
Update on the Molecular Pathology of Cutaneous Squamous Cell Carcinoma




Abstract
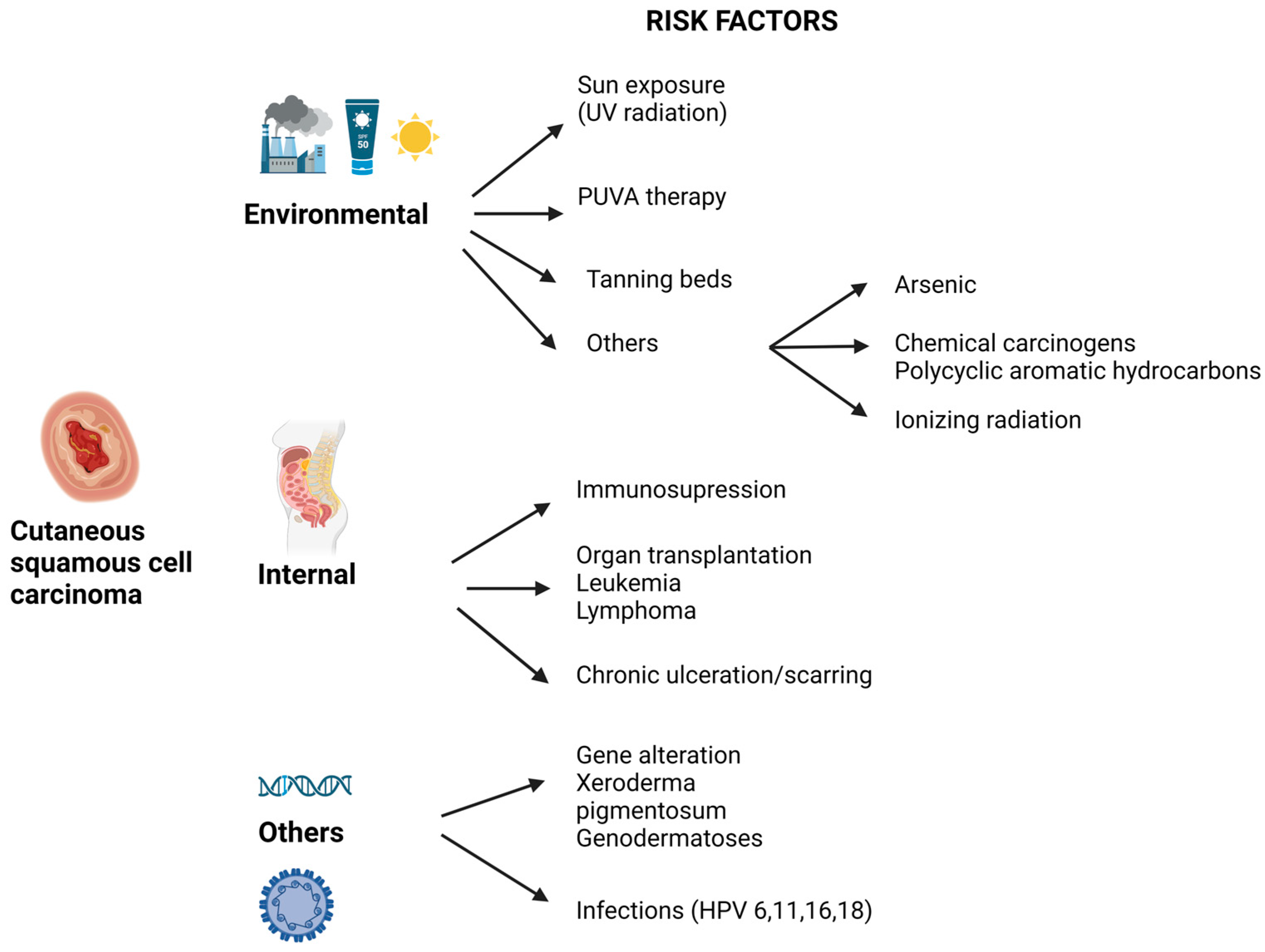
:1. Introduction
2. Mechanisms of SCC Carcinogenesis: Molecular Pathogenesis of SCC
3. Epigenetic Aspects of SCC: The Role of Epigenetics in Diagnosis, Metastatic Profile, and Prognosis

{kind=link}
| Epigenetic Changes | Involved Gene | Involved Protein | Role of the Protein | Results |
|---|---|---|---|---|
| DNA methylation of CpG, leading to loss of function | CDH1 gene promoter (E-cadherin gene promoter) | E-cadherin | Intercellular adhesion molecule | Hypermethylation of this gene is more frequent in invasive forms of SCC compared to AK and normal population. Hypermethylation of CDH1 gene promoter is present in 95% of cutaneous squamous cell carcinoma samples [63,64]. |
| DNA methylation of CpG, leading to loss of function | CDH13 gene promoter (T-cadherin gene promoter) | T-cadherin | Cell migration; Phenotypic changes; Calcium ion transport | Hypermethylation of T-cadherin gene promoter is associated with phenotypic cellular changes, both in SCC and in actinic keratoses that will evolve into invasive SCC [65,66]. |
| DNA methylation of CpG, leading to loss of function | CDKN2A gene | p16 (ARF) | Tumor suppressor genes—cell cycle regulator proteins | Hypermethylation is associated with a statistically significant decrease in the synthesis of aforementioned proteins (identified by immunohistochemical studies) (p < 0.001), with the inactivation of p16 and p14 [64]. |
| DNA methylation of CpG, leading to loss of function | CDKN2A gene | p14 (INK4A) | ||
| DNA methylation of CpG, leading to loss of function | Retinoblastoma protein 1 (RB1) gene | RB1 | ||
| DNA methylation of CpG, leading to loss of function | SFRP1-5 promoter gene | SFRP1-5 glycoproteins | Modulatory effects on Wnt pathway | Hypermethylation of the promoter region of these genes leads to a decrease in the expression of SFRP1-5 proteins in cSCC. It can be used as a marker of cSCC [67]. |
| DNA methylation of CpG, leading to loss of function | FRZB (frizzled-related protein) promoter gene | FRZB protein | Modulatory effects on Wnt pathway—involved in cell growth and differentiation | Hypermethylation of the promoter of this gene is significantly higher in metastatic cSCC compared to non-metastatic forms (p = 0.00004). It can be used as a marker of subtypes with aggressive evolution [68]. |
