.; Qiu, A.; Zhuang, S.; Liu, N. Blockade of Autophagy Prevents the Development and Progression of Peritoneal Fibrosis. Front. Pharmacol. 2021, 12, 724141. [Google Scholar] [CrossRef]
- Miyake, T.; Sakai, N.; Tamai, A.; Sato, K.; Kamikawa, Y.; Miyagawa, T.; Ogura, H.; Yamamura, Y.; Oshima, M.; Nakagawa, S.; et al. Trehalose ameliorates peritoneal fibrosis by promoting Snail degradation and inhibiting mesothelial-to-mesenchymal transition in mesothelial cells. Sci. Rep. 2020, 10, 14292. [Google Scholar] [CrossRef]
- Yang, L.; Fan, Y.; Zhang, X.; Liu, J.; Ma, J. Effect of 1,25(OH)2D3 on high glucose-induced autophagy inhibition in peritoneum. Mol. Med. Rep. 2017, 16, 7080–7085. [Google Scholar] [CrossRef]
- Yang, L.; Fan, Y.; Zhang, X.; Huang, W.; Ma, J. 1,25(OH)2D3 treatment attenuates high glucose-induced peritoneal epithelial to mesenchymal transition in mice. Mol. Med. Rep. 2017, 16, 3817–3824. [Google Scholar] [CrossRef] [Green Version]
- Stavenuiter, A.W.; Farhat, K.; Vila Cuenca, M.; Schilte, M.N.; Keuning, E.D.; Paauw, N.J.; ter Wee, P.M.; Beelen, R.H.; Vervloet, M.G. Protective Effects of Paricalcitol on Peritoneal Remodeling during Peritoneal Dialysis. BioMed Res. Int. 2015, 2015, 468574. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Mateo, G.T.; Fernandez-Millara, V.; Bellon, T.; Liappas, G.; Ruiz-Ortega, M.; Lopez-Cabrera, M.; Selgas, R.; Aroeira, L.S. Paricalcitol reduces peritoneal fibrosis in mice through the activation of regulatory T cells and reduction in IL-17 production. PLoS ONE 2014, 9, e108477. [Google Scholar] [CrossRef]
- Xu, T.; Xie, J.Y.; Wang, W.M.; Ren, H.; Chen, N. Impact of rapamycin on peritoneal fibrosis and transport function. Blood Purif. 2012, 34, 48–57. [Google Scholar] [CrossRef]
- Gonzalez-Mateo, G.T.; Aguirre, A.R.; Loureiro, J.; Abensur, H.; Sandoval, P.; Sanchez-Tomero, J.A.; del Peso, G.; Jimenez-Heffernan, J.A.; Ruiz-Carpio, V.; Selgas, R.; et al. Rapamycin Protects from Type-I Peritoneal Membrane Failure Inhibiting the Angiogenesis, Lymphangiogenesis, and Endo-MT. BioMed Res. Int. 2015, 2015, 989560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, S.; Li, M.; Xie, X.; Xie, Z.; Zhou, Q.; Tian, Y.; Lin, W.; Zhang, X.; Jiang, H.; Shou, Z.; et al. Rapamycin inhibits epithelial-to-mesenchymal transition of peritoneal mesothelium cells through regulation of Rho GTPases. FEBS J. 2016, 283, 2309–2325. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jiang, C.M.; Feng, Y.; Zhu, W.; Jin, B.; Xia, Y.Y.; Zhang, Q.Y.; Xu, P.F.; Zhang, M. Rapamycin inhibits peritoneal fibrosis by modifying lipid homeostasis in the peritoneum. Am. J. Transl. Res. 2019, 11, 1473–1485. [Google Scholar] [PubMed]
- Ramil-Gomez, O.; Lopez-Pardo, M.; Fernandez-Rodriguez, J.A.; Rodriguez-Carmona, A.; Perez-Lopez, T.; Vaamonde-Garcia, C.; Perez-Fontan, M.; Lopez-Armada, M.J. Involvement of Mitochondrial Dysfunction in the Inflammatory Response in Human Mesothelial Cells from Peritoneal Dialysis Effluent. Antioxidants 2022, 11, 2184. [Google Scholar] [CrossRef] [PubMed]
- Roumeliotis, S.; Dounousi, E.; Salmas, M.; Eleftheriadis, T.; Liakopoulos, V. Unfavorable Effects of Peritoneal Dialysis Solutions on the Peritoneal Membrane: The Role of Oxidative Stress. Biomolecules 2020, 10, 768. [Google Scholar] [CrossRef]
