
Identification of TIAM1 as a Potential Synthetic-Lethal-like Gene in a Defined Subset of Hepatocellular Carcinoma


 ,
,
Abstract
:1. Introduction
2. Results
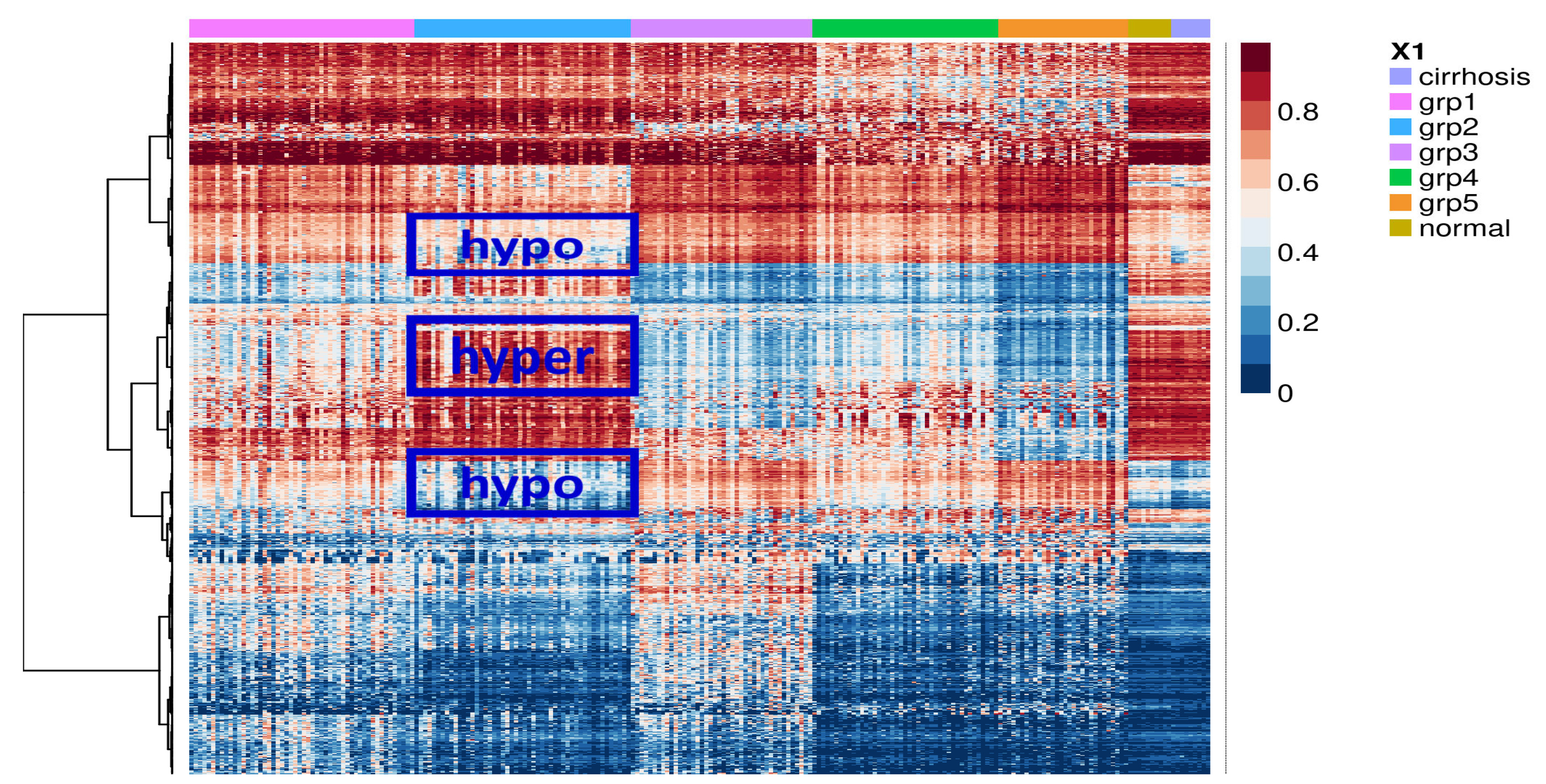
2.1. Identification of Potential Molecular Subgroups in HCC
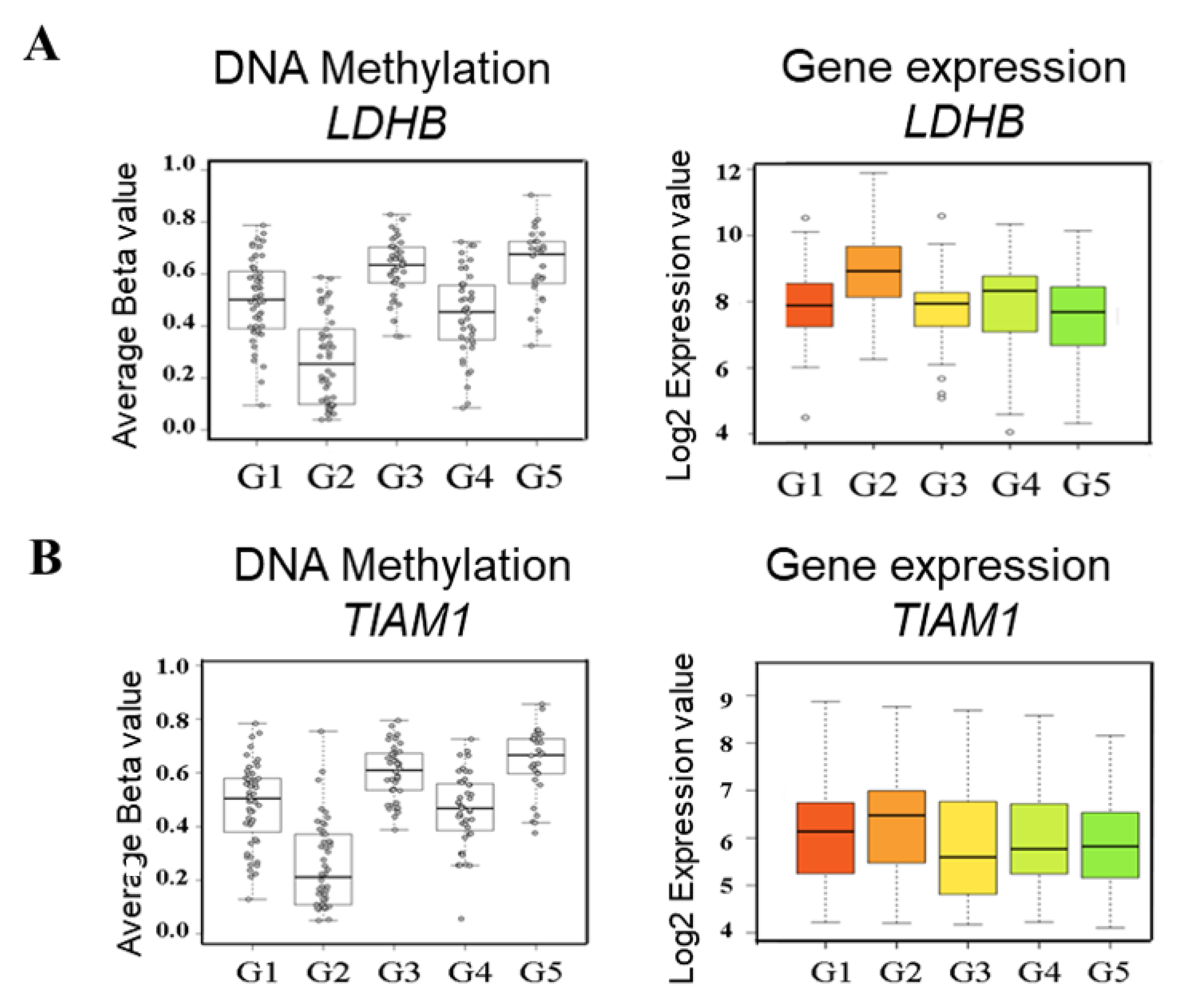
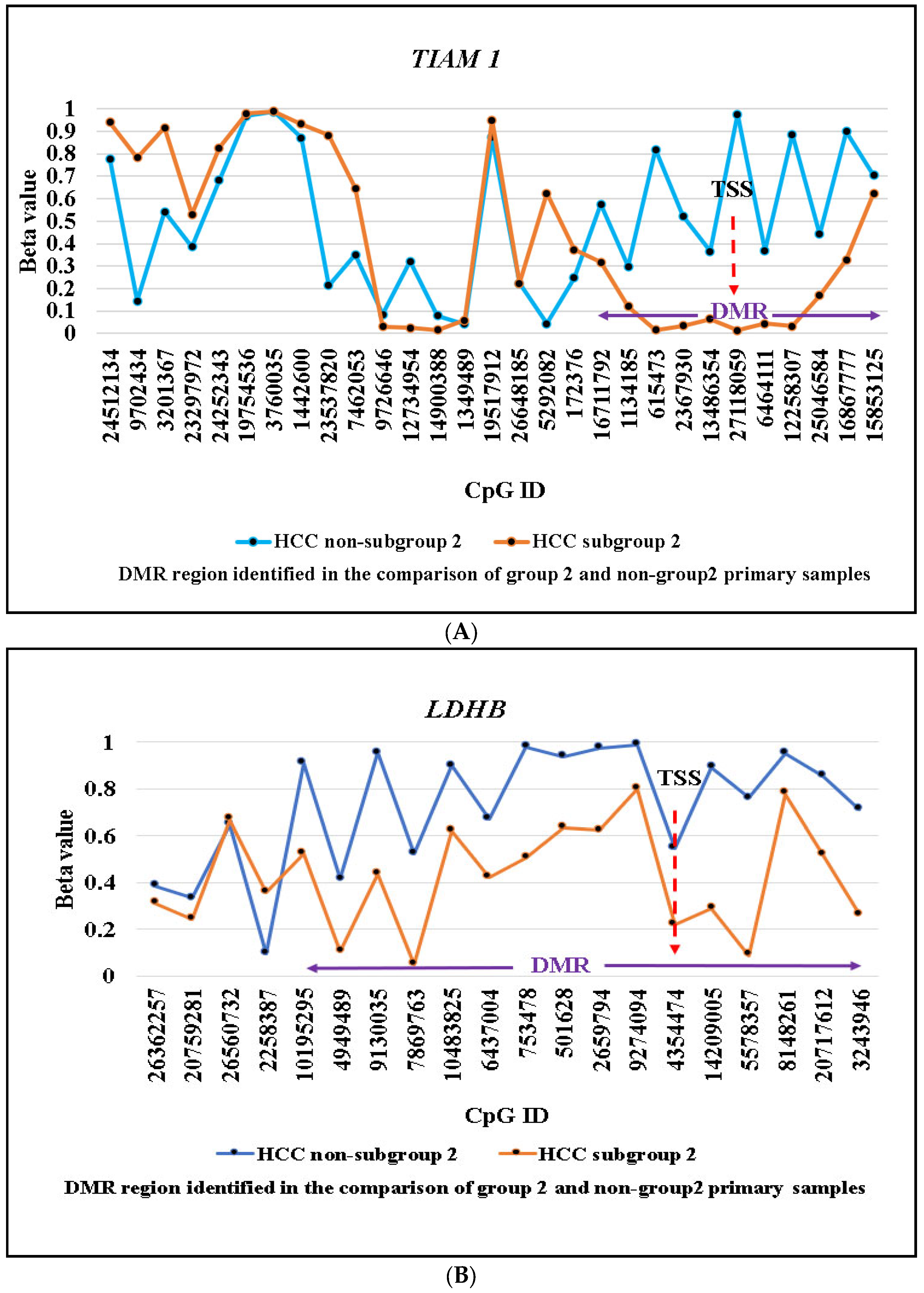
2.2. Identification of Subgroup-2-Specific SSV Gene Candidates
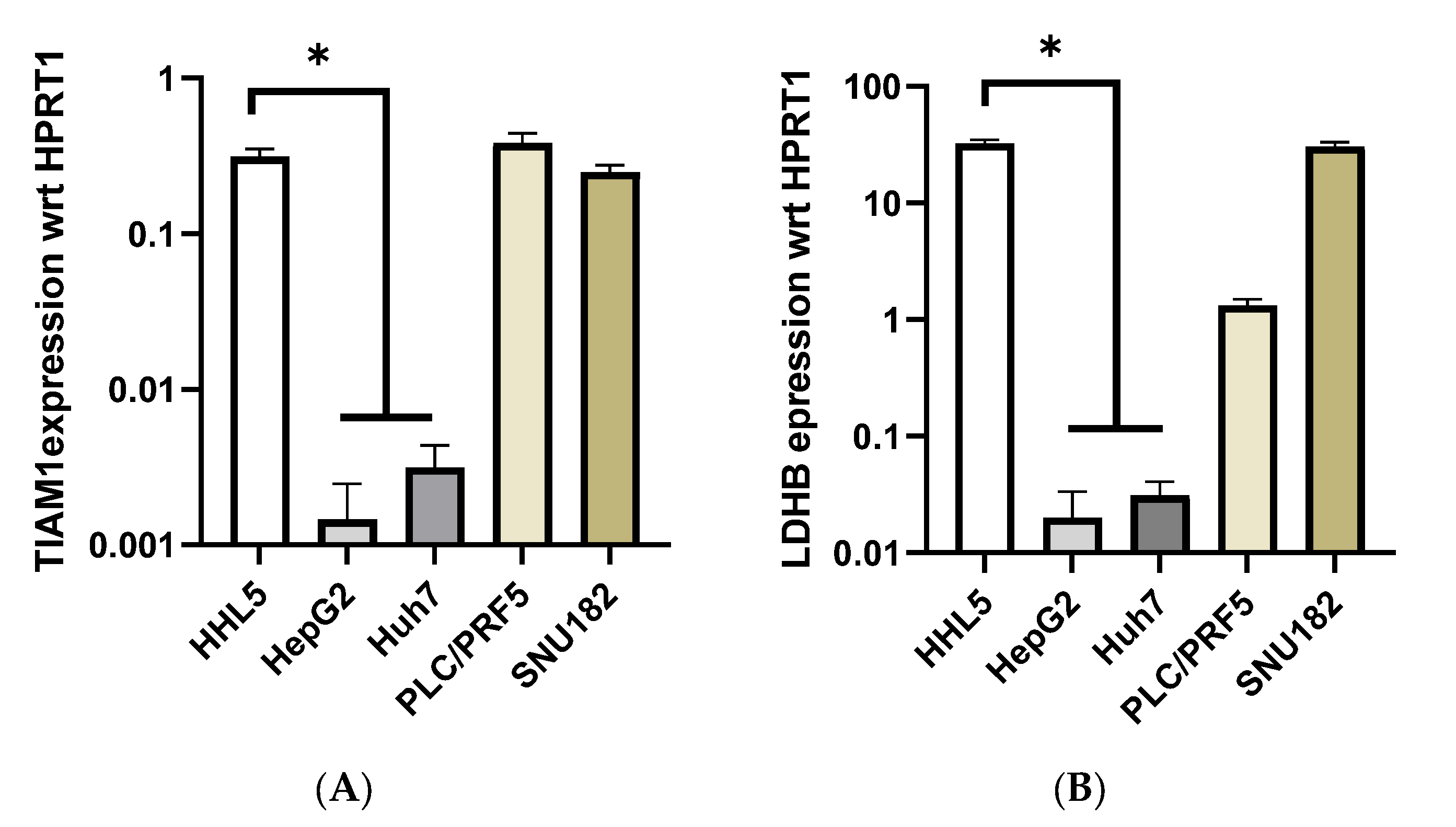
2.3. Identification of Cell Line Models Based on Methylation/Expression of the Identified SSV Gene Candidates
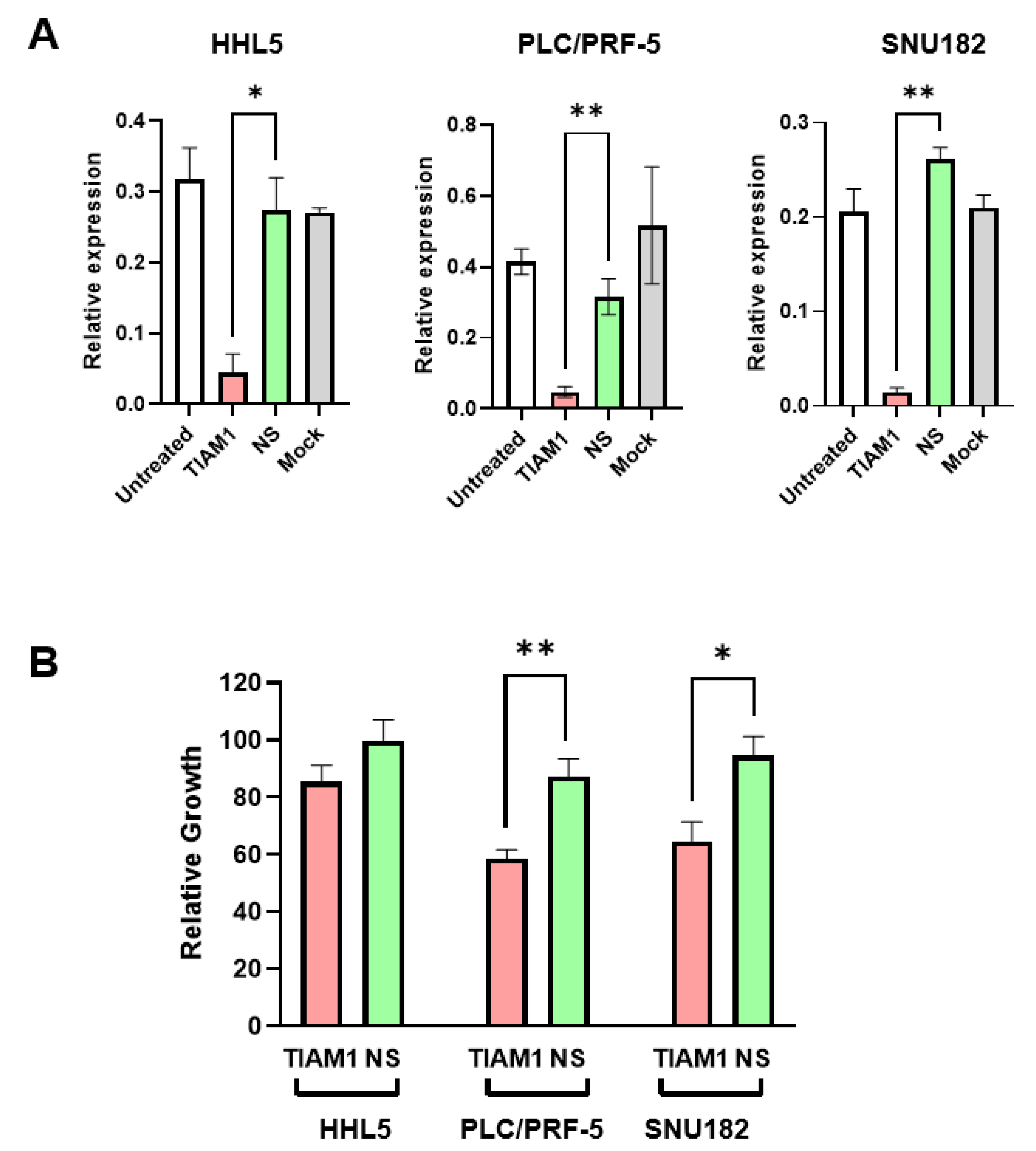
2.4. siRNA-Mediated Knockdown of TIAM1 Inhibits Growth in HCC Subgroup-2-like Cell Line Models
2.5. HCC Subgroup-2-like Cell Line Models Show Increased Sensitivity to the TIAM1/RAC1 Inhibitor NSC23766
3. Discussion
4. Materials and Methods
4.1. Cell Culture
Assessment of Sensitivity to RAC1 Inhibitor NSC23766
4.2. siRNA Transfection of Mammalian Cells
4.3. RNA Extraction and qRT-PCR
4.4. Genome-Wide DNA Methylation Analysis in HCC Cell Lines
