

The Tricky Connection between Extracellular Vesicles and Mitochondria in Inflammatory-Related Diseases

 , ,
, ,  ,
, 
 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
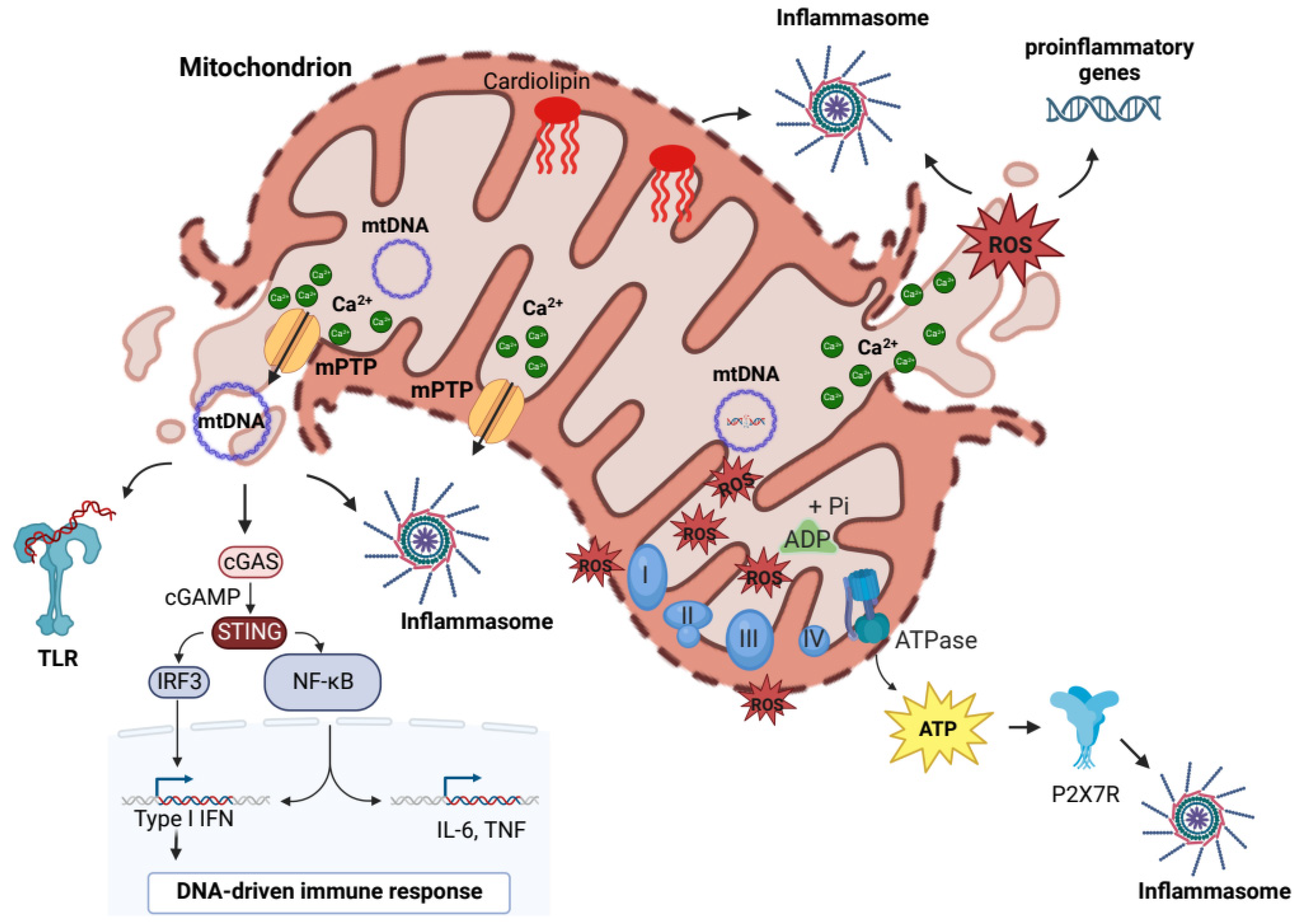
2. “Mitochondrial Damage-Associated Molecular Patterns” Drive the Mito-Inflammation
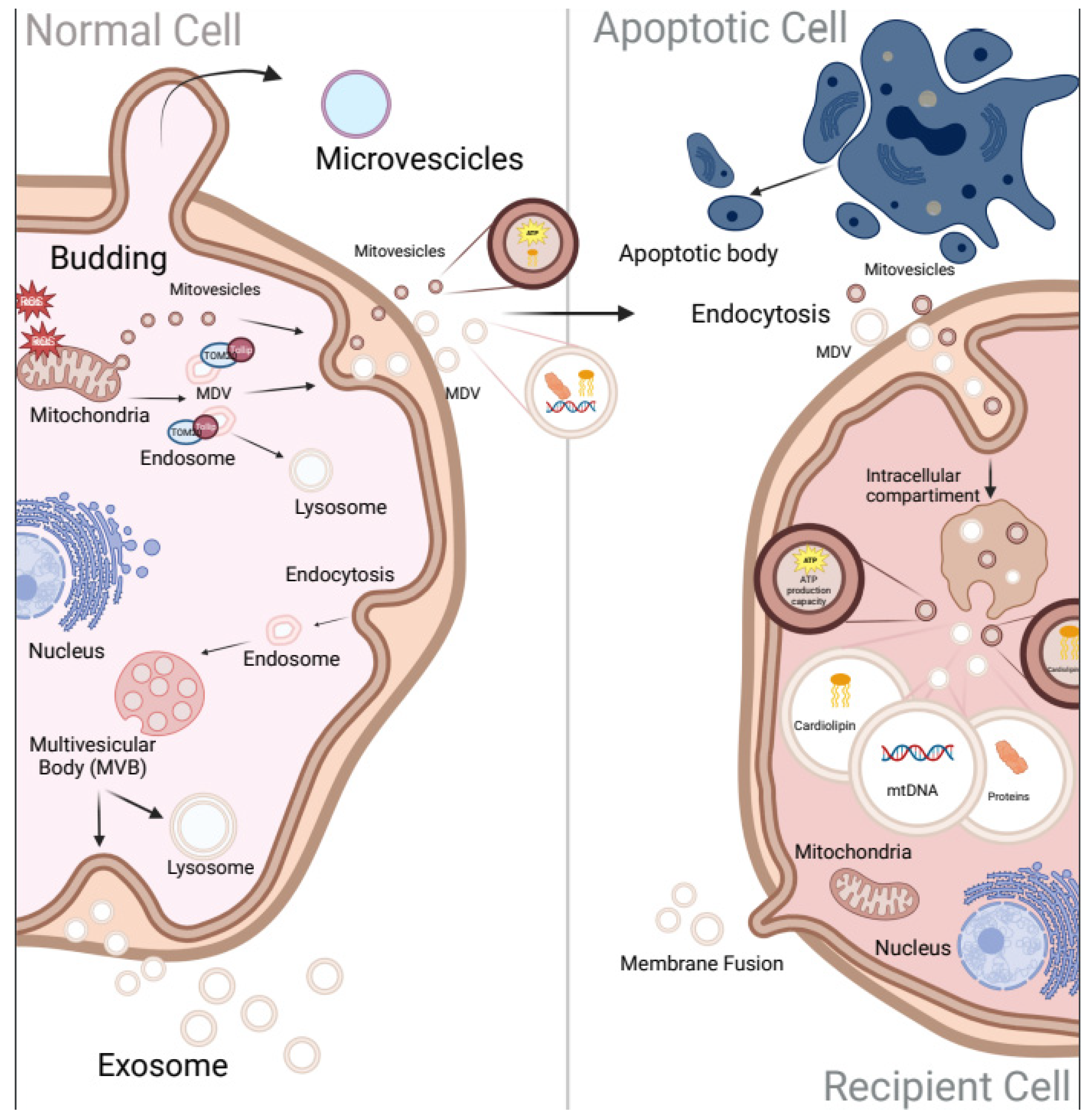
3. The New Era of Extracellular Vesicles Communication
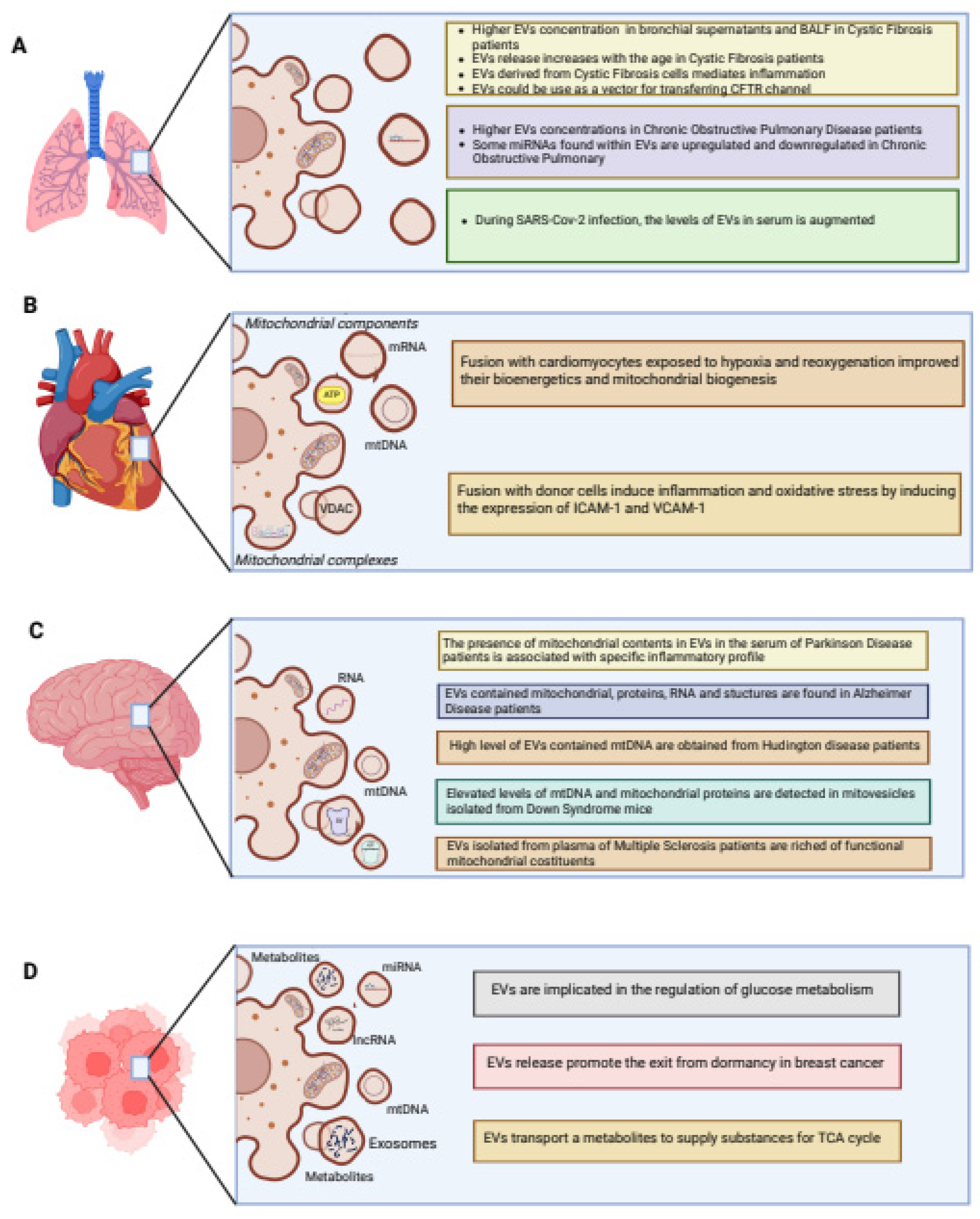
4. The Contribution of Mitochondria and EVs in Inflammatory-Related Diseases
4.1. Pulmonary System
4.2. Cardiovascular System
4.3. Nervous System
4.4. Cancer
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Kuhlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Colombini, M. Voltage gating in the mitochondrial channel, VDAC. J. Membr. Biol. 1989, 111, 103–111. [Google Scholar] [CrossRef]
- Nicholls, D.G. The influence of respiration and ATP hydrolysis on the proton-electrochemical gradient across the inner membrane of rat-liver mitochondria as determined by ion distribution. Eur. J. Biochem. 1974, 50, 305–315. [Google Scholar] [CrossRef]
- Ionov, A.; Fisher, A.A.; Shubich, M.G. The diagnostic information value of determining the cytochemical properties of the neutrophils from the blood and synovial fluid of patients with rheumatoid arthritis and osteoarthrosis deformans. Ter. Arkhiv 1990, 62, 51–55. [Google Scholar]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef]
- Danese, A.; Patergnani, S.; Bonora, M.; Wieckowski, M.R.; Previati, M.; Giorgi, C.; Pinton, P. Calcium regulates cell death in cancer: Roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim. Biophys. Acta Bioenerg. 2017, 1858, 615–627. [Google Scholar] [CrossRef]
- Giorgi, C.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Poletti, F.; Rimessi, A.; et al. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion 2012, 12, 77–85. [Google Scholar] [PubMed]
- Marchi, S.; Giorgi, C.; Galluzzi, L.; Pinton, P. Ca2+ Fluxes and Cancer. Mol. Cell 2020, 78, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Wenceslau, C.F.; McCarthy, C.G.; Szasz, T.; Spitler, K.; Goulopoulou, S.; Webb, R.C.; Working Group on DAMPs in Cardiovascular Disease. Mitochondrial damage-associated molecular patterns and vascular function. Eur. Heart J. 2014, 35, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef]
- Patergnani, S.; Vitto, V.A.M.; Pinton, P.; Rimessi, A. Mitochondrial Stress Responses and “Mito-Inflammation” in Cystic Fibrosis. Front. Pharmacol. 2020, 11, 581114. [Google Scholar] [CrossRef]
- Neuspiel, M.; Schauss, A.C.; Braschi, E.; Zunino, R.; Rippstein, P.; Rachubinski, R.A.; Andrade-Navarro, M.A.; McBride, H.M. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. CB 2008, 18, 102–108. [Google Scholar] [CrossRef]
- Soubannier, V.; Rippstein, P.; Kaufman, B.A.; Shoubridge, E.A.; McBride, H.M. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS ONE 2012, 7, e52830. [Google Scholar] [CrossRef]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef]
- D’Acunzo, P.; Perez-Gonzalez, R.; Kim, Y.; Hargash, T.; Miller, C.; Alldred, M.J.; Erdjument-Bromage, H.; Penikalapati, S.C.; Pawlik, M.; Saito, M.; et al. Mitovesicles are a novel population of extracellular vesicles of mitochondrial origin altered in Down syndrome. Sci. Adv. 2021, 7, eabe5085. [Google Scholar] [CrossRef]
- Todkar, K.; Chikhi, L.; Desjardins, V.; El-Mortada, F.; Pepin, G.; Germain, M. Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat. Commun. 2021, 12, 1971. [Google Scholar] [CrossRef]
- Patergnani, S.; Morciano, G.; Carinci, M.; Leo, S.; Pinton, P.; Rimessi, A. The “mitochondrial stress responses”: The “Dr. Jekyll and Mr. Hyde” of neuronal disorders. Neural Regen. Res. 2022, 17, 2563–2575. [Google Scholar] [PubMed]
- Garg, A.D.; Nowis, D.; Golab, J.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Immunogenic cell death, DAMPs and anticancer therapeutics: An emerging amalgamation. Biochim. Biophys. Acta 2010, 1805, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Dela Cruz, C.S.; Kang, M.J. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 2018, 41, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Fischer, F.; Hamann, A.; Osiewacz, H.D. Mitochondrial quality control: An integrated network of pathways. Trends Biochem. Sci. 2012, 37, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.; Zimmermann, M.; Reichert, A.S. Controlling quality and amount of mitochondria by mitophagy: Insights into the role of ubiquitination and deubiquitination. Biol. Chem. 2016, 397, 637–647. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nunez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef]
- Amari, L.; Germain, M. Mitochondrial Extracellular Vesicles—Origins and Roles. Front. Mol. Neurosci. 2021, 14, 767219. [Google Scholar] [CrossRef]
- Garcia, N.; Garcia, J.J.; Correa, F.; Chavez, E. The permeability transition pore as a pathway for the release of mitochondrial DNA. Life Sci. 2005, 76, 2873–2880. [Google Scholar] [CrossRef]
- Garg, M.; Johri, S.; Chakraborty, K. Immunomodulatory role of mitochondrial DAMPs: A missing link in pathology? FEBS J. 2022. [Google Scholar] [CrossRef]
- Nakayama, H.; Otsu, K. Mitochondrial DNA as an inflammatory mediator in cardiovascular diseases. Biochem. J. 2018, 475, 839–852. [Google Scholar] [CrossRef]
- Marchi, S.; Guilbaud, E.; Tait, S.W.G.; Yamazaki, T.; Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 2023, 23, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Gebert, N.; Joshi, A.S.; Kutik, S.; Becker, T.; McKenzie, M.; Guan, X.L.; Mooga, V.P.; Stroud, D.A.; Kulkarni, G.; Wenk, M.R.; et al. Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: Implications for Barth syndrome. Curr. Biol. CB 2009, 19, 2133–2139. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Greenberg, M.L. Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 3–7. [Google Scholar] [CrossRef]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef]