- Hung, K.Y.; Liu, S.Y.; Yang, T.C.; Liao, T.L.; Kao, S.H. High-dialysate-glucose-induced oxidative stress and mitochondrial-mediated apoptosis in human peritoneal mesothelial cells. Oxid. Med. Cell. Longev. 2014, 2014, 642793. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Li, J.; Feng, B.; Bi, Z.; Zhu, G.; Zhang, Y.; Li, X. Activation of AMPK-PGC-1alpha pathway ameliorates peritoneal dialysis related peritoneal fibrosis in mice by enhancing mitochondrial biogenesis. Ren. Fail. 2022, 44, 1545–1557. [Google Scholar] [CrossRef]
- Inoue, H.; Torigoe, K.; Torigoe, M.; Muta, K.; Obata, Y.; Suzuki, T.; Suzuki, C.; Abe, T.; Koji, T.; Mukae, H.; et al. Mitochonic acid-5 ameliorates chlorhexidine gluconate-induced peritoneal fibrosis in mice. Med. Mol. Morphol. 2022, 55, 27–40. [Google Scholar] [CrossRef]
- Lu, H.; Chen, W.; Liu, W.; Si, Y.; Zhao, T.; Lai, X.; Kang, Z.; Sun, X.; Guo, Z. Molecular hydrogen regulates PTEN-AKT-mTOR signaling via ROS to alleviate peritoneal dialysis-related peritoneal fibrosis. FASEB J. 2020, 34, 4134–4146. [Google Scholar] [CrossRef] [Green Version]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Balzer, M.S.; Rong, S.; Menne, J.; von Vietinghoff, S.; Dong, L.; Gueler, F.; Jang, M.S.; Xu, G.; Timrott, K.; et al. Protein kinase C alpha inhibition prevents peritoneal damage in a mouse model of chronic peritoneal exposure to high-glucose dialysate. Kidney Int. 2016, 89, 1253–1267. [Google Scholar] [CrossRef] [PubMed]
- Morgado-Pascual, J.L.; Marchant, V.; Rodrigues-Diez, R.; Dolade, N.; Suarez-Alvarez, B.; Kerr, B.; Valdivielso, J.M.; Ruiz-Ortega, M.; Rayego-Mateos, S. Epigenetic Modification Mechanisms Involved in Inflammation and Fibrosis in Renal Pathology. Mediators Inflamm. 2018, 2018, 2931049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Shi, Y.; Tao, M.; Zhuang, S.; Liu, N. Peritoneal fibrosis and epigenetic modulation. Perit. Dial. Int. 2021, 41, 168–178. [Google Scholar] [CrossRef]
- Kim, K.H.; Ryu, H.M.; Oh, S.H.; Oh, E.J.; Ahn, J.S.; Lee, J.H.; Choi, J.Y.; Cho, J.H.; Kim, C.D.; Kim, Y.L.; et al. Effect of DNA demethylation in experimental encapsulating peritoneal sclerosis. Ther. Apher. Dial. 2014, 18, 628–636. [Google Scholar] [CrossRef]
- Maeda, K.; Doi, S.; Nakashima, A.; Nagai, T.; Irifuku, T.; Ueno, T.; Masaki, T. Inhibition of H3K9 methyltransferase G9a ameliorates methylglyoxal-induced peritoneal fibrosis. PLoS ONE 2017, 12, e0173706. [Google Scholar] [CrossRef] [Green Version]
- Tamura, R.; Doi, S.; Nakashima, A.; Sasaki, K.; Maeda, K.; Ueno, T.; Masaki, T. Inhibition of the H3K4 methyltransferase SET7/9 ameliorates peritoneal fibrosis. PLoS ONE 2018, 13, e0196844. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Tao, M.; Wang, Y.; Zang, X.; Ma, X.; Qiu, A.; Zhuang, S.; Liu, N. Genetic or pharmacologic blockade of enhancer of zeste homolog 2 inhibits the progression of peritoneal fibrosis. J. Pathol. 2020, 250, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Liu, K.; Liang, Y.; Chen, Y.; Chen, Y.; Gong, Y. Histone acetyltransferase inhibitor C646 reverses epithelial to mesenchymal transition of human peritoneal mesothelial cells via blocking TGF-beta1/Smad3 signaling pathway in vitro. Int. J. Clin. Exp. Pathol. 2015, 8, 2746–2754. [Google Scholar]
