1. Introduction
Over-activation of Toll-like receptor 4 (TLR4) is the key molecular pathology of Gram-negative bacterial infection-induced sepsis, in which regulation of mitochondria remains elusive. Lipopolysaccharide (LPS)-activated inflammatory macrophages (M1) constitute the first line of host defense against invading Gram-negative bacteria [
1]. Specifically, activated M1 macrophages produce abundant pro-inflammatory mediators, including IL-6, TNFα, IL-1, nitric oxide (NO), and reactive oxygen species (ROS), to kill the invading pathogens [
2]. Nevertheless, if M1-mediated inflammation exerts excessive intensity, a cytokine storm can develop, thus causing the occurrence of severe sepsis [
3]. Conversely, M2 macrophages express anti-inflammatory cytokines, such as IL-10, thus inducing an LPS-tolerant state in the host as the infection persists [
4]. The metabolic state influenced by mitochondrial function plays a key role in macrophage M1/M2 polarization. Specifically, M1 macrophages block tricarboxylic acid cycle (TAC) and oxidative phosphorylation. They activate glycolysis and pentose phosphate metabolism as a substitute. M2 macrophages have intact TAC and oxidative phosphorylation. They oxidate pyruvate and fatty acid to produce adenosine triphosphate (ATP) [
5,
6]. In addition, a prior study revealed that mitochondrial dysfunction could impede the reprogramming of M1 macrophages to M2 macrophages, whereas M2 macrophages, which have intact mitochondria, were more readily repolarized to M1 macrophages upon LPS and IFNγ induction [
7].
SAM and HD domain-containing deoxynucleoside triphosphate triphosphohydrolase 1 (SAMHD1), a regulatory molecule of the innate immune system found in a wide range of cells, is especially highly expressed in dendritic cells and macrophages (including microglia, Kupffer cells, Langerhans cells, and other macrophages localized in specific tissues). SAMHD1 is a deoxy-ribonucleoside triphosphate hydrolase (dNTPase), whose activity is responsible for hydrolysis of dNTPs into deoxynucleosides (dN) and inorganic triphosphates (PPPi) [
8]. Accordingly, this molecule is critical for regulating deoxy-ribonucleoside triphosphate (dNTP) metabolism and maintaining homeostasis of the intracellular dNTP pool [
9]. Furthermore, proper quantity and balance of the dNTP pool plays a pivotal role in DNA replication and genome stability. For example, an insufficient dNTP pool results in replication stress [
10], while excessive or disturbed dNTP pools may lead to mismatched base pairing and DNA polymerase stall during replication [
11,
12]. As reported,
Samhd1 deficiency can contribute to dNTP pool imbalance and genomic instability, which may be associated with elevation in IFN-α level and the occurrence of Aicardi–Goutières syndrome [
13]. In non-proliferating cells (such as resting CD4
+ T cells and macrophages), SAMHD1 blocks the reverse transcription of viruses by decreasing the intracellular dNTP concentration [
14]. Previous studies have unveiled that SAMHD1 has the ability to repress the replication of many RNA and DNA viruses such as human immunodeficiency virus 1 (HIV-1), equine infectious anemia (EIA) virus, and human papillomavirus virus 16 (HPV-16) [
15,
16,
17]. Additionally, SAMHD1 can maintain genome stability by recruiting Mre11, a DNA double-strand break repairing protein, to stalled replication forks [
18], or by reducing the activity of long interspersed element 1 (LINE-1) retrotransposons through multiple pathways independent of dNTPase [
19,
20,
21].