- Rimessi, A.; Previati, M.; Nigro, F.; Wieckowski, M.R.; Pinton, P. Mitochondrial reactive oxygen species and inflammation: Molecular mechanisms, diseases and promising therapies. Int. J. Biochem. Cell Biol. 2016, 81 Pt B, 281–293. [Google Scholar] [CrossRef]
- Nohl, H.; Gille, L.; Staniek, K. Intracellular generation of reactive oxygen species by mitochondria. Biochem. Pharmacol. 2005, 69, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Perrone, M.; Gafa, R.; Nicoli, F.; Bonora, M.; Morciano, G.; Boncompagni, C.; Marchi, S.; Lebiedzinska-Arciszewska, M.; Vezzani, B.; et al. PML at mitochondria-associated membranes governs a trimeric complex with NLRP3 and P2X7R that modulates the tumor immune microenvironment. Cell Death Differ. 2023, 30, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, H.; Parthasarathi, K.; Quadri, S.; Issekutz, A.C.; Bhattacharya, J. Mechano-oxidative coupling by mitochondria induces proinflammatory responses in lung venular capillaries. J. Clin. Investig. 2003, 111, 691–699. [Google Scholar] [CrossRef]
- Mansfield, K.D.; Guzy, R.D.; Pan, Y.; Young, R.M.; Cash, T.P.; Schumacker, P.T.; Simon, M.C. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005, 1, 393–399. [Google Scholar] [CrossRef]
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- Beltran, B.; Mathur, A.; Duchen, M.R.; Erusalimsky, J.D.; Moncada, S. The effect of nitric oxide on cell respiration: A key to understanding its role in cell survival or death. Proc. Natl. Acad. Sci. USA 2000, 97, 14602–14607. [Google Scholar] [CrossRef]
- Biasutto, L.; Azzolini, M.; Szabo, I.; Zoratti, M. The mitochondrial permeability transition pore in AD 2016: An update. Biochim. Biophys. Acta 2016, 1863, 2515–2530. [Google Scholar] [CrossRef]
- Feno, S.; Butera, G.; Vecellio Reane, D.; Rizzuto, R.; Raffaello, A. Crosstalk between Calcium and ROS in Pathophysiological Conditions. Oxidative Med. Cell. Longev. 2019, 2019, 9324018. [Google Scholar] [CrossRef]
- Dong, Z.; Shanmughapriya, S.; Tomar, D.; Siddiqui, N.; Lynch, S.; Nemani, N.; Breves, S.L.; Zhang, X.; Tripathi, A.; Palaniappan, P.; et al. Mitochondrial Ca2+ Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol. Cell 2017, 65, 1014–1028 e7. [Google Scholar] [CrossRef]
- Sivaramakrishnan, V.; Fountain, S.J. Evidence for Extracellular ATP as a Stress Signal in a Single-Celled Organism. Eukaryot. Cell 2015, 14, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.; Chiozzi, P.; Falzoni, S.; Dal Susino, M.; Melchiorri, L.; Baricordi, O.R.; Di Virgilio, F. Extracellular ATP triggers IL-1 beta release by activating the purinergic P2Z receptor of human macrophages. J. Immunol. 1997, 159, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Corriden, R.; Inoue, Y.; Yip, L.; Hashiguchi, N.; Zinkernagel, A.; Nizet, V.; Insel, P.A.; Junger, W.G. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 2006, 314, 1792–1795. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.N.; Leitinger, N. Purinergic and calcium signaling in macrophage function and plasticity. Front. Immunol. 2014, 5, 580. [Google Scholar] [CrossRef] [PubMed]
- Stahl, P.D.; Raposo, G. Extracellular Vesicles: Exosomes and Microvesicles, Integrators of Homeostasis. Physiology 2019, 34, 169–177. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e18. [Google Scholar] [CrossRef]
- Heijnen, H.F.; Schiel, A.E.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated platelets release two types of membrane vesicles: Microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood 1999, 94, 3791–3799. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Caruso, S.; Poon, I.K.H. Apoptotic Cell-Derived Extracellular Vesicles: More Than Just Debris. Front. Immunol. 2018, 9, 1486. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, M.; Chen, Z.; Xu, L.; Chang, M.; Wang, K.; Deng, C.; Gu, Y.; Zhou, S.; Shen, Y.; et al. Biogenesis and function of extracellular vesicles in pathophysiological processes of skeletal muscle atrophy. Biochem. Pharmacol. 2022, 198, 114954. [Google Scholar] [CrossRef] [PubMed]
- Grenier-Pleau, I.; Abraham, S.A. Extracellular vesicles tell all: How vesicle-mediated cellular communication shapes hematopoietic stem cell biology with increasing age. Exp. Hematol. 2021, 101–102, 7–15. [Google Scholar] [CrossRef]
- Fitzgerald, W.; Freeman, M.L.; Lederman, M.M.; Vasilieva, E.; Romero, R.; Margolis, L. A System of Cytokines Encapsulated in ExtraCellular Vesicles. Sci. Rep. 2018, 8, 8973. [Google Scholar] [CrossRef]
- Xie, M.; Xiong, W.; She, Z.; Wen, Z.; Abdirahman, A.S.; Wan, W.; Wen, C. Immunoregulatory Effects of Stem Cell-Derived Extracellular Vesicles on Immune Cells. Front. Immunol. 2020, 11, 13. [Google Scholar] [CrossRef]
- Sun, X.; Meng, H.; Wan, W.; Xie, M.; Wen, C. Application potential of stem/progenitor cell-derived extracellular vesicles in renal diseases. Stem Cell Res. Ther. 2019, 10, 8. [Google Scholar] [CrossRef]
- Shao, H.; Im, H.; Castro, C.M.; Breakefield, X.; Weissleder, R.; Lee, H. New Technologies for Analysis of Extracellular Vesicles. Chem. Rev. 2018, 118, 1917–1950. [Google Scholar] [CrossRef]
- Lener, T.; Gimona, M.; Aigner, L.; Borger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; Del Portillo, H.A.; et al. Applying extracellular vesicles based therapeutics in clinical trials—An ISEV position paper. J. Extracell. Vesicles 2015, 4, 30087. [Google Scholar] [CrossRef]