- Rossi, L.; Battistelli, C.; de Turris, V.; Noce, V.; Zwergel, C.; Valente, S.; Moioli, A.; Manzione, A.; Palladino, M.; Bordoni, V.; et al. HDAC1 inhibition by MS-275 in mesothelial cells limits cellular invasion and promotes MMT reversal. Sci. Rep. 2018, 8, 8492. [Google Scholar] [CrossRef] [Green Version]
- Bontempi, G.; Terri, M.; Garbo, S.; Montaldo, C.; Mariotti, D.; Bordoni, V.; Valente, S.; Zwergel, C.; Mai, A.; Marchetti, A.; et al. Restoration of WT1/miR-769-5p axis by HDAC1 inhibition promotes MMT reversal in mesenchymal-like mesothelial cells. Cell Death Dis. 2022, 13, 965. [Google Scholar] [CrossRef] [PubMed]
- Io, K.; Nishino, T.; Obata, Y.; Kitamura, M.; Koji, T.; Kohno, S. SAHA Suppresses Peritoneal Fibrosis in Mice. Perit. Dial. Int. 2015, 35, 246–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Liu, N.; Gu, H.; Wang, H.; Shi, Y.; Ma, X.; Ma, S.; Ni, J.; Tao, M.; Qiu, A.; et al. Histone deacetylase 6 inhibition counteracts the epithelial-mesenchymal transition of peritoneal mesothelial cells and prevents peritoneal fibrosis. Oncotarget 2017, 8, 88730–88750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Li, J.; Chen, H.; Hu, Y.; Tang, L.; Zhou, X.; Tao, M.; Lv, Z.; Chen, S.; Qiu, A.; et al. Pharmacologic Inhibition of Histone Deacetylase 6 Prevents the Progression of Chlorhexidine Gluconate-Induced Peritoneal Fibrosis by Blockade of M2 Macrophage Polarization. Front. Immunol. 2022, 13, 899140. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, L.; Gou, R.; Tang, L.; Liu, P. Noncoding RNAs in peritoneal fibrosis: Background, Mechanism, and Therapeutic Approach. BioMed Pharm. 2020, 129, 110385. [Google Scholar] [CrossRef]
- Morishita, Y.; Yoshizawa, H.; Watanabe, M.; Imai, R.; Imai, T.; Hirahara, I.; Akimoto, T.; Ookawara, S.; Muto, S.; Nagata, D. MicroRNA expression profiling in peritoneal fibrosis. Transl. Res. 2016, 169, 47–66. [Google Scholar] [CrossRef]
- Li, X.; Liu, H.; Sun, L.; Zhou, X.; Yuan, X.; Chen, Y.; Liu, F.; Liu, Y.; Xiao, L. MicroRNA-302c modulates peritoneal dialysis-associated fibrosis by targeting connective tissue growth factor. J. Cell. Mol. Med. 2019, 23, 2372–2383. [Google Scholar] [CrossRef] [Green Version]
- Grant, R.A.; Morales-Nebreda, L.; Markov, N.S.; Swaminathan, S.; Querrey, M.; Guzman, E.R.; Abbott, D.A.; Donnelly, H.K.; Donayre, A.; Goldberg, I.A.; et al. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature 2021, 590, 635–641. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Shankar, P.; Singh, J.; Joshi, A.; Malhotra, A.G.; Shrivas, A.; Goel, G.; Gupta, P.; Yadav, J.; Saigal, S.; Singh, S.; et al. Organ Involvement in COVID-19: A Molecular Investigation of Autopsied Patients. Microorganisms 2022, 10, 1333. [Google Scholar] [CrossRef]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Bai, X.; Wang, J.; Yu, Q.; Liu, W.; Pu, J.; Wang, X.; Hu, J.; Xu, D.; Li, X.; et al. Clinical characteristics of coronavirus disease 2019 in Gansu province, China. Ann. Palliat. Med. 2020, 9, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Arentz, M.; Yim, E.; Klaff, L.; Lokhandwala, S.; Riedo, F.X.; Chong, M.; Lee, M. Characteristics and Outcomes of 21 Critically Ill Patients With COVID-19 in Washington State. JAMA 2020, 323, 1612–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronco, C.; Navalesi, P.; Vincent, J.L. Coronavirus epidemic: Preparing for extracorporeal organ support in intensive care. Lancet Respir. Med. 2020, 8, 240–241. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Qian, L.; Sun, R.; Huang, B.; Dong, X.; Xiao, Q.; Zhang, Q.; Lu, T.; Yue, L.; Chen, S.; et al. Multi-organ proteomic landscape of COVID-19 autopsies. Cell 2021, 184, 775–791.e14. [Google Scholar] [CrossRef] [PubMed]
