Genetics of Plant Metabolism

{kind=link}
{kind=link}
{kind=link}
Conflicts of Interest
References
- Valluru, R.; Reynolds, M.P.; Salse, J. Genetic and Molecular Bases of Yield-Associated Traits: A Translational Biology Approach between Rice and Wheat. Theor. Appl. Genet. 2014, 127, 1463–1489. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Liu, C.; Tan, Y.; Jiang, J.; Chen, F.; Xiong, G.; Chen, S. Five Post-Translational Modification Residues of CmPT2 Play Key Roles in Yeast and Rice. Int. J. Mol. Sci. 2023, 24, 2025. [Google Scholar] [CrossRef] [PubMed]
- Mochida, K.; Shinozaki, K. Advances in Omics and Bioinformatics Tools for Systems Analyses of Plant Functions. Plant Cell Physiol. 2011, 52, 2017–2038. [Google Scholar] [CrossRef] [PubMed]
- Napier, J.D.; Heckman, R.W.; Juenger, T.E. Gene-by-Environment Interactions in Plants: Molecular Mechanisms, Environmental Drivers, and Adaptive Plasticity. Plant Cell 2023, 35, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Lemus, E.; Davies, M.J. Effect of Crowding, Compartmentalization and Nanodomains on Protein Modification and Redox Signaling—Current State and Future Challenges. Free Radic. Biol. Med. 2023, 196, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kovrigin, E.L.; Eletr, Z. NMR Studies of Escherichia Coli Acyl Carrier Protein: Dynamic and Structural Differences of the Apo- and Holo-Forms. Biochem. Biophys. Res. Commun. 2006, 341, 776–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Famiani, F.; Walker, R.P.; Szló Té, L.; Chen, Z.-H.; Proietti1, P.; Leegood, R.C. An Immunohistochemical Study of the Compartmentation of Metabolism during the Development of Grape (Vitis vinifera L.) Berries. J. Exp. Bot. 2000, 51, 675–683. [Google Scholar] [PubMed]
- Cultrera, N.G.M.; Alagna, F.; Mariotti, R.; de Marchis, F.; Pompa, A.; Bellucci, M.; Baldoni, L. Isolation and Molecular Characterization of Three Acyl Carrier Protein Genes in Olive (Olea europaea L.). Tree Genet. Genomes 2014, 10, 895–909. [Google Scholar] [CrossRef]
