
Molecular Mechanisms of AMPA Receptor Trafficking in the Nervous System


Abstract
:1. Introduction
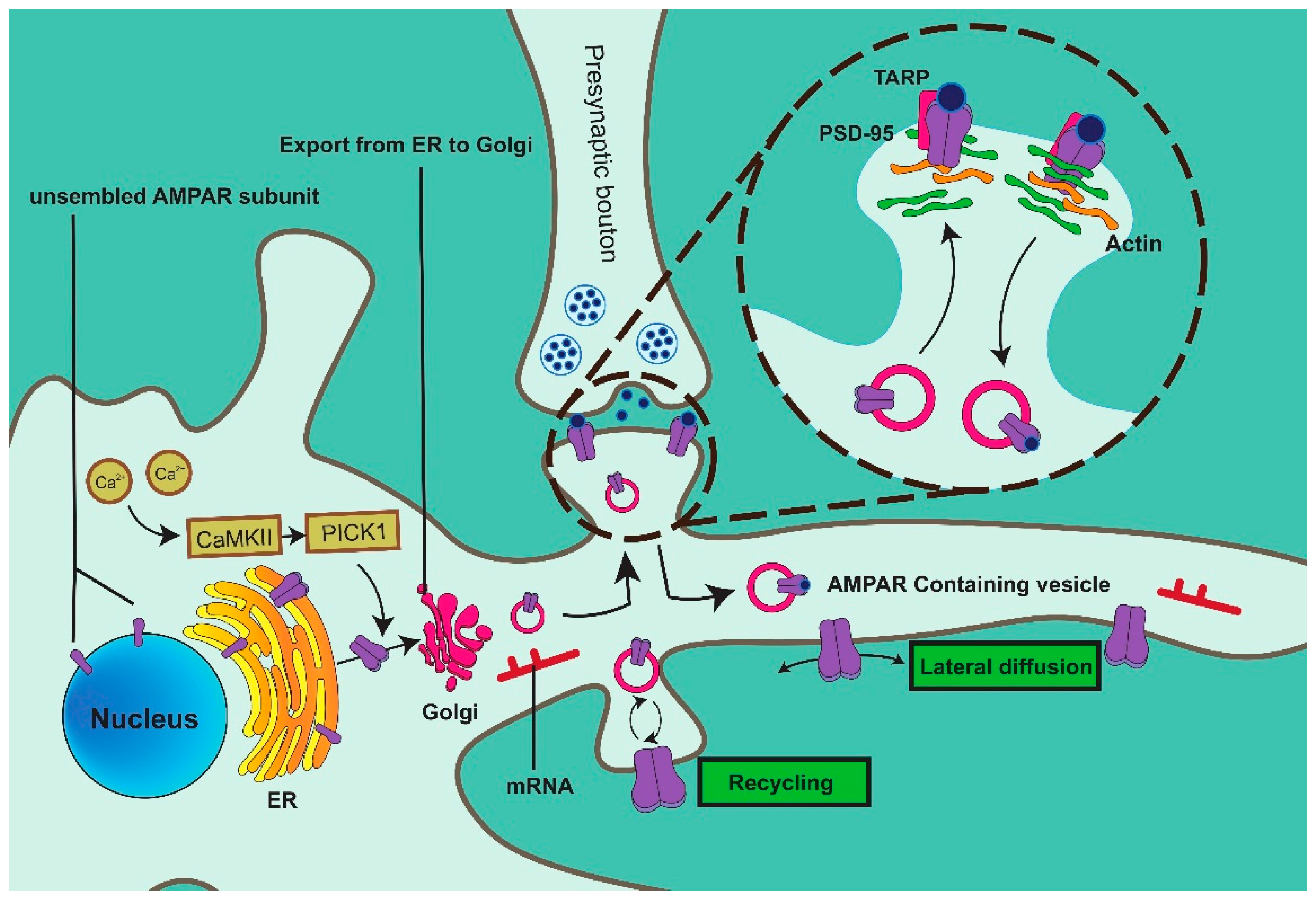
2. AMPAR Vesicle Formation
2.1. Rab11
2.2. CaMKII
3. AMPAR Vesicle Trafficking
3.1. MAP
3.2. Actin
4. AMPAR Anchoring at Postsynaptic Sites
4.1. Stargazin
4.2. SAP-97
4.3. PKA
4.4. PKC and GRIP 1/2
4.5. EPH41L1
5. AMPAR Vesicle Fusion
5.1. SNARE Complex
5.2. AP-2
6. The Actin Cytoskeleton That Facilitates AMPAR Trafficking
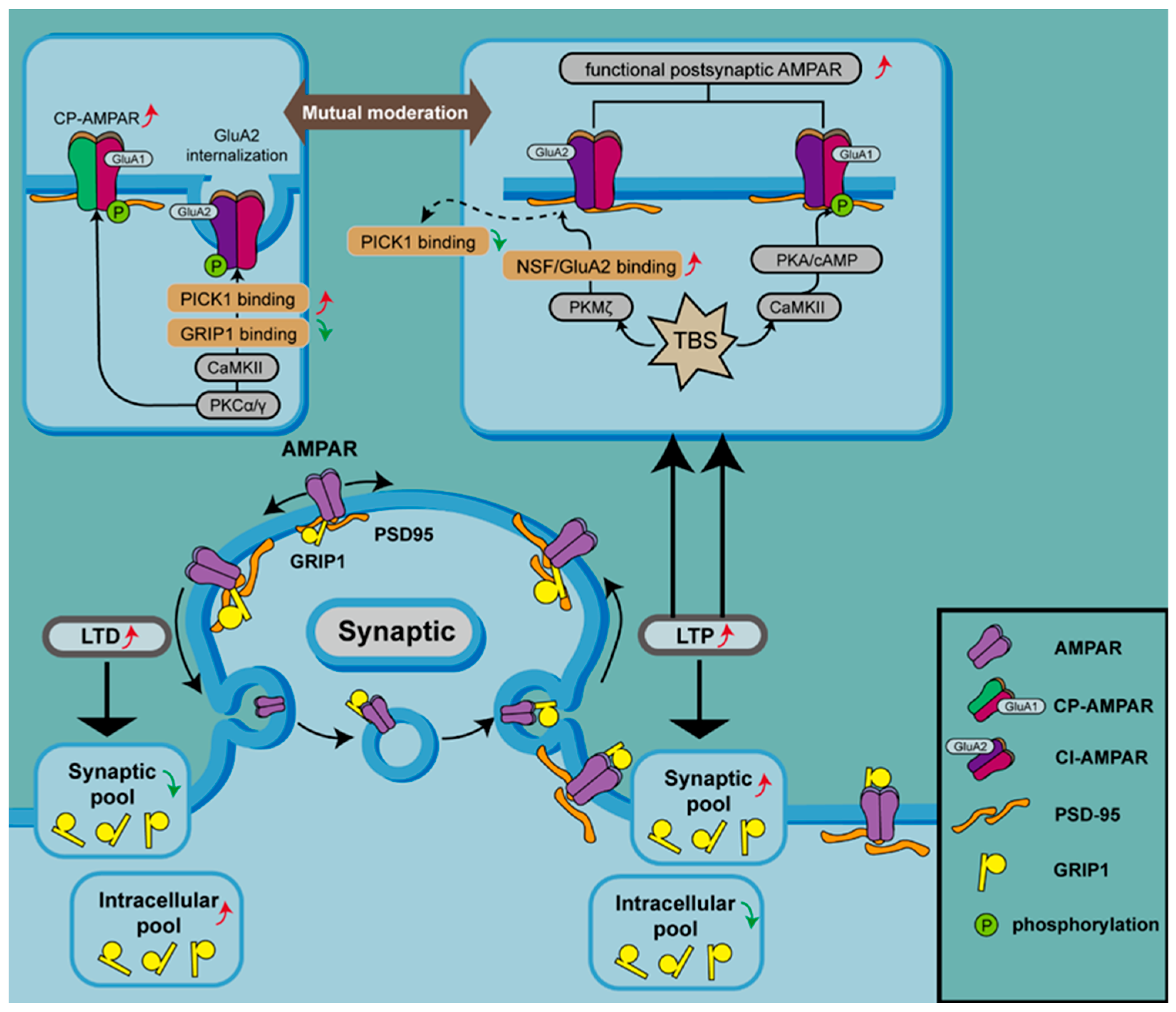
7. AMPAR Trafficking in Neurological Diseases
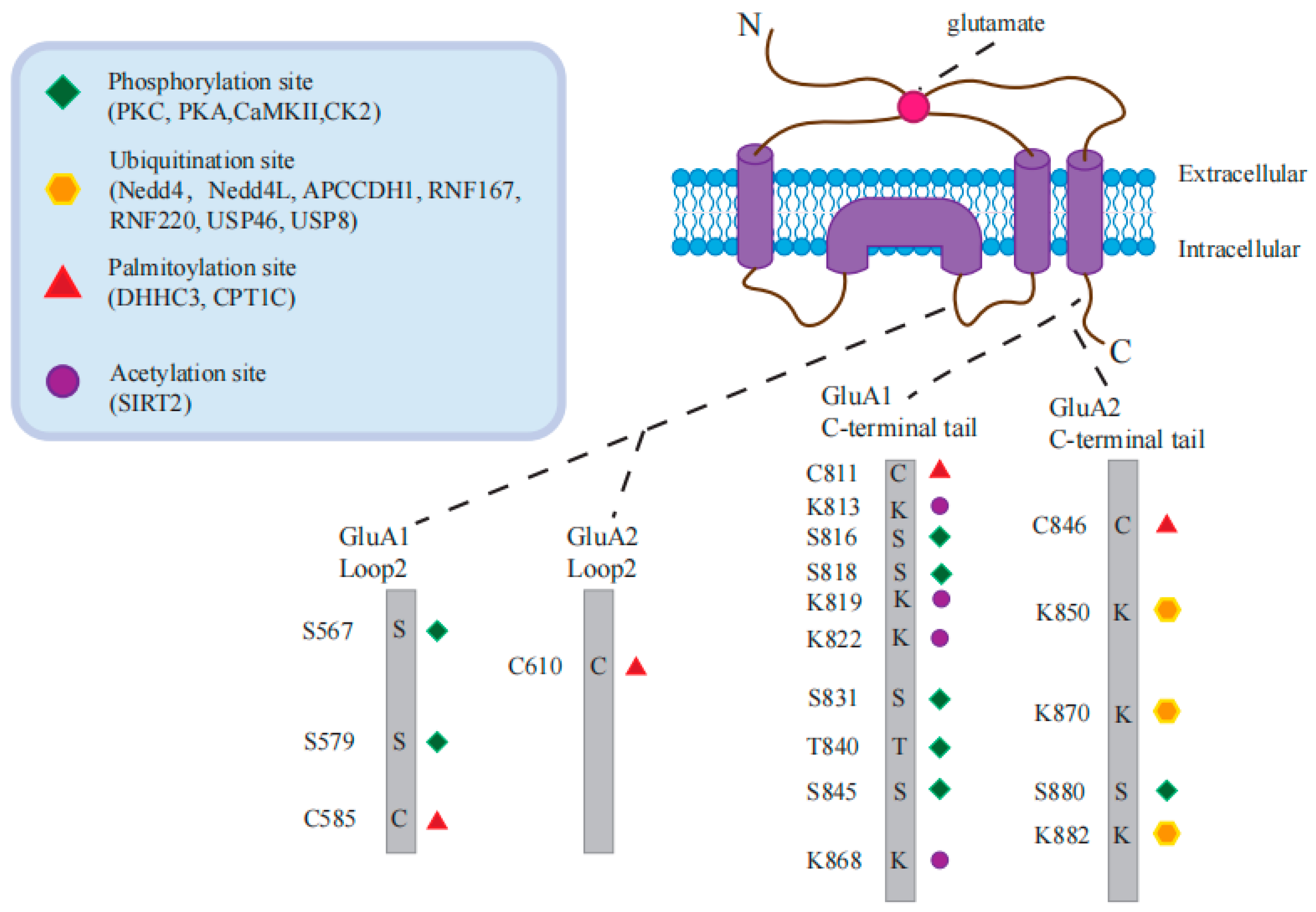
8. AMPARs PTM
8.1. Phosphorylation in AMPAR Trafficking
8.2. Ubiquitination in AMPAR Trafficking
8.3. Palmitoylation in AMPAR Trafficking
8.4. Acetylation in AMPAR Trafficking
8.5. Crosstalk between AMPAR PTMs
9. AMPAR PTMs in Neurological Diseases
10. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Wang, C.S.; Kavalali, E.T.; Monteggia, L.M. BDNF signaling in context: From synaptic regulation to psychiatric disorders. Cell 2022, 185, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Heine, M.; Groc, L.; Frischknecht, R.; Béïque, J.C.; Lounis, B.; Rumbaugh, G.; Huganir, R.L.; Cognet, L.; Choquet, D. Surface mobility of postsynaptic AMPARs