- McVey, M.J.; Maishan, M.; Blokland, K.E.C.; Bartlett, N.; Kuebler, W.M. Extracellular vesicles in lung health, disease, and therapy. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L977–L989. [Google Scholar] [CrossRef] [PubMed]
- Peruzzotti-Jametti, L.; Bernstock, J.D.; Willis, C.M.; Manferrari, G.; Rogall, R.; Fernandez-Vizarra, E.; Williamson, J.C.; Braga, A.; van den Bosch, A.; Leonardi, T.; et al. Neural stem cells traffic functional mitochondria via extracellular vesicles. PLoS Biol. 2021, 19, e3001166. [Google Scholar] [CrossRef] [PubMed]
- Budden, C.F.; Gearing, L.J.; Kaiser, R.; Standke, L.; Hertzog, P.J.; Latz, E. Inflammasome-induced extracellular vesicles harbour distinct RNA signatures and alter bystander macrophage responses. J. Extracell. Vesicles 2021, 10, e12127. [Google Scholar] [CrossRef] [PubMed]
- Soubannier, V.; McLelland, G.L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. CB 2012, 22, 135–141. [Google Scholar] [CrossRef]
- Ryan, T.A.; Phillips, E.O.; Collier, C.L.; Jb Robinson, A.; Routledge, D.; Wood, R.E.; Assar, E.A.; Tumbarello, D.A. Tollip coordinates Parkin-dependent trafficking of mitochondrial-derived vesicles. EMBO J. 2020, 39, e102539. [Google Scholar] [CrossRef]
- Phinney, D.G.; Di Giuseppe, M.; Njah, J.; Sala, E.; Shiva, S.; St Croix, C.M.; Stolz, D.B.; Watkins, S.C.; Di, Y.P.; Leikauf, G.D.; et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat. Commun. 2015, 6, 8472. [Google Scholar] [CrossRef]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef]
- D’Souza, A.; Burch, A.; Dave, K.M.; Sreeram, A.; Reynolds, M.J.; Dobbins, D.X.; Kamte, Y.S.; Zhao, W.; Sabatelle, C.; Joy, G.M.; et al. Microvesicles transfer mitochondria and increase mitochondrial function in brain endothelial cells. J. Control. Release Off. J. Control. Release Soc. 2021, 338, 505–526. [Google Scholar]
- Bartel, S.; Deshane, J.; Wilkinson, T.; Gabrielsson, S. Extracellular Vesicles as Mediators of Cellular Cross Talk in the Lung Microenvironment. Front. Med. 2020, 7, 326. [Google Scholar] [CrossRef]
- Kulshreshtha, A.; Ahmad, T.; Agrawal, A.; Ghosh, B. Proinflammatory role of epithelial cell-derived exosomes in allergic airway inflammation. J. Allergy Clin. Immunol. 2013, 131, 1194–1203, 1203.e1–14. [Google Scholar] [CrossRef]
- Kadota, T.; Fujita, Y.; Yoshioka, Y.; Araya, J.; Kuwano, K.; Ochiya, T. Emerging role of extracellular vesicles as a senescence-associated secretory phenotype: Insights into the pathophysiology of lung diseases. Mol. Asp. Med. 2018, 60, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Aravamudan, B.; Thompson, M.A.; Pabelick, C.M.; Prakash, Y.S. Mitochondria in lung diseases. Expert Rev. Respir. Med. 2013, 7, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Prakash, Y.S. Airway smooth muscle in airway reactivity and remodeling: What have we learned? Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L912–L933. [Google Scholar] [CrossRef] [PubMed]
- Delmotte, P.; Yang, B.; Thompson, M.A.; Pabelick, C.M.; Prakash, Y.S.; Sieck, G.C. Inflammation alters regional mitochondrial Ca2+ in human airway smooth muscle cells. Am. J. Physiol. Cell Physiol. 2012, 303, C244–C256. [Google Scholar] [CrossRef] [PubMed]
- Wiegman, C.H.; Michaeloudes, C.; Haji, G.; Narang, P.; Clarke, C.J.; Russell, K.E.; Bao, W.; Pavlidis, S.; Barnes, P.J.; Kanerva, J.; et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015, 136, 769–780. [Google Scholar] [CrossRef]
- Gibson, R.L.; Burns, J.L.; Ramsey, B.W. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 918–951. [Google Scholar] [CrossRef]
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic fibrosis. Lancet 2021, 397, 2195–2211. [Google Scholar] [CrossRef]
- Rimessi, A.; Bezzerri, V.; Patergnani, S.; Marchi, S.; Cabrini, G.; Pinton, P. Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat. Commun. 2015, 6, 6201. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Legendre, C.; Mooij, M.J.; Adams, C.; O’Gara, F. Impaired expression of hypoxia-inducible factor-1alpha in cystic fibrosis airway epithelial cells—A role for HIF-1 in the pathophysiology of CF? J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2011, 10, 286–290. [Google Scholar] [CrossRef]
- Levy, H.; Murphy, A.; Zou, F.; Gerard, C.; Klanderman, B.; Schuemann, B.; Lazarus, R.; Garcia, K.C.; Celedon, J.C.; Drumm, M.; et al. IL1B polymorphisms modulate cystic fibrosis lung disease. Pediatr. Pulmonol. 2009, 44, 580–593. [Google Scholar] [CrossRef] [PubMed]
- McNamara, N.; Gallup, M.; Sucher, A.; Maltseva, I.; McKemy, D.; Basbaum, C. AsialoGM1 and TLR5 cooperate in flagellin-induced nucleotide signaling to activate Erk1/2. Am. J. Respir. Cell Mol. Biol. 2006, 34, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Saadane, A.; Eastman, J.; Berger, M.; Bonfield, T.L. Parthenolide inhibits ERK and AP-1 which are dysregulated and contribute to excessive IL-8 expression and secretion in cystic fibrosis cells. J. Inflamm. 2011, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, X.; Zhang, Y.H.; Strokes, D.C.; Naren, A.P. Lumacaftor/ivacaftor combination for cystic fibrosis patients homozygous for Phe508del-CFTR. Drugs Today 2016, 52, 229–237. [Google Scholar]
- Donaldson, S.H.; Pilewski, J.M.; Griese, M.; Cooke, J.; Viswanathan, L.; Tullis, E.; Davies, J.C.; Lekstrom-Himes, J.A.; Wang, L.T.; Group, V.X.S. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am. J. Respir. Crit. Care Med. 2018, 197, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Baixauli, F.; Lopez-Otin, C.; Mittelbrunn, M. Exosomes and autophagy: Coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 2014, 5, 403. [Google Scholar] [CrossRef]