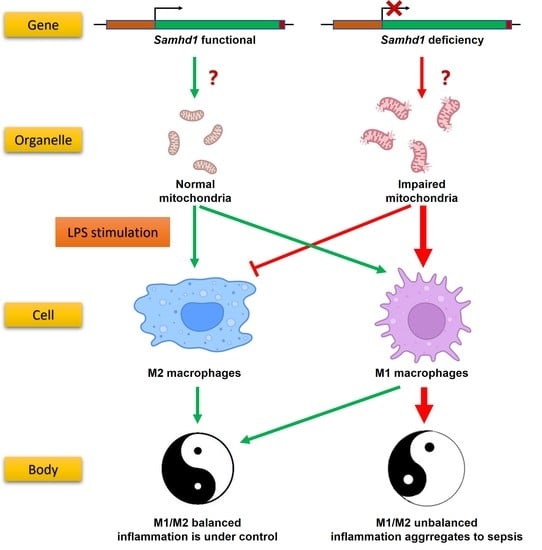
Our preliminary experiments showed that SAMHD1 expression was significantly elevated in macrophages upon LPS stimulation. Therefore, we hypothesized that SAMHD1 was functional in this process. Through in vivo and in vitro LPS-stimulated models of Samhd1−/− mice and macrophages, we found that SAMHD1 was able to decrease TLR4 signaling through maintaining the mitochondrial state, which was dependent on its dNTPase function and regulated by the phosphorylation site T634. Deficiency of Samhd1 led to acute inflammation and M1 polarization of macrophages. We also proved that SAMHD1 interacted with voltage-dependent anion channel-1 (VDAC1), which happens to be an outer mitochondrial membrane (OMM)-localized protein that is vital to mitochondria, and deficiency of Samhd1 resulted in down-regulation of VDAC1. This indicated that SAMHD1 may exert its mitochondria-regulating function through VDAC1.
3. Discussion
The pathogenesis of sepsis has not been fully elucidated. The identification of LPS from bacterial cell walls by TLRs on the surface of macrophages generates a natural immune response, which is widely recognized as the basic model for the study of sepsis at present [
27]. When macrophages are hyperpolarized towards M1 phenotype and anti-inflammatory M2 macrophages fail to balance the pro-inflammatory effects in the early stage of bacterial infection, it will promote uncontrolled local inflammation to systemic sepsis [
28]. Energy metabolic pathways regulated by the functional state of mitochondrion are determinants of macrophage M1 or M2 polarization [
5,
6,
29]. Therefore, mitochondrion is expected to be a key target for altering the course of sepsis.
It has been reported that SAMHD1 has dNTPase activity, which can hydrolyze dNTPs to dN and PPPi [
8]. It is also implicated in the maintenance of intracellular dNTP pool homeostasis [
9] and genomic stability [
10,
11,
12,
13] as well as antiviral activity [
14]. However, little is known about whether SAMHD1 is involved in bacterial infection and sepsis. In our study, the results unveiled that SAMHD1 was upregulated in LPS-stimulated wild-type macrophages, suggesting that it may play a role in the pathological process of sepsis. In LPS-induced
Samhd1−/− mice and
Samhd1−/− peritoneal macrophage models, our data elucidated that
Samhd1 deficiency promoted the activation of the LPS-TLR4 pathway, triggering stronger systemic/local inflammation as well as higher lethality.
Samhd1−/− peritoneal macrophages polarized to M1 more upon M1 induction and the situation is on the contrary upon M2 induction. We hypothesized that the difference in polarization between
Samhd1−/− and control macrophages might be attributed to their different energetic metabolism. The JC-1 assay elaborated that the mitochondrial membrane potential of
Samhd1−/− macrophages was lower than control macrophages both in resting and M1-polarized states. Meanwhile, mitochondrial stress assay and glycolysis stress assay demonstrated that the mitochondrial function of
Samhd1−/− macrophages was impaired and compensated with glycolysis. Intriguingly, despite the damage in mitochondria of
Samhd1−/− macrophages, there was no significant apoptosis of
Samhd1−/− macrophages in M0, M1, and M2 states, indicating that mechanisms such as mitochondrial autophagy may exist in the cells to maintain their survival.