4.5. Identification of Molecular Subgroups Using Non-Negative Matrix Factorisation
4.6. Identification of Subtype-Specific Vulnerability or Synthetic-Lethal-like Gene Candidates in HCC
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrick, J.L.; Florio, A.A.; Znaor, A.; Ruggieri, D.; Laversanne, M.; Alvarez, C.S.; Ferlay, J.; Valery, P.C.; Bray, F.; McGlynn, K.A. International trends in hepatocellular carcinoma incidence, 1978–2012. Int. J. Cancer 2020, 147, 317–330. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lin, Y.; Zhang, J.; Zhang, Y.; Li, Y.; Liu, Z.; Li, Q.; Luo, M.; Liang, R.; Ye, J. Molecular targeted and immune checkpoint therapy for advanced hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 447. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: What’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef]
- Boussios, S.; Rassy, E.; Moschetta, M.; Ghose, A.; Adeleke, S.; Sanchez, E.; Sheriff, M.; Chargari, C.; Pavlidis, N. BRCA Mutations in Ovarian and Prostate Cancer: Bench to Bedside. Cancers 2022, 14, 3888. [Google Scholar] [CrossRef]
- Schwalbe, E.C.; Lafta, F.; Barrow, T.M.; Strathdee, G. Integration of genome-level data to allow identification of subtype-specific vulnerability genes as novel therapeutic targets. Oncogene 2021, 40, 5213–5223. [Google Scholar] [CrossRef]
- Esteller, M. Aberrant DNA methylation as a cancer-inducing mechanism. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 629–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.D.; Seung, H.S. Learning the parts of objects by non-negative matrix factorization. Nature 1999, 401, 788–791. [Google Scholar] [CrossRef]
- Villanueva, A.; Portela, A.; Sayols, S.; Battiston, C.; Hoshida, Y.; Méndez-González, J.; Imbeaud, S.; Letouzé, E.; Hernandez-Gea, V.; Cornella, H.; et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology 2015, 61, 1945–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef] [Green Version]