- Amaral, M.D.; Balch, W.E. Hallmarks of therapeutic management of the cystic fibrosis functional landscape. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2015, 14, 687–699. [Google Scholar] [CrossRef]
- Useckaite, Z.; Ward, M.P.; Trappe, A.; Reilly, R.; Lennon, J.; Davage, H.; Matallanas, D.; Cassidy, H.; Dillon, E.T.; Brennan, K.; et al. Increased extracellular vesicles mediate inflammatory signalling in cystic fibrosis. Thorax 2020, 75, 449–458. [Google Scholar] [CrossRef]
- Cook-Mills, J.M.; Marchese, M.E.; Abdala-Valencia, H. Vascular cell adhesion molecule-1 expression and signaling during disease: Regulation by reactive oxygen species and antioxidants. Antioxid. Redox Signal. 2011, 15, 1607–1638. [Google Scholar] [CrossRef]
- Forrest, O.A.; Dobosh, B.; Ingersoll, S.A.; Rao, S.; Rojas, A.; Laval, J.; Alvarez, J.A.; Brown, M.R.; Tangpricha, V.; Tirouvanziam, R. Neutrophil-derived extracellular vesicles promote feed-forward inflammasome signaling in cystic fibrosis airways. J. Leukoc. Biol. 2022, 112, 707–716. [Google Scholar] [CrossRef]
- Vituret, C.; Gallay, K.; Confort, M.P.; Ftaich, N.; Matei, C.I.; Archer, F.; Ronfort, C.; Mornex, J.F.; Chanson, M.; Di Pietro, A.; et al. Transfer of the Cystic Fibrosis Transmembrane Conductance Regulator to Human Cystic Fibrosis Cells Mediated by Extracellular Vesicles. Hum. Gene Ther. 2016, 27, 166–183. [Google Scholar] [CrossRef] [PubMed]
- Rabe, K.F.; Watz, H. Chronic obstructive pulmonary disease. Lancet 2017, 389, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- American Thoracic, S.; European Respiratory, S. American Thoracic Society/European Respiratory Society statement: Standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am. J. Respir. Crit. Care Med. 2003, 168, 818–900. [Google Scholar]
- Di Gioia, S.; Daniello, V.; Conese, M. Extracellular Vesicles’ Role in the Pathophysiology and as Biomarkers in Cystic Fibrosis and COPD. Int. J. Mol. Sci. 2022, 24, 228. [Google Scholar] [CrossRef]
- Takahashi, T.; Kobayashi, S.; Fujino, N.; Suzuki, T.; Ota, C.; He, M.; Yamada, M.; Suzuki, S.; Yanai, M.; Kurosawa, S.; et al. Increased circulating endothelial microparticles in COPD patients: A potential biomarker for COPD exacerbation susceptibility. Thorax 2012, 67, 1067–1074. [Google Scholar] [CrossRef]
- Savarimuthu Francis, S.M.; Davidson, M.R.; Tan, M.E.; Wright, C.M.; Clarke, B.E.; Duhig, E.E.; Bowman, R.V.; Hayward, N.K.; Fong, K.M.; Yang, I.A. MicroRNA-34c is associated with emphysema severity and modulates SERPINE1 expression. BMC Genom. 2014, 15, 88. [Google Scholar] [CrossRef]
- Corsello, T.; Kudlicki, A.S.; Garofalo, R.P.; Casola, A. Cigarette Smoke Condensate Exposure Changes RNA Content of Extracellular Vesicles Released from Small Airway Epithelial Cells. Cells 2019, 8, 1652. [Google Scholar] [CrossRef]
- Serban, K.A.; Rezania, S.; Petrusca, D.N.; Poirier, C.; Cao, D.; Justice, M.J.; Patel, M.; Tsvetkova, I.; Kamocki, K.; Mikosz, A.; et al. Structural and functional characterization of endothelial microparticles released by cigarette smoke. Sci. Rep. 2016, 6, 31596. [Google Scholar] [CrossRef]
- Tahyra, A.S.C.; Calado, R.T.; Almeida, F. The Role of Extracellular Vesicles in COVID-19 Pathology. Cells 2022, 11, 2496. [Google Scholar] [CrossRef]
- Xia, X.; Yuan, P.; Liu, Y.; Wang, Y.; Cao, W.; Zheng, J.C. Emerging roles of extracellular vesicles in COVID-19, a double-edged sword? Immunology 2021, 163, 416–430. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.G.; Cao, Y.; Yang, J.; Lee, J.H.; Choi, H.S.; Jin, Y. Lung epithelial cell-derived extracellular vesicles activate macrophage-mediated inflammatory responses via ROCK1 pathway. Cell Death Dis. 2015, 6, e2016. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zhang, D.; Laskin, D.L.; Jin, Y. Functional Evidence of Pulmonary Extracellular Vesicles in Infectious and Noninfectious Lung Inflammation. J. Immunol. 2018, 201, 1500–1509. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, S.; Bihl, J. Exosome-Mediated Transfer of ACE2 (Angiotensin-Converting Enzyme 2) from Endothelial Progenitor Cells Promotes Survival and Function of Endothelial Cell. Oxidative Med. Cell. Longev. 2020, 2020, 4213541. [Google Scholar] [CrossRef]
- Gunasekaran, M.; Bansal, S.; Ravichandran, R.; Sharma, M.; Perincheri, S.; Rodriguez, F.; Hachem, R.; Fisher, C.E.; Limaye, A.P.; Omar, A.; et al. Respiratory viral infection in lung transplantation induces exosomes that trigger chronic rejection. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 2020, 39, 379–388. [Google Scholar] [CrossRef]
- Dwivedi, V.; Yaniv, K.; Sharon, M. Beyond cells: The extracellular circulating 20S proteasomes. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166041. [Google Scholar] [CrossRef]
- Balbi, C.; Burrello, J.; Bolis, S.; Lazzarini, E.; Biemmi, V.; Pianezzi, E.; Burrello, A.; Caporali, E.; Grazioli, L.G.; Martinetti, G.; et al. Circulating extracellular vesicles are endowed with enhanced procoagulant activity in SARS-CoV-2 infection. EBioMedicine 2021, 67, 103369. [Google Scholar] [CrossRef]
- Puhm, F.; Flamand, L.; Boilard, E. Platelet extracellular vesicles in COVID-19: Potential markers and makers. J. Leukoc. Biol. 2022, 111, 63–74. [Google Scholar] [CrossRef]
- Hassanpour, M.; Rezaie, J.; Nouri, M.; Panahi, Y. The role of extracellular vesicles in COVID-19 virus infection. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2020, 85, 104422. [Google Scholar] [CrossRef]