Subsequently, rescue experiments were conducted by overexpressing full-length mouse SAMHD1 in
Samhd1−/− RAW264.7 cells. The results further clarified that SAMHD1 reduced secretion of inflammatory cytokines, maintained mitochondrial stability, and inhibited hyperpolarization toward the M1 phenotype in LPS-induced macrophages. Meanwhile, we also overexpressed several mutants of full-length SAMHD1 in
Samhd1−/− RAW264.7 cells, among which the H238A mutant, which associated with the loss of dNTPase activity [
24], could not reverse the above phenotypes, illustrating that SAMHD1′s inhibition of LPS-TLR4 inflammation and protection of mitochondria were dNTPase-dependent. Mouse SAMHD1 T634 has been identified as a phosphorylation site homologous to human SAMHD1 T592, the function of which is unclear. Although the phosphor-mimetic T634D mutant has been demonstrated to cause the loss of HIV-1 restriction in non-dividing cells, it does not affect murine leukemia virus infection in dividing cells [
24]. Accordingly, we overexpressed the phospho-ablative alanine residue mutant T634A or the phospho-mimetic aspartic acid residue mutant T634D in
Samhd1−/− RAW264.7 cells, respectively, and found that overexpression of the T634D mutant, but not the T634A mutant, exerted similar effects to overexpression of full-length SAMHD1. This result illustrates that the phosphorylation of T634 is a potential target for regulating the LPS-TLR4 inflammation. Of note, our rescue experiments were performed with immortalized RAW264.7 cells as a cell model. In this type of cell, CDK activity that regulates T634 phosphorylation may be different from that of terminally differentiated macrophages, which warrants further investigation.
Finally, VDAC1 was screened as the candidate molecule that interacted with SAMHD1 by IP-MS. Co-IP and immunofluorescent assay further verified that SAMHD1 could directly interact with VDAC1. Interestingly, VDAC1, which mainly localized on OMM, is the only known pathway of OMM that mediates the passage of ions, nucleotides, and other high-molecular-weight metabolites (such as pyruvate, malate, succinate, NADH/NAD as well as heme and cholesterol) [
26]. In a former study, human SAMHD1 was found to be able to regulate mitochondrial dNTP pools, thus influenced the copy number of mitochondrial DNA in fibroblasts [
30]. Though different in species and cell types, it is consistent with our research results that SAMHD1 is involved in mitochondrial maintenance in a dNTPase-dependent manner. We proved that
Samhd1 deficiency in macrophages could result in the down regulation of VDAC1 expression. Because VDAC1 is also a commonly recognized loading control of mitochondria for many quantitative assays [
31], down regulation of VDAC1 means a reduction in total mitochondrial quantity, which is consistent with our results that mitochondria in
Samhd1−/− macrophages are damaged. However, whether SAMHD1-VDAC1 interaction is indispensable and how it works in this process remains to be further investigated. Other research shows that SAMHD1 may inhibits activation of NF-κB during virus infection by directly binding to NF-κB1/2 and reducing phosphorylation of the NF-κB inhibitory protein IκBα [
32] or through a negative regulation of TRAF6-TAK1 checkpoint [
33]. SAMHD1 also inhibits IFN-I induction pathway by interacting with inhibitor-κB kinase ε (IKKε) and IFN regulatory factor 7 (IRF7) [
32]. These results are consistent with ours (
Figure 2), but also raise questions about which mechanism of SAMHD1 plays the leading role in its regulation of the TLR4 pathway. Though downregulation of mitochondrial function may be the result of TLR4 signaling in
Samhd1−/− macrophages upon LPS stimulation, our results support the hypothesis that it is the mitochondrial dysfunction caused by
Samhd1 deficiency that leads to intensified TLR4 activation. We observed that impaired function of mitochondria already existed in
Samhd1−/− macrophages before LPS was loaded (
Figure 4). However, more work needs to be performed to further reveal how these mechanisms of SAMHD1 cooperate and interact with each other in regulation of TLR4 pathway.
In summary, our study unravels that SAMHD1 negatively regulates LPS-TLR4 pathway. SAMHD1 expression is elevated upon LPS-stimulation. Samhd1 deficiency increases mitochondrial damage, which enhances LPS-TLR4 pathway and makes macrophages more M1 polarized, and SAMHD1-VDAC1 interaction may be involved in this process. Consistently, a higher level of inflammatory cytokine secretion aggravates local and systemic tissue damage and thus raises lethality in LPS-induced sepsis. Therefore, SAMHD1 is a potential target molecule against sepsis. In addition, we identified H238/D239 and T634 on mouse SAMHD1 as two key targets that regulate its anti-sepsis function.