| DNA methylation of CpG, leading to loss of function | Death-associated protein kinase 1 (DAPK1) gene | DAPK1 | Tumor suppressor activity—protein involved in apoptosis and autophagy | DAPK1 hypermethylation is much more frequent in invasive forms of cSCC [66]. |
4. Tumor Heterogeneity: Cancer Stem Cells and Therapy Resistance
5. Novel Therapeutic Approaches in Cutaneous SCC (Targeted Therapies and Immunotherapies)
5.1. Surgery
5.2. Radiation Therapy
5.3. Systemic Therapy
5.3.1. Chemotherapy
5.3.2. Targeted Therapy
Anti-PD-1 Agents
- Cemiplimab
- Nivolumab
- Pembrolizumab
Anti-CTLA-4 Antibody
Anti CTLA-4 Antibodies Combined with PD-1 Antibodies
Epidermal Growth Factor Receptor Inhibitors (iEGFR)
- Cetuximab
5.4. Novel Approaches
5.4.1. Radiotherapy Associated with Immunotherapy
5.4.2. Oncolytic Viruses
5.5. Transplant Recipients
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sanchez-Danes, A.; Blanpain, C. Deciphering the cells of origin of squamous cell carcinomas. Nat. Rev. Cancer 2018, 18, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Stang, A.; Khil, L.; Kajuter, H.; Pandeya, N.; Schmults, C.D.; Ruiz, E.S.; Karia, P.S.; Green, A.C. Incidence and mortality for cutaneous squamous cell carcinoma: Comparison across three continents. J. Eur. Acad. Dermatol. Venereol. 2019, 33 (Suppl. 8), 6–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Que, S.K.T.; Zwald, F.O.; Schmults, C.D. Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J. Am. Acad. Dermatol. 2018, 78, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonerandi, J.J.; Beauvillain, C.; Caquant, L.; Chassagne, J.F.; Chaussade, V.; Clavere, P.; Desouches, C.; Garnier, F.; Grolleau, J.L.; Grossin, M.; et al. Guidelines for the diagnosis and treatment of cutaneous squamous cell carcinoma and precursor lesions. J. Eur. Acad. Dermatol. Venereol. 2011, 25 (Suppl. 5), 1–51. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; He, Y.Y. Ultraviolet radiation-induced non-melanoma skin cancer: Regulation of DNA damage repair and inflammation. Genes Dis. 2014, 1, 188–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Agency for Research on Cancer. WHO Classification of Tumour. Available online: https://tumourclassification.iarc.who.int/welcome/#preview (accessed on 13 January 2023).