- Beyerstedt, S.; Casaro, E.B.; Rangel, E.B. COVID-19: Angiotensin-converting enzyme 2 (ACE2) expression and tissue susceptibility to SARS-CoV-2 infection. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 905–919. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Diao, B.; Wang, C.; Wang, R.; Feng, Z.; Zhang, J.; Yang, H.; Tan, Y.; Wang, H.; Wang, C.; Liu, L.; et al. Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 infection. Nat. Commun. 2021, 12, 2506. [Google Scholar] [CrossRef]
- Pan, X.W.; Xu, D.; Zhang, H.; Zhou, W.; Wang, L.H.; Cui, X.G. Identification of a potential mechanism of acute kidney injury during the COVID-19 outbreak: A study based on single-cell transcriptome analysis. Intensive Care Med. 2020, 46, 1114–1116. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Luo, R.; Wang, K.; Zhang, M.; Wang, Z.; Dong, L.; Li, J.; Yao, Y.; Ge, S.; Xu, G. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020, 97, 829–838. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brunink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, R. Pandemic potential of 2019-nCoV. Lancet Infect. Dis. 2020, 20, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Yao, L.; Wei, T.; Tian, F.; Jin, D.Y.; Chen, L.; Wang, M. Presumed Asymptomatic Carrier Transmission of COVID-19. JAMA 2020, 323, 1406–1407. [Google Scholar] [CrossRef] [Green Version]
- Xiong, F.; Tang, H.; Liu, L.; Tu, C.; Tian, J.B.; Lei, C.T.; Liu, J.; Dong, J.W.; Chen, W.L.; Wang, X.H.; et al. Clinical Characteristics of and Medical Interventions for COVID-19 in Hemodialysis Patients in Wuhan, China. J. Am. Soc. Nephrol. 2020, 31, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Manganaro, M.; Baldovino, S. on behalf of The Working group of the Piedmont and Aosta Valley Section of the SIN. First considerations on the SARS-CoV-2 epidemic in the Dialysis Units of Piedmont and Aosta Valley, Northern Italy. J. Nephrol. 2020, 33, 393–395. [Google Scholar] [CrossRef]
- Rodriguez-Chagolla, J.M.; Vasquez Jimenez, E.; Herrera Arellano, L.; Villa Torres, A.; Acosta Garcia, N.; Aleman Quimbiulco, D.; Armeaga Aguilar, S.; Madero, M. Peritoneal Dialysis Is an Option for Acute Kidney Injury Management in Patients with COVID-19. Blood Purif. 2021, 50, 283–289. [Google Scholar] [CrossRef]
- Jiang, H.J.; Tang, H.; Xiong, F.; Chen, W.L.; Tian, J.B.; Sun, J.; Dong, J.W.; Wang, X.H.; Jin, X.F.; Ding, Y.Q.; et al. COVID-19 in Peritoneal Dialysis Patients. Clin. J. Am. Soc. Nephrol. 2020, 16, 121–123. [Google Scholar] [CrossRef]
- Gagliardi, I.; Patella, G.; Michael, A.; Serra, R.; Provenzano, M.; Andreucci, M. COVID-19 and the Kidney: From Epidemiology to Clinical Practice. J. Clin. Med. 2020, 9, 2506. [Google Scholar] [CrossRef] [PubMed]
- Matusali, G.; Trionfetti, F.; Bordoni, V.; Nardacci, R.; Falasca, L.; Colombo, D.; Terri, M.; Montaldo, C.; Castilletti, C.; Mariotti, D.; et al. Pleural Mesothelial Cells Modulate the Inflammatory/Profibrotic Response During SARS-CoV-2 Infection. Front. Mol. Biosci. 2021, 8, 752616. [Google Scholar] [CrossRef]