tunes synaptic transmission. Science 2008, 320, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Penn, A.C.; Zhang, C.L.; Georges, F.; Royer, L.; Breillat, C.; Hosy, E.; Petersen, J.D.; Humeau, Y.; Choquet, D. Hippocampal LTP and contextual learning require surface diffusion of AMPA receptors. Nature 2017, 549, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Diering, G.H.; Huganir, R.L. The AMPA Receptor Code of Synaptic Plasticity. Neuron 2018, 100, 314–329. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Rao, P.; Clark, S.; Mitra, J.; Ha, T.; Gouaux, E. Hippocampal AMPA receptor assemblies and mechanism of allosteric inhibition. Nature 2021, 594, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Choquet, D.; Hosy, E. AMPA receptor nanoscale dynamic organization and synaptic plasticities. Curr. Opin. Neurobiol. 2020, 63, 137–145. [Google Scholar] [CrossRef]
- Chen, M.W.; Zhu, H.; Xiong, C.H.; Li, J.B.; Zhao, L.X.; Chen, H.Z.; Qiu, Y. PKC and Ras are Involved in M1 Muscarinic Receptor-Mediated Modulation of AMPA Receptor GluA1 Subunit. Cell. Mol. Neurobiol. 2020, 40, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, L.; Regoni, M.; Ratti, E.; Valtorta, F.; Sassone, J. Presynaptic AMPA Receptors in Health and Disease. Cells 2021, 10, 2260. [Google Scholar] [CrossRef]