- Criscione, V.D.; Weinstock, M.A.; Naylor, M.F.; Luque, C.; Eide, M.J.; Bingham, S.F.; Department of Veteran Affairs Topical Tretinoin Chemoprevention Trial Group. Actinic keratoses: Natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer 2009, 115, 2523–2530. [Google Scholar] [CrossRef]
- Sartini, D.; Campagna, R.; Lucarini, G.; Pompei, V.; Salvolini, E.; Mattioli-Belmonte, M.; Molinelli, E.; Brisigotti, V.; Campanati, A.; Bacchetti, T.; et al. Differential immunohistochemical expression of paraoxonase-2 in actinic keratosis and squamous cell carcinoma. Hum. Cell 2021, 34, 1929–1931. [Google Scholar] [CrossRef]
- Righi, V.; Reggiani, C.; Tarentini, E.; Mucci, A.; Paganelli, A.; Cesinaro, A.M.; Mataca, E.; Kaleci, S.; Ferrari, B.; Meleti, M.; et al. Metabolomic Analysis of Actinic Keratosis and SCC Suggests a Grade-Independent Model of Squamous Cancerization. Cancers 2021, 13, 5560. [Google Scholar] [CrossRef]
- Ratushny, V.; Gober, M.D.; Hick, R.; Ridky, T.W.; Seykora, J.T. From keratinocyte to cancer: The pathogenesis and modeling of cutaneous squamous cell carcinoma. J. Clin. Investig. 2012, 122, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Friedberg, E.C.; Wood, R.D.; Walker, G.C.; Schultz, R.A.; Siede, W.; Ellenberger, T.; ProQuest. DNA Repair and Mutagenesis, 2nd ed.; ASM Press: Washington, DC, USA, 2006. [Google Scholar]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 3667. [Google Scholar] [CrossRef] [PubMed]
- Campagna, R.; Pozzi, V.; Sartini, D.; Salvolini, E.; Brisigotti, V.; Molinelli, E.; Campanati, A.; Offidani, A.; Emanuelli, M. Beyond Nicotinamide Metabolism: Potential Role of Nicotinamide N-Methyltransferase as a Biomarker in Skin Cancers. Cancers 2021, 13, 4943. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.L.; Nairn, R.S. The biology of the (6–4) photoproduct. Photochem. Photobiol. 1989, 49, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.E.; Rudolph, J.A.; Simon, J.A.; Lin, A.; McKenna, G.J.; Baden, H.P.; Halperin, A.J.; Ponten, J. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 1991, 88, 10124–10128. [Google Scholar] [CrossRef] [Green Version]
- Brash, D.E. UV signature mutations. Photochem. Photobiol. 2015, 91, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef] [Green Version]
- Hedberg, M.; Seykora, J.T. Clarifying Progress on the Genomic Landscape of Actinic Keratosis. J. Investig. Dermatol. 2021, 141, 1622–1624. [Google Scholar] [CrossRef]
- Thomson, J.; Bewicke-Copley, F.; Anene, C.A.; Gulati, A.; Nagano, A.; Purdie, K.; Inman, G.J.; Proby, C.M.; Leigh, I.M.; Harwood, C.A.; et al. The Genomic Landscape of Actinic Keratosis. J. Investig. Dermatol. 2021, 141, 1664–1674. [Google Scholar] [CrossRef]
- Chitsazzadeh, V.; Coarfa, C.; Drummond, J.A.; Nguyen, T.; Joseph, A.; Chilukuri, S.; Charpiot, E.; Adelmann, C.H.; Ching, G.; Nguyen, T.N.; et al. Cross-species identification of genomic drivers of squamous cell carcinoma development across preneoplastic intermediates. Nat. Commun. 2016, 7, 12601. [Google Scholar] [CrossRef]
- Quintana, R.M.; Dupuy, A.J.; Bravo, A.; Casanova, M.L.; Alameda, J.P.; Page, A.; Sanchez-Viera, M.; Ramirez, A.; Navarro, M. A transposon-based analysis of gene mutations related to skin cancer development. J. Investig. Dermatol. 2013, 133, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Ashford, B.G.; Clark, J.; Gupta, R.; Iyer, N.G.; Yu, B.; Ranson, M. Reviewing the genetic alterations in high-risk cutaneous squamous cell carcinoma: A search for prognostic markers and therapeutic targets. Head Neck 2017, 39, 1462–1469. [Google Scholar] [CrossRef] [PubMed]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstrahle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 1661–1662. [Google Scholar] [CrossRef]