- Bobrovitz, N.; Ware, H.; Ma, X.; Li, Z.; Hosseini, R.; Cao, C.; Selemon, A.; Whelan, M.; Premji, Z.; Issa, H.; et al. Protective effectiveness of previous SARS-CoV-2 infection and hybrid immunity against the omicron variant and severe disease: A systematic review and meta-regression. Lancet Infect. Dis. 2023. [Google Scholar] [CrossRef]
- Babel, N.; Hugo, C.; Westhoff, T.H. Vaccination in patients with kidney failure: Lessons from COVID-19. Nat. Rev. Nephrol. 2022, 18, 708–723. [Google Scholar] [CrossRef]
- Caillard, S.; Thaunat, O.; Benotmane, I.; Masset, C.; Blancho, G. Antibody Response to a Fourth Messenger RNA COVID-19 Vaccine Dose in Kidney Transplant Recipients: A Case Series. Ann. Intern. Med. 2022, 175, 455–456. [Google Scholar] [CrossRef] [PubMed]
- Sattler, A.; Schrezenmeier, E.; Weber, U.A.; Potekhin, A.; Bachmann, F.; Straub-Hohenbleicher, H.; Budde, K.; Storz, E.; Pross, V.; Bergmann, Y.; et al. Impaired humoral and cellular immunity after SARS-CoV-2 BNT162b2 (tozinameran) prime-boost vaccination in kidney transplant recipients. J. Clin. Investig. 2021, 131, e150175. [Google Scholar] [CrossRef] [PubMed]
- Fava, A.; Donadeu, L.; Sabe, N.; Pernin, V.; Gonzalez-Costello, J.; Llado, L.; Meneghini, M.; Charmetant, X.; Garcia-Romero, E.; Cachero, A.; et al. SARS-CoV-2-specific serological and functional T cell immune responses during acute and early COVID-19 convalescence in solid organ transplant patients. Am. J. Transplant. 2021, 21, 2749–2761. [Google Scholar] [CrossRef] [PubMed]
- Charmetant, X.; Espi, M.; Benotmane, I.; Barateau, V.; Heibel, F.; Buron, F.; Gautier-Vargas, G.; Delafosse, M.; Perrin, P.; Koenig, A.; et al. Infection or a third dose of mRNA vaccine elicits neutralizing antibody responses against SARS-CoV-2 in kidney transplant recipients. Sci. Transl. Med. 2022, 14, eabl6141. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trionfetti, F.; Marchant, V.; González-Mateo, G.T.; Kawka, E.; Márquez-Expósito, L.; Ortiz, A.; López-Cabrera, M.; Ruiz-Ortega, M.; Strippoli, R. Novel Aspects of the Immune Response Involved in the Peritoneal Damage in Chronic Kidney Disease Patients under Dialysis. Int. J. Mol. Sci. 2023, 24, 5763. https://doi.org/10.3390/ijms24065763
Trionfetti F, Marchant V, González-Mateo GT, Kawka E, Márquez-Expósito L, Ortiz A, López-Cabrera M, Ruiz-Ortega M, Strippoli R. Novel Aspects of the Immune Response Involved in the Peritoneal Damage in Chronic Kidney Disease Patients under Dialysis. International Journal of Molecular Sciences. 2023; 24(6):5763. https://doi.org/10.3390/ijms24065763
Chicago/Turabian StyleTrionfetti, Flavia, Vanessa Marchant, Guadalupe T. González-Mateo, Edyta Kawka, Laura Márquez-Expósito, Alberto Ortiz, Manuel López-Cabrera, Marta Ruiz-Ortega, and Raffaele Strippoli. 2023. "Novel Aspects of the Immune Response Involved in the Peritoneal Damage in Chronic Kidney Disease Patients under Dialysis" International Journal of Molecular Sciences 24, no. 6: 5763. https://doi.org/10.3390/ijms24065763
APA StyleTrionfetti, F., Marchant, V., González-Mateo, G. T., Kawka, E., Márquez-Expósito, L., Ortiz, A., López-Cabrera, M., Ruiz-Ortega, M., & Strippoli, R. (2023). Novel Aspects of the Immune Response Involved in the Peritoneal Damage in Chronic Kidney Disease Patients under Dialysis. International Journal of Molecular Sciences, 24(6), 5763. https://doi.org/10.3390/ijms24065763