- Huganir, R.L.; Nicoll, R.A. AMPARs and synaptic plasticity: The last 25 years. Neuron 2013, 80, 704–717. [Google Scholar] [CrossRef]
- Roth, R.H.; Cudmore, R.H.; Tan, H.L.; Hong, I.; Zhang, Y.; Huganir, R.L. Cortical Synaptic AMPA Receptor Plasticity during Motor Learning. Neuron 2020, 105, 895–908.e5. [Google Scholar] [CrossRef]
- Guo, C.; Wen, D.; Zhang, Y.; Mustaklem, R.; Mustaklem, B.; Zhou, M.; Ma, T.; Ma, Y.Y. Amyloid-beta oligomers in the nucleus accumbens decrease motivation via insertion of calcium-permeable AMPA receptors. Mol. Psychiatry 2022, 27, 2146–2157. [Google Scholar] [CrossRef] [PubMed]
- Salpietro, V.; SYNAPS Study Group; Dixon, C.L.; Guo, H.; Bello, O.D.; Vandrovcova, J.; Efthymiou, S.; Maroofian, R.; Heimer, G.; Burglen, L.; et al. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat. Commun. 2019, 10, 3094. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Vazquez, H.; Roach, R.L.; Nip, K.; Chanda, S.; Sathler, M.F.; Garver, T.; Danzman, R.A.; Moseley, M.C.; Roberts, J.P.; Koch, O.N.; et al. The autism-associated loss of delta-catenin functions disrupts social behavior. Proc. Natl. Acad. Sci. USA 2023, 120, e2300773120. [Google Scholar] [CrossRef] [PubMed]