- Meek, D.W.; Anderson, C.W. Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 2009, 1, a000950. [Google Scholar] [CrossRef] [Green Version]
- Zilfou, J.T.; Lowe, S.W. Tumor suppressive functions of p53. Cold Spring Harb. Perspect. Biol. 2009, 1, a001883. [Google Scholar] [CrossRef]
- Sherr, C.J. Divorcing ARF and p53: An unsettled case. Nat. Rev. Cancer 2006, 6, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Yasuda, H. Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 1999, 18, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Al-Rohil, R.N.; Tarasen, A.J.; Carlson, J.A.; Wang, K.; Johnson, A.; Yelensky, R.; Lipson, D.; Elvin, J.A.; Vergilio, J.A.; Ali, S.M.; et al. Evaluation of 122 advanced-stage cutaneous squamous cell carcinomas by comprehensive genomic profiling opens the door for new routes to targeted therapies. Cancer 2016, 122, 249–257. [Google Scholar] [CrossRef]
- Heppt, M.V.; Dykukha, I.; Graziadio, S.; Salido-Vallejo, R.; Chapman-Rounds, M.; Edwards, M. Comparative Efficacy and Safety of Tirbanibulin for Actinic Keratosis of the Face and Scalp in Europe: A Systematic Review and Network Meta-Analysis of Randomized Controlled Trials. J. Clin. Med. 2022, 11, 1654. [Google Scholar] [CrossRef]
- Morris, L.G.; Kaufman, A.M.; Gong, Y.; Ramaswami, D.; Walsh, L.A.; Turcan, S.; Eng, S.; Kannan, K.; Zou, Y.; Peng, L.; et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat. Genet. 2013, 45, 253–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Watt, S.A.; Purdie, K.J.; den Breems, N.Y.; Dimon, M.; Arron, S.T.; McHugh, A.T.; Xue, D.J.; Dayal, J.H.; Proby, C.M.; Harwood, C.A.; et al. Novel CARD11 Mutations in Human Cutaneous Squamous Cell Carcinoma Lead to Aberrant NF-kappaB Regulation. Am. J. Pathol. 2015, 185, 2354–2363. [Google Scholar] [CrossRef] [PubMed]
- Seykora, J.T.; Mei, L.; Dotto, G.P.; Stein, P.L. ‘Srcasm: A novel Src activating and signaling molecule. J. Biol. Chem. 2002, 277, 2812–2822. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Marshall, C.; Mei, L.; Dzubow, L.; Schmults, C.; Dans, M.; Seykora, J. Srcasm modulates EGF and Src-kinase signaling in keratinocytes. J. Biol. Chem. 2005, 280, 6036–6046. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Marshall, C.; Mei, L.; Gelfand, J.; Seykora, J.T. Srcasm corrects Fyn-induced epidermal hyperplasia by kinase down-regulation. J. Biol. Chem. 2007, 282, 1161–1169. [Google Scholar] [CrossRef] [Green Version]
- Malanchi, I.; Peinado, H.; Kassen, D.; Hussenet, T.; Metzger, D.; Chambon, P.; Huber, M.; Hohl, D.; Cano, A.; Birchmeier, W.; et al. Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature 2008, 452, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Deng, X.; Dai, Y.; Niu, X.; Zhou, M. miR-27a Downregulation Promotes Cutaneous Squamous Cell Carcinoma Progression via Targeting EGFR. Front. Oncol. 2019, 9, 1565. [Google Scholar] [CrossRef] [Green Version]
- Jost, M.; Kari, C.; Rodeck, U. The EGF receptor—An essential regulator of multiple epidermal functions. Eur. J. Dermatol. 2000, 10, 505–510. [Google Scholar]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, aal2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Aboud, N.M.; Tupper, C.; Jialal, I. Genetics, Epigenetic Mechanism. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penta, D.; Somashekar, B.S.; Meeran, S.M. Epigenetics of skin cancer: Interventions by selected bioactive phytochemicals. Photodermatol. Photoimmunol. Photomed. 2018, 34, 42–49. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locke, W.J.; Guanzon, D.; Ma, C.; Liew, Y.J.; Duesing, K.R.; Fung, K.Y.C.; Ross, J.P. DNA Methylation Cancer Biomarkers: Translation to the Clinic. Front. Genet. 2019, 10, 1150. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef] [PubMed]