4. Materials and Methods
4.1. Mouse Models of LPS-Induced Sepsis
All mice were housed under specific pathogen-free conditions and all experiments involving animals were performed in accordance with National Institute of Health Guide for the Care and Use of Laboratory Animals and approved by Scientific Investigation Board of Naval Medical University (Shanghai, China). The Samhd1−/− mice were generated using CRISPR-Cas9 technique by Cyagen Biosciences, Inc. (Suzhou, China). Specifically, the mouse Samhd1-specific gRNA (gRNA1: TGTTGACAGGAAGGGATCGCTGG; gRNA2: TTGTGGTGACCGTGAACTAAGGG), the donor vector containing loxP sites, and Cas9 mRNA were co-injected into fertilized mouse eggs for generating targeted conditional knockout offspring. Loxp sites are on both sides of exon 2 on Samhd1-encoding mRNA (NCBI Reference Sequence: NM_018851.4). F0 founder animals were identified by polymerase chain reaction (PCR) and then subjected to sequence analyses, which were mated to wild-type mice to test germline transmission and generate F1 animals. Target F1 mice were bred with tissue-specific lysozyme 2-Cre-deleted mice to generate F2 animals. Heterozygous Cre+ mice were bred with homozygous mice to generate Samhd1−/− mice (male, aged 6–8 weeks; the experimental group) and Samhd1fl/fl mice (male, aged 6–8 weeks; the control group). Genomic DNA was isolated from tails and analyzed by PCR amplification with the use of the following primers: 5′-ACACTAGTAGTCCCTTCTGAGGTG-3′ and 5′-TCTTTACCACAATCTGCCTGACA-3′. LoxP homozygote had bands with 200 bp, wild-type had bands with 139 bp, and heterozygote had bands with both 200 bp and 139 bp.
Mice were injected intraperitoneally with LPS (15 mg/kg) for 3 h to establish a model of LPS-induced sepsis or with PBS as sham operation. Three hours after LPS injection, mice were anesthetized with isoflurane, and orbital blood was rapidly collected, followed by euthanasia of mice by cervical dislocation. Subsequently, the lung tissues were obtained and preserved in 4% paraformaldehyde solution, or ground into single cells using a syringe pusher in a 70 μm filter for subsequent flow cytometry or quantitative PCR (qPCR) assays. In the assay of observing survival rate of mice, the dose of LPS injection was 25 mg/kg. The observation lasted for 60 h, and we took completely incapacitated as the endpoint. Then, mice were euthanized by cervical dislocation after anesthesia.
4.2. Cell Culture
Peritoneal macrophages were isolated from mice aged 6–8 weeks as previously described [
34]. RAW264.7 cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). All cells were cultured in a Dulbecco’s Modified Eagle Medium encompassing 10% fetal bovine serum (Gibco, Carlsbad, CA, USA) and 5% Penicillin-Streptomycin Solution (Sangon, Shanghai, China). Cells were stimulated with LPS (100 ng/mL) or treated with 20 ng/mL mIFNγ + 100 ng/mL LPS (M1 macrophage polarization) or with 20 ng/mL mIL-4 (M2 macrophage polarization) for indicated times.
CRISPR-Cas9-mediated ablation of
Samhd1 was achieved with CRISPR-Cas9 ribonucleoproteins (Haixing Bioscience, Zhaoqing, China) containing expression cassettes for hSpCas9 and chimeric guide RNA. To target exon 2 of
Samhd1, two guide RNA sequences (gRNA1: CCAATCGGAATCCATTTGGGGGG; gRNA2: AAAGTTACCGCCCTCTTTGTAGG) were selected through a website (
http://crispr.mit.edu. accessed on 12 July 2022). Plasmids containing the guide RNA sequences were electro-transfected into cells as instructed in the manuals of the Neon transfection system (Thermo Fisher, Waltham, MA, USA). After two days, single colonies were transferred into 96-well plates. To identify the presence of insertion or deletion (indels) in
Samhd1-targeted clones, genomic DNA was isolated using a Quick-DNA Miniprep kit (Zymo Research, Orange, CA, USA), and PCR amplification was conducted using 2 × Taq Master Mix (Vazyme, Nanjing, China) of primers flanking exon (forward: 5′-AGCCATTTAGGGAGGGGTAGG-3′ and reverse: 5′-TGTGACCCAGGCAAGTTTCT-3′). Plasmids were isolated from 8–10 single colonies and sequenced with Sanger sequencing (GENEWIZ, Beijing, China). Clones with mutations in both alleles were selected for subsequent experiments.