- Kautsar, S.A.; Suarez Duran, H.G.; Blin, K.; Osbourn, A.; Medema, M.H. PlantiSMASH: Automated Identification, Annotation and Expression Analysis of Plant Biosynthetic Gene Clusters. Nucleic Acids Res. 2017, 45, W55–W63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedayati, V.; Mousavi, A.; Razavi, K.; Cultrera, N.; Alagna, F.; Mariotti, R.; Hosseini-Mazinani, M.; Baldoni, L. Polymorphisms in the AOX2 Gene Are Associated with the Rooting Ability of Olive Cuttings. Plant Cell Rep. 2015, 34, 1151–1164. [Google Scholar] [CrossRef]
- Cultrera, N.G.M.; Sarri, V.; Lucentini, L.; Ceccarelli, M.; Alagna, F.; Mariotti, R.; Mousavi, S.; Ruiz, C.G.; Baldoni, L. High Levels of Variation within Gene Sequences of Olea europaea L. Front. Plant. Sci. 2019, 9, 1932. [Google Scholar] [CrossRef] [Green Version]
- Contreras, C.; Mariotti, R.; Mousavi, S.; Baldoni, L.; Guerrero, C.; Roka, L.; Cultrera, N.; Pierantozzi, P.; Maestri, D.; Gentili, L.; et al. Characterization and Validation of Olive FAD and SAD Gene Families: Expression Analysis in Different Tissues and during Fruit Development. Mol. Biol. Rep. 2020, 47, 4345–4355. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.; Regni, L.; Bocchini, M.; Mariotti, R.; Cultrera, N.G.M.; Mancuso, S.; Googlani, J.; Chakerolhosseini, M.R.; Guerrero, C.; Albertini, E.; et al. Physiological, Epigenetic and Genetic Regulation in Some Olive Cultivars under Salt Stress. Sci. Rep. 2019, 9, 1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Famiani, F.; Cultrera, N.G.M.; Battistelli, A.; Casulli, V.; Proietti, P.; Standardi, A.; Chen, Z.H.; Leegood, R.C.; Walker, R.P. Phosphoenolpyruvate Carboxykinase and Its Potential Role in the Catabolism of Organic Acids in the Flesh of Soft Fruit during Ripening. J. Exp. Bot. 2005, 56, 2959–2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galla, G.; Barcaccia, G.; Ramina, A.; Collani, S.; Alagna, F.; Baldoni, L.; Cultrera, N.G.M.; Martinelli, F.; Sebastiani, L.; Tonutti, P. Computational Annotation of Genes Differentially Expressed along Olive Fruit Development. BMC Plant. Biol. 2009, 9, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alagna, F.; Mariotti, R.; Panara, F.; Caporali, S.; Urbani, S.; Veneziani, G.; Esposto, S.; Taticchi, A.; Rosati, A.; Rao, R.; et