- Strub, T.; Ballotti, R.; Bertolotto, C. The “ART” of Epigenetics in Melanoma: From histone “Alterations, to Resistance and Therapies”. Theranostics 2020, 10, 1777–1797. [Google Scholar] [CrossRef]
- Hervas-Marin, D.; Higgins, F.; Sanmartin, O.; Lopez-Guerrero, J.A.; Bano, M.C.; Igual, J.C.; Quilis, I.; Sandoval, J. Genome wide DNA methylation profiling identifies specific epigenetic features in high-risk cutaneous squamous cell carcinoma. PLoS ONE 2019, 14, e0223341. [Google Scholar] [CrossRef]
- Ehrlich, M.; Lacey, M. DNA hypomethylation and hemimethylation in cancer. Adv. Exp. Med. Biol. 2013, 754, 31–56. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, D.H.; Shin, M.H.; Shin, H.S.; Kim, M.K.; Chung, J.H. UV-induced DNA methyltransferase 1 promotes hypermethylation of tissue inhibitor of metalloproteinase 2 in the human skin. J. Dermatol. Sci. 2018, 91, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Pacella, G.; Capell, B.C. Epigenetic and metabolic interplay in cutaneous squamous cell carcinoma. Exp. Dermatol. 2021, 30, 1115–1125. [Google Scholar] [CrossRef]
- Lohcharoenkal, W.; Harada, M.; Loven, J.; Meisgen, F.; Landen, N.X.; Zhang, L.; Lapins, J.; Mahapatra, K.D.; Shi, H.; Nissinen, L.; et al. MicroRNA-203 Inversely Correlates with Differentiation Grade, Targets c-MYC, and Functions as a Tumor Suppressor in cSCC. J. Investig. Dermatol. 2016, 136, 2485–2494. [Google Scholar] [CrossRef] [Green Version]
- Lohcharoenkal, W.; Li, C.; Das Mahapatra, K.; Lapins, J.; Homey, B.; Sonkoly, E.; Pivarcsi, A. MiR-130a Acts as a Tumor Suppressor MicroRNA in Cutaneous Squamous Cell Carcinoma and Regulates the Activity of the BMP/SMAD Pathway by Suppressing ACVR1. J. Investig. Dermatol. 2021, 141, 1922–1931. [Google Scholar] [CrossRef]
- Zhang, J.; Cao, Z.; Yang, G.; You, L.; Zhang, T.; Zhao, Y. MicroRNA-27a (miR-27a) in Solid Tumors: A Review Based on Mechanisms and Clinical Observations. Front. Oncol. 2019, 9, 893. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Sancha, N.; Corchado-Cobos, R.; Perez-Losada, J.; Canueto, J. MicroRNA Dysregulation in Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 2181. [Google Scholar] [CrossRef] [Green Version]
- Yin, S.; Lin, X. MicroRNA-21 Contributes to Cutaneous Squamous Cell Carcinoma Progression via Mediating TIMP3/PI3K/AKT Signaling Axis. Int. J. Gen. Med. 2021, 14, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lin, Y.; Jiang, T.; Gao, C.; Wang, D.; Wang, X.; Wei, Y.; Liu, T.; Zhu, L.; Wang, P.; et al. Up-regulation of TIMP-3 and RECK decrease the invasion and metastasis ability of colon cancer. Arab. J. Gastroenterol. 2019, 20, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Shen, R.; Yan, Y.; Deng, L. miR-186 promotes tumor growth in cutaneous squamous cell carcinoma by inhibiting apoptotic protease activating factor-1. Exp. Ther. Med. 2018, 16, 4010–4018. [Google Scholar] [CrossRef] [PubMed]
- Canueto, J.; Cardenoso-Alvarez, E.; Garcia-Hernandez, J.L.; Galindo-Villardon, P.; Vicente-Galindo, P.; Vicente-Villardon, J.L.; Alonso-Lopez, D.; De Las Rivas, J.; Valero, J.; Moyano-Sanz, E.; et al. MicroRNA (miR)-203 and miR-205 expression patterns identify subgroups of prognosis in cutaneous squamous cell carcinoma. Br. J. Dermatol. 2017, 177, 168–178. [Google Scholar] [CrossRef] [Green Version]
- Mulvaney, P.M.; Schmults, C.D. Molecular prediction of metastasis in cutaneous squamous cell carcinoma. Curr. Opin. Oncol. 2020, 32, 129–136. [Google Scholar] [CrossRef]