- Brebner, K.; Wong, T.P.; Liu, L.; Liu, Y.; Campsall, P.; Gray, S.; Phelps, L.; Phillips, A.G.; Wang, Y.T. Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science 2005, 310, 1340–1343. [Google Scholar] [CrossRef] [PubMed]
- Dewar, D.; Chalmers, D.T.; Graham, D.I.; McCulloch, J. Glutamate metabotropic and AMPA binding sites are reduced in Alzheimer’s disease: An autoradiographic study of the hippocampus. Brain Res. 1991, 553, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Baglietto-Vargas, D.; Prieto, G.A.; Limon, A.; Forner, S.; Rodriguez-Ortiz, C.J.; Ikemura, K.; Ager, R.R.; Medeiros, R.; Trujillo-Estrada, L.; Martini, A.C.; et al. Impaired AMPA signaling and cytoskeletal alterations induce early synaptic dysfunction in a mouse model of Alzheimer’s disease. Aging Cell 2018, 17, e12791. [Google Scholar] [CrossRef] [PubMed]
- Hettinger, J.C.; Lee, H.; Bu, G.; Holtzman, D.M.; Cirrito, J.R. AMPA-ergic regulation of amyloid-beta levels in an Alzheimer’s disease mouse model. Mol. Neurodegener. 2018, 13, 22. [Google Scholar] [CrossRef]
- Eiro, T.; Miyazaki, T.; Hatano, M.; Nakajima, W.; Arisawa, T.; Takada, Y.; Kimura, K.; Sano, A.; Nakano, K.; Mihara, T.; et al. Dynamics of AMPA receptors regulate epileptogenesis in patients with epilepsy. Cell Rep. Med. 2023, 4, 101020. [Google Scholar] [CrossRef]