al. Olive Phenolic Compounds: Metabolic and Transcriptional Profiling during Fruit Development. BMC Plant Biol. 2012, 12, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alagna, F.; Cirilli, M.; Galla, G.; Carbone, F.; Daddiego, L.; Facella, P.; Lopez, L.; Colao, C.; Mariotti, R.; Cultrera, N.; et al. Transcript Analysis and Regulative Events during Flower Development in Olive (Olea europaea L.). PLoS ONE 2016, 11, e0152943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladman, N.; Goodwin, S.; Chougule, K.; Richard McCombie, W.; Ware, D. Era of Gapless Plant Genomes: Innovations in Sequencing and Mapping Technologies Revolutionize Genomics and Breeding. Curr. Opin. Biotechnol. 2023, 79, 102886. [Google Scholar] [CrossRef] [PubMed]
- Alagna, F.; Reed, J.; Calderini, O.; Thimmappa, R.; Cultrera, N.G.; Cattivelli, A.; Tagliazucchi, D.; Mousavi, S.; Mariotti, R.; Osbourn, A.; et al. OeBAS and CYP716C67 catalyze the biosynthesis of health-beneficial triterpenoids in olive (Olea europaea L.) fruits. New Phytologist. 2023; online ahead of print. [Google Scholar] [CrossRef]
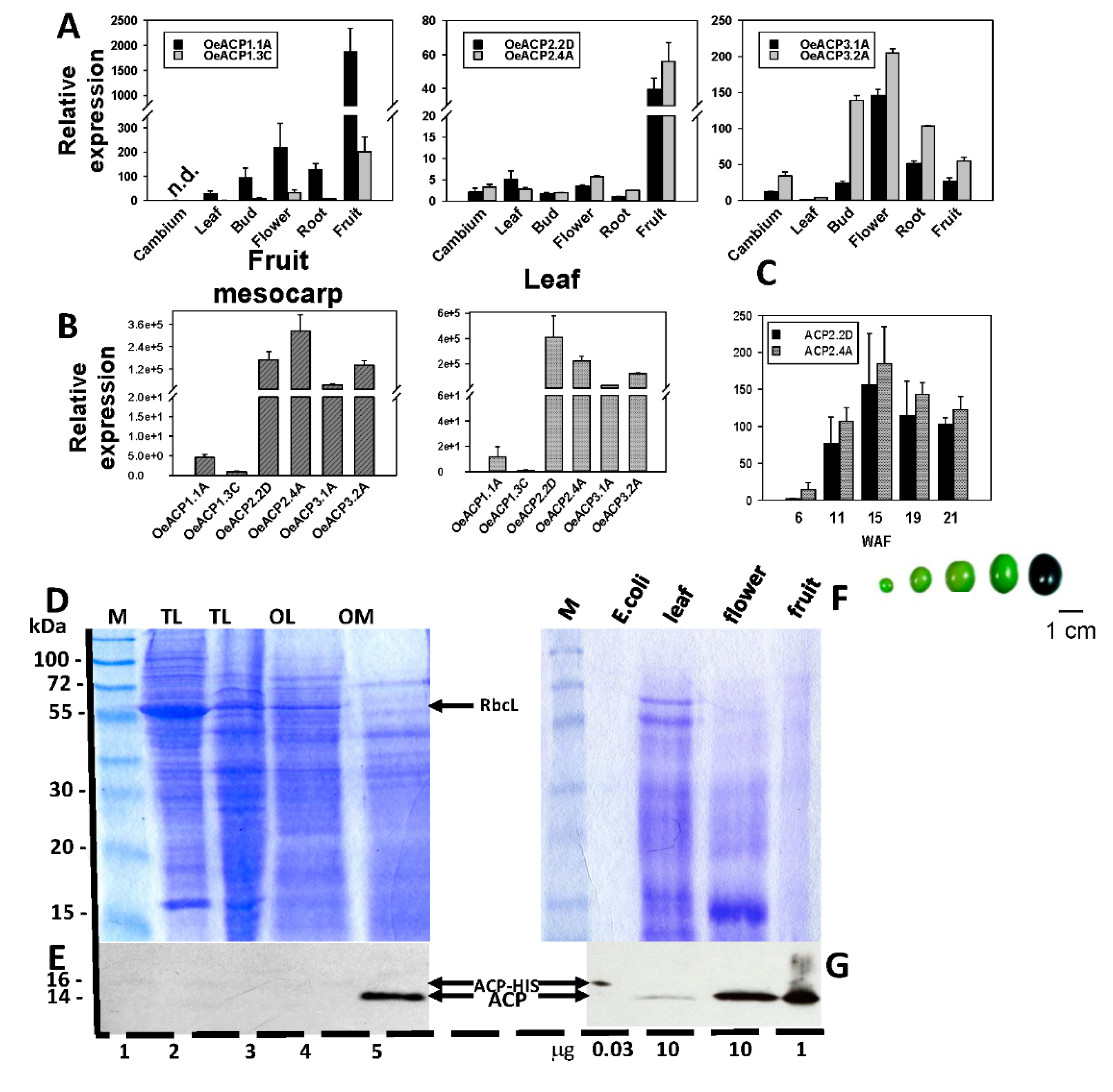
- de Marchis, F.; Valeri, M.C.; Pompa, A.; Bouveret, E.; Alagna, F.; Grisan, S.; Stanzione, V.; Mariotti, R.; Cultrera, N.; Baldoni, L.; et al. Overexpression of the Olive Acyl Carrier Protein Gene (OeACP1) Produces Alterations in Fatty Acid Composition of Tobacco Leaves. Transgenic Res. 2016, 25, 45–61. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cultrera, N.G.M. Genetics of Plant Metabolism. Int. J. Mol. Sci. 2023, 24, 6890. https://doi.org/10.3390/ijms24086890
Cultrera NGM. Genetics of Plant Metabolism. International Journal of Molecular Sciences. 2023; 24(8):6890. https://doi.org/10.3390/ijms24086890
Chicago/Turabian StyleCultrera, Nicolò G. M. 2023. "Genetics of Plant Metabolism" International Journal of Molecular Sciences 24, no. 8: 6890. https://doi.org/10.3390/ijms24086890
APA StyleCultrera, N. G. M. (2023). Genetics of Plant Metabolism. International Journal of Molecular Sciences, 24(8), 6890. https://doi.org/10.3390/ijms24086890