- Lalchungnunga, H.; Hao, W.; Maris, J.M.; Asgharzadeh, S.; Henrich, K.-O.; Westermann, F.; Tweddle, D.A.; Schwalbe, E.C.; Strathdee, G. Genome wide DNA methylation analysis identifies novel molecular subgroups and predicts survival in neuroblastoma. Br. J. Cancer 2022, 127, 2006–2015. [Google Scholar] [CrossRef] [PubMed]
- Schwalbe, E.C.; Hayden, J.T.; Rogers, H.A.; Miller, S.; Lindsey, J.C.; Hill, R.M.; Nicholson, S.L.; Kilday, J.P.; Adamowicz-Brice, M.; Storer, L.; et al. Histologically defined central nervous system primitive neuro-ectodermal tumours (CNS-PNETs) display heterogeneous DNA methylation profiles and show relationships to other paediatric brain tumour types. Acta Neuropathol. 2013, 126, 943–946. [Google Scholar] [CrossRef] [Green Version]
- Bid, H.K.; Roberts, R.D.; Manchanda, P.K.; Houghton, P.J. RAC1: An emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol. Cancer Ther. 2013, 12, 1925–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De, P.; Aske, J.C.; Dey, N. RAC1 Takes the Lead in Solid Tumors. Cells 2019, 8, 382. [Google Scholar] [CrossRef] [Green Version]