4.3. RNA Quantification
Total RNA was collected with RNA extraction kits (Fastagen, Shanghai, China), 200 ng of which was subjected to reverse transcription with reverse transcription-PCR kits (Vazyme, Nanjing, China). qPCR was performed according to the protocols of Taq Pro Universal SYBR qPCR Master Mix (Vazyme, Nanjing, China). Data were normalized to β-actin expression. Relative changes in gene and mRNA expression were analyzed using the 2-ΔΔCt method. Primers are listed in
Supplementary Table S1.
4.4. ELISA
IL-6, TNFα, and IFNβ in cell culture supernatants were measured using corresponding ELISA kits (R&D, Minneapolis, MN, USA). Blood was collected from the orbit of mice and stored at 4 °C overnight. After blood was centrifuged at 4 °C for 10 min, the supernatants were obtained as the specimen. Afterward, the levels of the cytokines were detected with corresponding ELISA kits (R&D, Minneapolis, MN, USA), and lactate levels were tested with an L-Lactate Assay Kit (Abcam, Cambridge, UK).
4.5. Immunoprecipitation (IP) and Immunoblot Analysis
Cells were lysed with cell lysis buffer, followed by the measurement of protein concentration with bicinchoninic acid kits (Thermo Fisher Scientific, Waltham, MA, USA). IP and immunoblot assays were performed as previously described [
34]. Primary antibodies are presented in
Supplementary Table S2.
4.6. Immunofluorescent Confocal Microscopy
After culturing, treating, fixing, blocking, permeating, and rinsing, cells were incubated on round glasses with primary antibodies (listed in
Supplementary Table S2) and fluorochrome-conjugated secondary antibodies according to the manufacturer’s instructions. The round glasses were then loaded onto the slides with 4′,6-diamidino-2-phenylindole (DAPI)-containing mounting medium (Yeasen, Shanghai, China) and observed with a laser confocal microscope (Leica Microsystems, Wetzlar, German).
4.7. Flow Cytometry and Apoptosis Assay
Flow cytometry data were obtained from an ID7000 full-spectrum flow cytometer (Sony, Tokyo, Japan) and analyzed using the Flowjo software v10.8.1 (BD, Franklin Lakes, NJ, USA). Fluorescence signal channels are listed in
Table 1. Gating strategy is shown in
Supplementary Figure S1. Single positive controls and fluorescence-minus-one (FMO) controls are listed in
Supplementary Figure S2 and Supplementary Figure S3, respectively.
Apoptosis was detected as directed in the manuals of Annexin V Apoptosis Kits (BD, Franklin Lakes, NJ, USA). In brief, cells were digested with trypsin, washed with PBS once and resuspended with Annexin V binding buffer containing FITC conjugated Annexin V antibody and PI. After incubating in the dark at room temperature for 15 min, cells were immediately resuspended using an appropriate amount of Annexin V binding buffer and detected by flow cytometer.
Polarization quantification of lung macrophages: Lung tissues were harvested, cut with scissors and digested with 100 U/mL type I collagenase (Absin, Shanghai, China) for 6 h at 37 °C. After filtering with 70 μm strainers, digestive residues were resuspended into single cells, among which lung macrophages were sorted by F4/80 positive selection magnetic beads (Stemcell, Vancouver, BC, Canada). Sorted macrophages were blocked with CD16/32 antibodies and then marked with conjugated antibodies including FITC-CD80, AF647-F4/80, BV421-CD11b, and PE-CD206, with the help of permeabilization reagents (Thermo Fisher Scientific, Waltham, MA, USA). The cells were then detected by flow cytometry. M1 and M2 macrophages were quantified among F4/80+ CD11b+ cells, according to the percentage of CD80+ cells and CD206+ cells, respectively.