- Gillespie, J.; Skeeles, L.E.; Allain, D.C.; Kent, M.N.; Peters, S.B.; Nagarajan, P.; Yu, L.; Teknos, T.N.; Olencki, T.; Toland, A.E. MicroRNA expression profiling in metastatic cutaneous squamous cell carcinoma. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1043–1045. [Google Scholar] [CrossRef]
- Hernandez-Ruiz, E.; Toll, A.; Garcia-Diez, I.; Andrades, E.; Ferrandiz-Pulido, C.; Masferrer, E.; Yebenes, M.; Jaka, A.; Gimeno, J.; Gimeno, R.; et al. The Polycomb proteins RING1B and EZH2 repress the tumoral pro-inflammatory function in metastasizing primary cutaneous squamous cell carcinoma. Carcinogenesis 2018, 39, 503–513. [Google Scholar] [CrossRef]
- Chiles, M.C.; Ai, L.; Zuo, C.; Fan, C.Y.; Smoller, B.R. E-cadherin promoter hypermethylation in preneoplastic and neoplastic skin lesions. Mod. Pathol. 2003, 16, 1014–1018. [Google Scholar] [CrossRef] [Green Version]
- Murao, K.; Kubo, Y.; Ohtani, N.; Hara, E.; Arase, S. Epigenetic abnormalities in cutaneous squamous cell carcinomas: Frequent inactivation of the RB1/p16 and p53 pathways. Br. J. Dermatol. 2006, 155, 999–1005. [Google Scholar] [CrossRef]
- Takeuchi, T.; Liang, S.B.; Matsuyoshi, N.; Zhou, S.; Miyachi, Y.; Sonobe, H.; Ohtsuki, Y. Loss of T-cadherin (CDH13, H-cadherin) expression in cutaneous squamous cell carcinoma. Lab. Investig. 2002, 82, 1023–1029. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Jiang, M.; Feng, Q.; Kiviat, N.B.; Stern, J.E.; Hawes, S.; Cherne, S.; Lu, H. Aberrant Methylation Changes Detected in Cutaneous Squamous Cell Carcinoma of Immunocompetent Individuals. Cell Biochem. Biophys. 2015, 72, 599–604. [Google Scholar] [CrossRef]
- Liang, J.; Kang, X.; Halifu, Y.; Zeng, X.; Jin, T.; Zhang, M.; Luo, D.; Ding, Y.; Zhou, Y.; Yakeya, B.; et al. Secreted frizzled-related protein promotors are hypermethylated in cutaneous squamous carcinoma compared with normal epidermis. BMC Cancer 2015, 15, 641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darr, O.A.; Colacino, J.A.; Tang, A.L.; McHugh, J.B.; Bellile, E.L.; Bradford, C.R.; Prince, M.P.; Chepeha, D.B.; Rozek, L.S.; Moyer, J.S. Epigenetic alterations in metastatic cutaneous carcinoma. Head Neck 2015, 37, 994–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- O’Connor, M.L.; Xiang, D.; Shigdar, S.; Macdonald, J.; Li, Y.; Wang, T.; Pu, C.; Wang, Z.; Qiao, L.; Duan, W. Cancer stem cells: A contentious hypothesis now moving forward. Cancer Lett. 2014, 344, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Haviv, I.; Campbell, I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009, 25, 30–38. [Google Scholar] [CrossRef]
- Rycaj, K.; Tang, D.G. Cell-of-Origin of Cancer versus Cancer Stem Cells: Assays and Interpretations. Cancer Res. 2015, 75, 4003–4011. [Google Scholar] [CrossRef] [Green Version]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cojoc, M.; Mabert, K.; Muders, M.H.; Dubrovska, A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Semin. Cancer Biol. 2015, 31, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Strait, A.; Jimeno, A.; Wang, X.J. Cancer Stem Cells in Squamous Cell Carcinoma. J. Invest. Dermatol. 2017, 137, 31–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Wu, T.; Liu, A.Y.; Ouyang, G. Differentiation and transdifferentiation potentials of cancer stem cells. Oncotarget 2015, 6, 39550–39563. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Naz, F.; Shi, M.; Sajid, S.; Yang, Z.; Yu, C. Cancer stem cells: A major culprit of intra-tumor heterogeneity. Am. J. Cancer Res. 2021, 11, 5782–5811. [Google Scholar]