- Marei, H.; Malliri, A. GEFs: Dual regulation of Rac1 signaling. Small GTPases 2017, 8, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Hans, S.; Zöller, D.; Hammer, J.; Stucke, J.; Spieß, S.; Kesavan, G.; Kroehne, V.; Eguiguren, J.S.; Ezhkova, D.; Petzold, A.; et al. Cre-Controlled CRISPR mutagenesis provides fast and easy conditional gene inactivation in zebrafish. Nat. Commun. 2021, 12, 1125. [Google Scholar] [CrossRef]
- Fortin, J.P.; Triche, T.J., Jr.; Hansen, K.D. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 2017, 33, 558–560. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-A.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; A Kibbe, W.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, A.S.; Lafta, F.M.; Schwalbe, E.C.; Nakjang, S.; Cockell, S.J.; Iliasova, A.; Enshaei, A.; Schwab, C.; Rand, V.; Clifford, S.C.; et al. Epigenetic landscape correlates with genetic subtype but does not predict outcome in childhood acute lymphoblastic leukemia. Epigenetics 2015, 10, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, P.; Kibbe, W.A.; Lin, S.M. lumi: A pipeline for processing Illumina microarray. Bioinformatics 2008, 24, 1547–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, T.J.; Buckley, M.J.; Statham, A.L.; Pidsley, R.; Samaras, K.; Lord, R.V.; Clark, S.J.; Molloy, P.L. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 2015, 8, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | IC50 (µM) | HCC Subgroup-2? | Average IC50 (µM) | p-Value * |
|---|---|---|---|---|
| PLC/PRF-5 | 24.8 ± 0.9 | Yes | 26.3 | - |
| SNU182 | 27.8 ± 5.2 | Yes | ||
| HepG2 | 57.1 ± 9.6 | No | 56.2 | 0.007 |
| Huh-7 | 49.9 ± 0.1 | No | ||
| HHL5 | 61.6 ± 5.9 | Non-HCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Permtermsin, C.; Lalchungnunga, H.; Nakjang, S.; Casement, J.; Ogle, L.F.; Reeves, H.L.; Strathdee, G.; Shukla, R. Identification of TIAM1 as a Potential Synthetic-Lethal-like Gene in a Defined Subset of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 6387. https://doi.org/10.3390/ijms24076387
Permtermsin C, Lalchungnunga H, Nakjang S, Casement J, Ogle LF, Reeves HL, Strathdee G, Shukla R. Identification of TIAM1 as a Potential Synthetic-Lethal-like Gene in a Defined Subset of Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2023; 24(7):6387. https://doi.org/10.3390/ijms24076387
Chicago/Turabian StylePermtermsin, Chalermsin, H Lalchungnunga, Sirintra Nakjang, John Casement, Laura Frances Ogle, Helen L. Reeves, Gordon Strathdee, and Ruchi Shukla. 2023. "Identification of TIAM1 as a Potential Synthetic-Lethal-like Gene in a Defined Subset of Hepatocellular Carcinoma" International Journal of Molecular Sciences 24, no. 7: 6387. https://doi.org/10.3390/ijms24076387
APA StylePermtermsin, C., Lalchungnunga, H., Nakjang, S., Casement, J., Ogle, L. F., Reeves, H. L., Strathdee, G., & Shukla, R. (2023). Identification of TIAM1 as a Potential Synthetic-Lethal-like Gene in a Defined Subset of Hepatocellular Carcinoma. International Journal of Molecular Sciences, 24(7), 6387. https://doi.org/10.3390/ijms24076387