- Fujita, Y.; Kadota, T.; Araya, J.; Ochiya, T.; Kuwano, K. Clinical Application of Mesenchymal Stem Cell-Derived Extracellular Vesicle-Based Therapeutics for Inflammatory Lung Diseases. J. Clin. Med. 2018, 7, 355. [Google Scholar] [CrossRef]
- Morrison, T.J.; Jackson, M.V.; Cunningham, E.K.; Kissenpfennig, A.; McAuley, D.F.; O’Kane, C.M.; Krasnodembskaya, A.D. Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer. Am. J. Respir. Crit. Care Med. 2017, 196, 1275–1286. [Google Scholar] [CrossRef]
- Fergie, N.; Todd, N.; McClements, L.; McAuley, D.; O’Kane, C.; Krasnodembskaya, A. Hypercapnic acidosis induces mitochondrial dysfunction and impairs the ability of mesenchymal stem cells to promote distal lung epithelial repair. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 5585–5598. [Google Scholar] [CrossRef] [PubMed]
- Spees, J.L.; Olson, S.D.; Ylostalo, J.; Lynch, P.J.; Smith, J.; Perry, A.; Peister, A.; Wang, M.Y.; Prockop, D.J. Differentiation, cell fusion, and nuclear fusion during ex vivo repair of epithelium by human adult stem cells from bone marrow stroma. Proc. Natl. Acad. Sci. USA 2003, 100, 2397–2402. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Kumar, M.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [PubMed]
- Dutra Silva, J.; Su, Y.; Calfee, C.S.; Delucchi, K.L.; Weiss, D.; McAuley, D.F.; O’Kane, C.; Krasnodembskaya, A.D. Mesenchymal stromal cell extracellular vesicles rescue mitochondrial dysfunction and improve barrier integrity in clinically relevant models of ARDS. Eur. Respir. J. 2021, 58, 2002978. [Google Scholar] [CrossRef]
- Li, C.; Cheung, M.K.H.; Han, S.; Zhang, Z.; Chen, L.; Chen, J.; Zeng, H.; Qiu, J. Mesenchymal stem cells and their mitochondrial transfer: A double-edged sword. Biosci. Rep. 2019, 39, BSR20182417. [Google Scholar] [CrossRef]
- Li, X.; Michaeloudes, C.; Zhang, Y.; Wiegman, C.H.; Adcock, I.M.; Lian, Q.; Mak, J.C.W.; Bhavsar, P.K.; Chung, K.F. Mesenchymal stem cells alleviate oxidative stress-induced mitochondrial dysfunction in the airways. J. Allergy Clin. Immunol. 2018, 141, 1634–1645.e5. [Google Scholar] [CrossRef]
- Tan, Y.L.; Eng, S.P.; Hafez, P.; Abdul Karim, N.; Law, J.X.; Ng, M.H. Mesenchymal Stromal Cell Mitochondrial Transfer as a Cell Rescue Strategy in Regenerative Medicine: A Review of Evidence in Preclinical Models. Stem Cells Transl. Med. 2022, 11, 814–827. [Google Scholar] [CrossRef]
- Buzas, E.I.; Gyorgy, B.; Nagy, G.; Falus, A.; Gay, S. Emerging role of extracellular vesicles in inflammatory diseases. Nat. Rev. Rheumatol. 2014, 10, 356–364. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Morciano, G.; Rimessi, A.; Patergnani, S.; Vitto, V.A.M.; Danese, A.; Kahsay, A.; Palumbo, L.; Bonora, M.; Wieckowski, M.R.; Giorgi, C.; et al. Calcium dysregulation in heart diseases: Targeting calcium channels to achieve a correct calcium homeostasis. Pharmacol. Res. 2022, 177, 106119. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, G.; Santoso, M.R.; Tada, Y.; Li, A.M.; Vaskova, E.; Jung, J.H.; O’Brien, C.; Egan, E.; Ye, J.; Yang, P.C. Mitochondria-Rich Extracellular Vesicles From Autologous Stem Cell-Derived Cardiomyocytes Restore Energetics of Ischemic Myocardium. J. Am. Coll. Cardiol. 2021, 77, 1073–1088. [Google Scholar] [CrossRef] [PubMed]
- Puhm, F.; Afonyushkin, T.; Resch, U.; Obermayer, G.; Rohde, M.; Penz, T.; Schuster, M.; Wagner, G.; Rendeiro, A.F.; Melki, I.; et al. Mitochondria Are a Subset of Extracellular Vesicles Released by Activated Monocytes and Induce Type I IFN and TNF Responses in Endothelial Cells. Circ. Res. 2019, 125, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.G.; Williams, J.C.; Davis, B.K.; Jacobson, K.; Doerschuk, C.M.; Ting, J.P.; Mackman, N. Monocytic microparticles activate endothelial cells in an IL-1beta-dependent manner. Blood 2011, 118, 2366–2374. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Vitto, V.A.M.; Bouhamida, E.; Giorgi, C.; Pinton, P. Mitochondrial Bioenergetics and Dynamism in the Failing Heart. Life 2021, 11, 436. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, D.; Biswas, B.; Manhas, A.; Singh, A.; Goyal, D.; Gaestel, M.; Jagavelu, K. Proinflammatory Effect of Endothelial Microparticles Is Mitochondria Mediated and Modulated Through MAPKAPK2 (MAPK-Activated Protein Kinase 2) Leading to Attenuation of Cardiac Hypertrophy. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1100–1112. [Google Scholar] [CrossRef] [PubMed]
- Pedriali, G.; Morciano, G.; Patergnani, S.; Cimaglia, P.; Morelli, C.; Mikus, E.; Ferrari, R.; Gasbarro, V.; Giorgi, C.; Wieckowski, M.R.; et al. Aortic Valve Stenosis and Mitochondrial Dysfunctions: Clinical and Molecular Perspectives. Int. J. Mol. Sci. 2020, 21, 4899. [Google Scholar] [CrossRef]
- Leroyer, A.S.; Isobe, H.; Leseche, G.; Castier, Y.; Wassef, M.; Mallat, Z.; Binder, B.R.; Tedgui, A.; Boulanger, C.M. Cellular origins and thrombogenic activity of microparticles isolated from human atherosclerotic plaques. J. Am. Coll. Cardiol. 2007, 49, 772–777. [Google Scholar] [CrossRef]
- Huber, J.; Vales, A.; Mitulovic, G.; Blumer, M.; Schmid, R.; Witztum, J.L.; Binder, B.R.; Leitinger, N. Oxidized membrane vesicles and blebs from apoptotic cells contain biologically active oxidized phospholipids that induce monocyte-endothelial interactions. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 101–107. [Google Scholar] [CrossRef]
- Buffolo, F.; Monticone, S.; Camussi, G.; Aikawa, E. Role of Extracellular Vesicles in the Pathogenesis of Vascular Damage. Hypertension 2022, 79, 863–873. [Google Scholar] [CrossRef]