Polarization quantification of peritoneal macrophages: Peritoneal macrophages were harvested, blocked with CD16/32 antibodies and then marked with conjugated antibodies including AF700-CD45.2, FITC-CD80, AF647-F4/80, BV421-CD11b, and PE-CD206, with the help of permeabilization reagents (Thermo Fisher Scientific, MA, USA). The cells were then detected by flow cytometry. M1 and M2 macrophages were quantified among CD45+ F4/80+ CD11b+ cells, according to the percentage of CD80+ cells and CD206+ cells, respectively.
Polarization quantification of RAW264.7 cell line: RAW264.7 cells were blocked with CD16/32 antibodies and then marked with conjugated antibodies including AF700-CD86, AF647-F4/80, and BV421-CD11b. The cells were then detected by flow cytometry. M1 macrophages were quantified among F4/80+ CD11b+ cells, according to the percentage of CD86+ cells.
4.8. Mitochondrial Membrane Potential Analysis
Macrophages were digested and incubated with 1X mitochondrial potential sensor JC-1 working solution (Yeasen, Shanghai, China) for 20 min in the dark at 37 °C. Then, macrophages were rinsed twice with JC-1 washing buffer as per the manufacturer’s instructions. The cells stained with JC-1 were loaded on a 96-well white-bottom plate. Signals were detected with a flow cytometer for flow cytometry diagrams or a fluorescence plate reader for quantification. Fluorescence signal channels of flow cytometry can be seen in
Table 1. Channels of fluorescence plate reader: cell fluorescence intensity in red channel (an Ex wavelength of 525 ± 10 nm and an Em wavelength of 590 ± 10 nm) and green channel (an Ex wavelength of 490 ± 10 nm and an Em wavelength of 535 ± 10 nm) were detected. Ratio of red to green fluorescence intensity was used as an index of mitochondrial membrane potential.
4.9. Mitochondrial Respiration and Glycolysis Measurements
This assay was performed using an XF Extracellular Flux Analyzer (Agilent, Palo Alto, CA, USA) based on the protocols of Glycolytic Stress Test and Mitochondrial Test Kits.
4.10. Co-Immunoprecipitation Coupled to Mass Spectrometry (IP-MS)
IP was performed in the non-denatured lysates of RAW264.7 cells with SAMHD1 antibodies and protein A/G-binding agarose beads. Then, the precipitating complexes were boiled after addition of sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer, followed by electrophoresing on Bis-Tris Gels. When the lanes were about 1 cm long, electrophoresing was stopped and the lanes were cut off from the gels for mass spectrometry to identify the proteins in the lanes.
4.11. Plasmid Constructs and Transfection
The recombinant plasmids expressing full-length wild-type mouse SAMHD1 were constructed. Specifically, cDNA from RAW264.7 cells was subjected to PCR-based amplification and then subcloned into the pMAXCloning eukaryotic expression vector with a seamless cloning kit (Vazyme, Nanjing, China). Mutants, H238A, T634A, and T634D, were generated based on the full-length wild-type mouse SAMHD1 pMAXCloning vector with a quick mutation kit (Beyotime, Shanghai, China). The recombinant plasmids were amplified with competent E. coli DH5α and were purified again with an endotoxin-free large plasmid extraction kit (Abclonal, Wuhan, China). Plasmids were electro-transfected into cells using the 4D-Nucleofector System (Lonza, Basel, Switzerland) as per manufacturer’s instructions. Expression efficiency of the plasmids at the protein level was validated by Western blotting (
Supplementary Figure S4).
4.12. Statistical Analysis
All quantitative data were summarized as mean ± standard deviation. Comparisons between two groups were analyzed using two-tailed unpaired Student’s t-test. The survival rate was analyzed with Log-rank test. Differences with a p-value < 0.05 were considered statistically significant.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}