- Iseghohi, S.O. Cancer stem cells may contribute to the difficulty in treating cancer. Genes Dis. 2016, 3, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.E.; Nambiar, R.; De Souza, C.; Nguyen, A.; Chien, J.; Lam, K.S. Targeting Epigenetic Modifiers of Tumor Plasticity and Cancer Stem Cell Behavior. Cells 2022, 11, 1403. [Google Scholar] [CrossRef]
- Liao, G.; Tang, J.; Bai, J. Early development of esophageal squamous cell cancer: Stem cells, cellular origins and early clone evolution. Cancer Lett. 2023, 555, 216047. [Google Scholar] [CrossRef]
- Argenziano, G.; Fargnoli, M.C.; Fantini, F.; Gattoni, M.; Gualdi, G.; Pastore, F.; Pellacani, G.; Quaglino, P.; Queirolo, P.; Troiani, T. Identifying candidates for immunotherapy with cemiplimab to treat advanced cutaneous squamous cell carcinoma: An expert opinion. Ther. Adv. Med. Oncol. 2022, 14, 17588359211066272. [Google Scholar] [CrossRef] [PubMed]
- Stratigos, A.J.; Garbe, C.; Dessinioti, C.; Lebbe, C.; Bataille, V.; Bastholt, L.; Dreno, B.; Concetta Fargnoli, M.; Forsea, A.M.; Frenard, C.; et al. European interdisciplinary guideline on invasive squamous cell carcinoma of the skin: Part 2. Treatment. Eur. J. Cancer 2020, 128, 83–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Foncillas, J.; Tejera-Vaquerizo, A.; Sanmartin, O.; Rojo, F.; Mestre, J.; Martin, S.; Azinovic, I.; Mesia, R. Update on Management Recommendations for Advanced Cutaneous Squamous Cell Carcinoma. Cancers 2022, 14, 629. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.O.; Cernea, S.S.; Weber, M.B.; Ferreira, I.G.; Sil, A.B. Advanced squamous cell carcinoma and immunotherapy: New therapeutic perspectives. Surg. Cosmet. Dermatol. 2021, 13, e20210023. [Google Scholar]
- Stratigos, A.J.; Garbe, C.; Dessinioti, C.; Lebbe, C.; Bataille, V.; Bastholt, L.; Dreno, B.; Fargnoli, M.C.; Forsea, A.M.; Frenard, C.; et al. European interdisciplinary guideline on invasive squamous cell carcinoma of the skin: Part 1. epidemiology, diagnostics and prevention. Eur. J. Cancer 2020, 128, 60–82. [Google Scholar] [CrossRef] [Green Version]
- Stanganelli, I.; Spagnolo, F.; Argenziano, G.; Ascierto, P.A.; Bassetto, F.; Bossi, P.; Donato, V.; Massi, D.; Massone, C.; Patuzzo, R.; et al. The Multidisciplinary Management of Cutaneous Squamous Cell Carcinoma: A Comprehensive Review and Clinical Recommendations by a Panel of Experts. Cancers 2022, 14, 377. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Squamous Cell Skin Cancer. Available online: https://www.nccn.org/professionals/physician_gls/pdf/squamous.pdf (accessed on 28 January 2023).
- Yanagi, T.; Kitamura, S.; Hata, H. Novel Therapeutic Targets in Cutaneous Squamous Cell Carcinoma. Front. Oncol. 2018, 8, 79. [Google Scholar] [CrossRef]
- Maubec, E. Update on the Management of Cutaneous Squamous Cell Carcinoma. Acta Derm. Venereol. 2020, 100, adv00143. [Google Scholar] [CrossRef]
- Alberti, A.; Bossi, P. Immunotherapy for Cutaneous Squamous Cell Carcinoma: Results and Perspectives. Front. Oncol. 2021, 11, 727027. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sancha, N.; Corchado-Cobos, R.; Bellido-Hernandez, L.; Roman-Curto, C.; Cardenoso-Alvarez, E.; Perez-Losada, J.; Orfao, A.; Canueto, J. Overcoming Resistance to Immunotherapy in Advanced Cutaneous Squamous Cell Carcinoma. Cancers 2021, 13, 5134. [Google Scholar] [CrossRef] [PubMed]
- Villegas-Romero, I.; Jiménez-Gallo, D.; Gutiérrez-Bayard, L.; Linares-Barrios, M. Carcinoma epidermoide cutáneo avanzado tratado con pembrolizumab. Actas Dermo-Sifiliogr 2021, 112, 672–675. [Google Scholar] [CrossRef]