- Rautou, P.E.; Leroyer, A.S.; Ramkhelawon, B.; Devue, C.; Duflaut, D.; Vion, A.C.; Nalbone, G.; Castier, Y.; Leseche, G.; Lehoux, S.; et al. Microparticles from human atherosclerotic plaques promote endothelial ICAM-1-dependent monocyte adhesion and transendothelial migration. Circ. Res. 2011, 108, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Gomez, I.; Ward, B.; Souilhol, C.; Recarti, C.; Ariaans, M.; Johnston, J.; Burnett, A.; Mahmoud, M.; Luong, L.A.; West, L.; et al. Neutrophil microvesicles drive atherosclerosis by delivering miR-155 to atheroprone endothelium. Nat. Commun. 2020, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Wadey, R.M.; Connolly, K.D.; Mathew, D.; Walters, G.; Rees, D.A.; James, P.E. Inflammatory adipocyte-derived extracellular vesicles promote leukocyte attachment to vascular endothelial cells. Atherosclerosis 2019, 283, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Kuravi, S.J.; Harrison, P.; Rainger, G.E.; Nash, G.B. Ability of Platelet-Derived Extracellular Vesicles to Promote Neutrophil-Endothelial Cell Interactions. Inflammation 2019, 42, 290–305. [Google Scholar] [CrossRef]
- Chimen, M.; Evryviadou, A.; Box, C.L.; Harrison, M.J.; Hazeldine, J.; Dib, L.H.; Kuravi, S.J.; Payne, H.; Price, J.M.J.; Kavanagh, D.; et al. Appropriation of GPIbalpha from platelet-derived extracellular vesicles supports monocyte recruitment in systemic inflammation. Haematologica 2020, 105, 1248–1261. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Chen, Q.; Fan, M.; Guo, J.; Liu, Y.; Ji, T.; Zhu, J.; Zhao, X. Platelet-derived microparticles promote phagocytosis of oxidized low-density lipoprotein by macrophages, potentially enhancing foam cell formation. Ann. Transl. Med. 2019, 7, 477. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Wang, X.; Liu, X.; Du, H.; Sun, C.; Shao, X.; Tian, J.; Gu, X.; Wang, H.; Tian, J.; et al. Adipose-Derived Exosomes Exert Proatherogenic Effects by Regulating Macrophage Foam Cell Formation and Polarization. J. Am. Heart Assoc. 2018, 7, e007442. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Han, J.; Wu, Y.; Li, S.; Wang, Q.; Lin, W.; Zhu, J. Exosomal MALAT1 derived from oxidized low-density lipoprotein-treated endothelial cells promotes M2 macrophage polarization. Mol. Med. Rep. 2018, 18, 509–515. [Google Scholar] [CrossRef]
- Morciano, G.; Patergnani, S.; Pedriali, G.; Cimaglia, P.; Mikus, E.; Calvi, S.; Albertini, A.; Giorgi, C.; Campo, G.; Ferrari, R.; et al. Impairment of mitophagy and autophagy accompanies calcific aortic valve stenosis favouring cell death and the severity of disease. Cardiovasc. Res. 2022, 118, 2548–2559. [Google Scholar] [CrossRef]
- Gardin, C.; Morciano, G.; Ferroni, L.; Mikus, E.; Tripodi, A.; Pin, M.; Tremoli, E.; Albertini, A.; Zavan, B. Biological Characterization of Human Autologous Pericardium Treated with the Ozaki Procedure for Aortic Valve Reconstruction. J. Clin. Med. 2021, 10, 3954. [Google Scholar] [CrossRef]
- Kawakami, R.; Katsuki, S.; Travers, R.; Romero, D.C.; Becker-Greene, D.; Passos, L.S.A.; Higashi, H.; Blaser, M.C.; Sukhova, G.K.; Buttigieg, J.; et al. S100A9-RAGE Axis Accelerates Formation of Macrophage-Mediated Extracellular Vesicle Microcalcification in Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1838–1853. [Google Scholar] [CrossRef] [PubMed]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [PubMed]
- Jetto, C.T.; Nambiar, A.; Manjithaya, R. Mitophagy and Neurodegeneration: Between the Knowns and the Unknowns. Front. Cell Dev. Biol. 2022, 10, 837337. [Google Scholar] [PubMed]
- Grunewald, A.; Rygiel, K.A.; Hepplewhite, P.D.; Morris, C.M.; Picard, M.; Turnbull, D.M. Mitochondrial DNA Depletion in Respiratory Chain-Deficient Parkinson Disease Neurons. Ann. Neurol. 2016, 79, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Podlesniy, P.; Figueiro-Silva, J.; Llado, A.; Antonell, A.; Sanchez-Valle, R.; Alcolea, D.; Lleo, A.; Molinuevo, J.L.; Serra, N.; Trullas, R. Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann. Neurol. 2013, 74, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Renz, A.; Berdel, W.E.; Kreuter, M.; Belka, C.; Schulze-Osthoff, K.; Los, M. Rapid extracellular release of cytochrome c is specific for apoptosis and marks cell death in vivo. Blood 2001, 98, 1542–1548. [Google Scholar] [CrossRef]
- Pullerits, R.; Bokarewa, M.; Jonsson, I.M.; Verdrengh, M.; Tarkowski, A. Extracellular cytochrome c, a mitochondrial apoptosis-related protein, induces arthritis. Rheumatology 2005, 44, 32–39. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Bajwa, E.; Klegeris, A. Cytochrome c can be released into extracellular space and modulate functions of human astrocytes in a toll-like receptor 4-dependent manner. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 129400. [Google Scholar]
- Pointer, C.B.; Wenzel, T.J.; Klegeris, A. Extracellular cardiolipin regulates select immune functions of microglia and microglia-like cells. Brain Res. Bull. 2019, 146, 153–163. [Google Scholar]
- Deus, C.M.; Tavares, H.; Beatriz, M.; Mota, S.; Lopes, C. Mitochondrial Damage-Associated Molecular Patterns Content in Extracellular Vesicles Promotes Early Inflammation in Neurodegenerative Disorders. Cells 2022, 11, 2364. [Google Scholar] [CrossRef]
- Beatriz, M.; Vilaça, R.; Anjo, S.I.; Manadas, B.; Januário, C.; Rego, A.C.; Lopes, C. Defective mitochondria-lysosomal axis enhances the release of extracellular vesicles containing mitochondrial DNA and proteins in Huntington’s disease. J. Extracell. Biol. 2022, 1, e65. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial-Derived Vesicles as Candidate Biomarkers in Parkinson’s Disease: Rationale, Design and Methods of the EXosomes in PArkiNson Disease (EXPAND) Study. Int. J. Mol. Sci. 2019, 20, 2373. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Guerra, F.; Calvani, R.; Marini, F.; Biancolillo, A.; Landi, G.; Beli, R.; Landi, F.; Bernabei, R.; Bentivoglio, A.R.; et al. Mitochondrial Signatures in Circulating Extracellular Vesicles of Older Adults with Parkinson’s Disease: Results from the EXosomes in PArkiNson’s Disease (EXPAND) Study. J. Clin. Med. 2020, 9, 504. [Google Scholar] [CrossRef] [PubMed]
- Pyle, A.; Brennan, R.; Kurzawa-Akanbi, M.; Yarnall, A.; Thouin, A.; Mollenhauer, B.; Burn, D.; Chinnery, P.F.; Hudson, G. Reduced cerebrospinal fluid mitochondrial DNA is a biomarker for early-stage Parkinson’s disease. Ann. Neurol. 2015, 78, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Lowes, H.; Pyle, A.; Santibanez-Koref, M.; Hudson, G. Circulating cell-free mitochondrial DNA levels in Parkinson’s disease are influenced by treatment. Mol. Neurodegener. 2020, 15, 10. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.E.; Burelle, Y.; et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef]
- McLelland, G.L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef]
- Torralba, D.; Baixauli, F.; Sanchez-Madrid, F. Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front. Cell Dev. Biol. 2016, 4, 107. [Google Scholar]
- Patergnani, S.; Fossati, V.; Bonora, M.; Giorgi, C.; Marchi, S.; Missiroli, S.; Rusielewicz, T.; Wieckowski, M.R.; Pinton, P. Mitochondria in Multiple Sclerosis: Molecular Mechanisms of Pathogenesis. Int. Rev. Cell Mol. Biol. 2017, 328, 49–103. [Google Scholar]
- Giorgi, C.; Bouhamida, E.; Danese, A.; Previati, M.; Pinton, P.; Patergnani, S. Relevance of Autophagy and Mitophagy Dynamics and Markers in Neurodegenerative Diseases. Biomedicines 2021, 9, 149. [Google Scholar] [PubMed]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Fainardi, E.; Casetta, I.; Granieri, E.; Pugliatti, M.; Pinton, P. Correlation between auto/mitophagic processes and magnetic resonance imaging activity in multiple sclerosis patients. J. Neuroinflamm. 2019, 16, 131. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Bonora, M.; Ingusci, S.; Previati, M.; Marchi, S.; Zucchini, S.; Perrone, M.; Wieckowski, M.R.; Castellazzi, M.; Pugliatti, M.; et al. Antipsychotic drugs counteract autophagy and mitophagy in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2020078118. [Google Scholar] [CrossRef] [PubMed]
- Ladakis, D.C.; Yao, P.J.; Vreones, M.; Blommer, J.; Kalaitzidis, G.; Sotirchos, E.S.; Fitzgerald, K.C.; Saidha, S.; Calabresi, P.A.; Kapogiannis, D.; et al. Mitochondrial measures in neuronally enriched extracellular vesicles predict brain and retinal atrophy in multiple sclerosis. Mult. Scler. 2022, 28, 2020–2026. [Google Scholar] [CrossRef]
- Villar-Vesga, J.; Henao-Restrepo, J.; Voshart, D.C.; Aguillon, D.; Villegas, A.; Castano, D.; Arias-Londono, J.D.; Zuhorn, I.S.; Ribovski, L.; Barazzuol, L.; et al. Differential Profile of Systemic Extracellular Vesicles From Sporadic and Familial Alzheimer’s Disease Leads to Neuroglial and Endothelial Cell Degeneration. Front. Aging Neurosci. 2020, 12, 587989. [Google Scholar]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Bosi, C.; Brombo, G.; Pugliatti, M.; Seripa, D.; Zuliani, G.; et al. Autophagy and mitophagy biomarkers are reduced in sera of patients with Alzheimer’s disease and mild cognitive impairment. Sci. Rep. 2019, 9, 20009. [Google Scholar] [CrossRef]
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Kim, K.M.; Meng, Q.; Perez de Acha, O.; Mustapic, M.; Cheng, A.; Eren, E.; Kundu, G.; Piao, Y.; Munk, R.; Wood, W.H., 3rd; et al. Mitochondrial RNA in Alzheimer’s Disease Circulating Extracellular Vesicles. Front. Cell Dev. Biol. 2020, 8, 581882. [Google Scholar] [CrossRef]
- Tsilioni, I.; Theoharides, T.C. Extracellular vesicles are increased in the serum of children with autism spectrum disorder, contain mitochondrial DNA, and stimulate human microglia to secrete IL-1beta. J. Neuroinflamm. 2018, 15, 239. [Google Scholar] [CrossRef]
- Danese, A.; Patergnani, S.; Maresca, A.; Peron, C.; Raimondi, A.; Caporali, L.; Marchi, S.; La Morgia, C.; Del Dotto, V.; Zanna, C.; et al. Pathological mitophagy disrupts mitochondrial homeostasis in Leber’s hereditary optic neuropathy. Cell Rep. 2022, 40, 111124. [Google Scholar] [PubMed]
- Subramaniam, M.D.; Chirayath, R.B.; Iyer, M.; Nair, A.P.; Vellingiri, B. Mesenchymal stem cells (MSCs) in Leber’s hereditary optic neuropathy (LHON): A potential therapeutic approach for future. Int. Ophthalmol. 2022, 42, 2949–2964. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [PubMed]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef]
- Chen, F.; Chen, J.; Yang, L.; Liu, J.; Zhang, X.; Zhang, Y.; Tu, Q.; Yin, D.; Lin, D.; Wong, P.P.; et al. Extracellular vesicle-packaged HIF-1alpha-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat. Cell Biol. 2019, 21, 498–510. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar]
- Wang, W.; Zhu, N.; Yan, T.; Shi, Y.N.; Chen, J.; Zhang, C.J.; Xie, X.J.; Liao, D.F.; Qin, L. The crosstalk: Exosomes and lipid metabolism. Cell Commun. Signal. CCS 2020, 18, 119. [Google Scholar]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 2016, 5, e10250. [Google Scholar]
- Clement, E.; Lazar, I.; Attane, C.; Carrie, L.; Dauvillier, S.; Ducoux-Petit, M.; Esteve, D.; Menneteau, T.; Moutahir, M.; Le Gonidec, S.; et al. Adipocyte extracellular vesicles carry enzymes and fatty acids that stimulate mitochondrial metabolism and remodeling in tumor cells. EMBO J. 2020, 39, e102525. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Xia, T.; Konno, H.; Konno, K.; Ruiz, P.; Barber, G.N. Inflammation-driven carcinogenesis is mediated through STING. Nat. Commun. 2014, 5, 5166. [Google Scholar] [CrossRef]