- Peris, K.; Piccerillo, A.; Del Regno, L.; Di Stefani, A. Treatment approaches of advanced cutaneous squamous cell carcinoma. J. Eur. Acad. Dermatol. Venereol. 2022, 36 (Suppl. 1), 19–22. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Kozakiewicz, P.; Grzybowska-Szatkowska, L. Application of molecular targeted therapies in the treatment of head and neck squamous cell carcinoma. Oncol. Lett. 2018, 15, 7497–7505. [Google Scholar] [CrossRef] [Green Version]
- Grob, J.J.; Mendoza, R.G.; Basset-Seguin, N.; Vornicova, O.; Schachter, J.; Joshi, A.; Meyer, N.; Grange, F.; Piulats, J.M.; Bauman, J.; et al. LBA72—Pembrolizumab for recurrent/metastatic cutaneous squamous cell carcinoma (cSCC): Efficacy and safety results from the phase II KEYNOTE-629 study. Ann. Oncol. 2019, 30, v908. [Google Scholar] [CrossRef]
- Wessely, A.; Steeb, T.; Leiter, U.; Garbe, C.; Berking, C.; Heppt, M.V. Immune Checkpoint Blockade in Advanced Cutaneous Squamous Cell Carcinoma: What Do We Currently Know in 2020? Int. J. Mol. Sci. 2020, 21, 9300. [Google Scholar] [CrossRef]
- Miller, D.M.; Faulkner-Jones, B.E.; Stone, J.R.; Drews, R.E. Complete pathologic response of metastatic cutaneous squamous cell carcinoma and allograft rejection after treatment with combination immune checkpoint blockade. JAAD Case Rep. 2017, 3, 412–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferris, R.L.; Licitra, L.; Fayette, J.; Even, C.; Blumenschein, G., Jr.; Harrington, K.J.; Guigay, J.; Vokes, E.E.; Saba, N.F.; Haddad, R.; et al. Nivolumab in Patients with Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck: Efficacy and Safety in CheckMate 141 by Prior Cetuximab Use. Clin. Cancer Res. 2019, 25, 5221–5230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaudie, H.; Viotti, J.; Combemale, P.; Dutriaux, C.; Dupin, N.; Robert, C.; Mortier, L.; Kaphan, R.; Duval-Modeste, A.B.; Dalle, S.; et al. Cetuximab is efficient and safe in patients with advanced cutaneous squamous cell carcinoma: A retrospective, multicentre study. Oncotarget 2020, 11, 378–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, M.C.; Miura, J.T.; Naqvi, S.M.H.; Kim, Y.; Holstein, A.; Lee, D.; Sarnaik, A.A.; Zager, J.S. Talimogene Laherparepvec (TVEC) for the Treatment of Advanced Melanoma: A Single-Institution Experience. Ann. Surg. Oncol. 2018, 25, 3960–3965. [Google Scholar] [CrossRef] [PubMed]
- Middleton, M.; Aroldi, F.; Sacco, J.; Milhem, M.M.; Curti, B.D.; Vanderwalde, A.M.; Baum, S.; Samson, A.; Pavlick, A.C.; Chesney, J.A.; et al. An open-label, single-arm, phase II clinical trial of RP1, an enhanced potency oncolytic herpes virus, combined with nivolumab in four solid tumor types: Initial results from the skin cancer cohorts. J. Clin. Oncol. 2020, 38, e22050. [Google Scholar] [CrossRef]

| Gene | Role |
|---|---|
| TP53 |
|
| NOTCH | |
| RAS |
|
| CDKN2A |
|
| PIK3CA |
|
| KNSTRN | |
| FAT1 |
|
| CARD11 | |
| SRCASM | |
| TP63 |
|
| EGFR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cozma, E.-C.; Banciu, L.M.; Soare, C.; Cretoiu, S.-M. Update on the Molecular Pathology of Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 6646. https://doi.org/10.3390/ijms24076646
Cozma E-C, Banciu LM, Soare C, Cretoiu S-M. Update on the Molecular Pathology of Cutaneous Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2023; 24(7):6646. https://doi.org/10.3390/ijms24076646
Chicago/Turabian StyleCozma, Elena-Codruta, Laura Madalina Banciu, Cristina Soare, and Sanda-Maria Cretoiu. 2023. "Update on the Molecular Pathology of Cutaneous Squamous Cell Carcinoma" International Journal of Molecular Sciences 24, no. 7: 6646. https://doi.org/10.3390/ijms24076646
APA StyleCozma, E. -C., Banciu, L. M., Soare, C., & Cretoiu, S. -M. (2023). Update on the Molecular Pathology of Cutaneous Squamous Cell Carcinoma. International Journal of Molecular Sciences, 24(7), 6646. https://doi.org/10.3390/ijms24076646