- Ahn, J.; Konno, H.; Barber, G.N. Diverse roles of STING-dependent signaling on the development of cancer. Oncogene 2015, 34, 5302–5308. [Google Scholar] [PubMed]
- Brokatzky, D.; Dorflinger, B.; Haimovici, A.; Weber, A.; Kirschnek, S.; Vier, J.; Metz, A.; Henschel, J.; Steinfeldt, T.; Gentle, I.E.; et al. A non-death function of the mitochondrial apoptosis apparatus in immunity. EMBO J. 2019, 38, e100907. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar]
- Yamazaki, T.; Kirchmair, A.; Sato, A.; Buque, A.; Rybstein, M.; Petroni, G.; Bloy, N.; Finotello, F.; Stafford, L.; Navarro Manzano, E.; et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat. Immunol. 2020, 21, 1160–1171. [Google Scholar] [PubMed]
- Han, C.; Liu, Z.; Zhang, Y.; Shen, A.; Dong, C.; Zhang, A.; Moore, C.; Ren, Z.; Lu, C.; Cao, X.; et al. Tumor cells suppress radiation-induced immunity by hijacking caspase 9 signaling. Nat. Immunol. 2020, 21, 546–554. [Google Scholar] [CrossRef]
- Rodriguez-Ruiz, M.E.; Buque, A.; Hensler, M.; Chen, J.; Bloy, N.; Petroni, G.; Sato, A.; Yamazaki, T.; Fucikova, J.; Galluzzi, L. Apoptotic caspases inhibit abscopal responses to radiation and identify a new prognostic biomarker for breast cancer patients. Oncoimmunology 2019, 8, e1655964. [Google Scholar] [CrossRef]
- Cosentino, K.; Hertlein, V.; Jenner, A.; Dellmann, T.; Gojkovic, M.; Pena-Blanco, A.; Dadsena, S.; Wajngarten, N.; Danial, J.S.H.; Thevathasan, J.V.; et al. The interplay between BAX and BAK tunes apoptotic pore growth to control mitochondrial-DNA-mediated inflammation. Mol. Cell 2022, 82, 933–949 e9. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Singel, K.L.; Grzankowski, K.S.; Khan, A.; Grimm, M.J.; D’Auria, A.C.; Morrell, K.; Eng, K.H.; Hylander, B.; Mayor, P.C.; Emmons, T.R.; et al. Mitochondrial DNA in the tumour microenvironment activates neutrophils and is associated with worse outcomes in patients with advanced epithelial ovarian cancer. Br. J. Cancer 2019, 120, 207–217. [Google Scholar] [PubMed]
- Keshari, R.S.; Jyoti, A.; Kumar, S.; Dubey, M.; Verma, A.; Srinag, B.S.; Krishnamurthy, H.; Barthwal, M.K.; Dikshit, M. Neutrophil extracellular traps contain mitochondrial as well as nuclear DNA and exhibit inflammatory potential. Cytometry. Part A J. Int. Soc. Anal. Cytol. 2012, 81, 238–247. [Google Scholar]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef]
- Itagaki, K.; Kaczmarek, E.; Lee, Y.T.; Tang, I.T.; Isal, B.; Adibnia, Y.; Sandler, N.; Grimm, M.J.; Segal, B.H.; Otterbein, L.E.; et al. Mitochondrial DNA released by trauma induces neutrophil extracellular traps. PLoS ONE 2015, 10, e0120549. [Google Scholar] [CrossRef]
- Yang, C.; Wang, Z.; Li, L.; Zhang, Z.; Jin, X.; Wu, P.; Sun, S.; Pan, J.; Su, K.; Jia, F.; et al. Aged neutrophils form mitochondria-dependent vital NETs to promote breast cancer lung metastasis. J. Immunother. Cancer 2021, 9, e002875. [Google Scholar] [CrossRef]
- Xu, M.M.; Pu, Y.; Han, D.; Shi, Y.; Cao, X.; Liang, H.; Chen, X.; Li, X.D.; Deng, L.; Chen, Z.J.; et al. Dendritic Cells but Not Macrophages Sense Tumor Mitochondrial DNA for Cross-priming through Signal Regulatory Protein alpha Signaling. Immunity 2017, 47, 363–373.e5. [Google Scholar] [CrossRef]
- Pazmandi, K.; Agod, Z.; Kumar, B.V.; Szabo, A.; Fekete, T.; Sogor, V.; Veres, A.; Boldogh, I.; Rajnavolgyi, E.; Lanyi, A.; et al. Oxidative modification enhances the immunostimulatory effects of extracellular mitochondrial DNA on plasmacytoid dendritic cells. Free Radic. Biol. Med. 2014, 77, 281–290. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Achreja, A.; Iessi, E.; Logozzi, M.; Mizzoni, D.; Di Raimo, R.; Nagrath, D.; Fais, S. The key role of extracellular vesicles in the metastatic process. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Cassim, S.; Pouyssegur, J. Tumor Microenvironment: A Metabolic Player that Shapes the Immune Response. Int. J. Mol. Sci. 2019, 21, 157. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Zhou, X.; Fang, M.; Li, H.; Su, P.; Tu, Y.; Zhang, L.; Zhou, F. Extracellular Vesicles in Cancer Immune Microenvironment and Cancer Immunotherapy. Adv. Sci. 2019, 6, 1901779. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Mambro, T.; Pellielo, G.; Agyapong, E.D.; Carinci, M.; Chianese, D.; Giorgi, C.; Morciano, G.; Patergnani, S.; Pinton, P.; Rimessi, A. The Tricky Connection between Extracellular Vesicles and Mitochondria in Inflammatory-Related Diseases. Int. J. Mol. Sci. 2023, 24, 8181. https://doi.org/10.3390/ijms24098181
Di Mambro T, Pellielo G, Agyapong ED, Carinci M, Chianese D, Giorgi C, Morciano G, Patergnani S, Pinton P, Rimessi A. The Tricky Connection between Extracellular Vesicles and Mitochondria in Inflammatory-Related Diseases. International Journal of Molecular Sciences. 2023; 24(9):8181. https://doi.org/10.3390/ijms24098181
Chicago/Turabian StyleDi Mambro, Tommaso, Giulia Pellielo, Esther Densu Agyapong, Marianna Carinci, Diego Chianese, Carlotta Giorgi, Giampaolo Morciano, Simone Patergnani, Paolo Pinton, and Alessandro Rimessi. 2023. "The Tricky Connection between Extracellular Vesicles and Mitochondria in Inflammatory-Related Diseases" International Journal of Molecular Sciences 24, no. 9: 8181. https://doi.org/10.3390/ijms24098181
APA StyleDi Mambro, T., Pellielo, G., Agyapong, E. D., Carinci, M., Chianese, D., Giorgi, C., Morciano, G., Patergnani, S., Pinton, P., & Rimessi, A. (2023). The Tricky Connection between Extracellular Vesicles and Mitochondria in Inflammatory-Related Diseases. International Journal of Molecular Sciences, 24(9), 8181. https://doi.org/10.3390/ijms24098181