- Henley, J.M.; Nair, J.D.; Seager, R.; Yucel, B.P.; Woodhall, G.; Henley, B.S.; Talandyte, K.; Needs, H.I.; Wilkinson, K.A. Kainate and AMPA receptors in epilepsy: Cell biology, signalling pathways and possible crosstalk. Neuropharmacology 2021, 195, 108569. [Google Scholar] [CrossRef]
- Zhu, J.; Yang, Y.; Ma, W.; Wang, Y.; Chen, L.; Xiong, H.; Yin, C.; He, Z.; Fu, W.; Xu, R.; et al. Antiepilepticus Effects of Tetrahedral Framework Nucleic Acid via Inhibition of Gliosis-Induced Downregulation of Glutamine Synthetase and Increased AMPAR Internalization in the Postsynaptic Membrane. Nano Lett. 2022, 22, 2381–2390. [Google Scholar] [CrossRef]
- Hanada, T. Ionotropic Glutamate Receptors in Epilepsy: A Review Focusing on AMPA and NMDA Receptors. Biomolecules 2020, 10, 464. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrova, L.R.; Phillips, A.G.; Wang, Y.T. Antidepressant effects of ketamine and the roles of AMPA glutamate receptors and other mechanisms beyond NMDA receptor antagonism. J. Psychiatry Neurosci. 2017, 42, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Akinfiresoye, L.; Tizabi, Y. Antidepressant effects of AMPA and ketamine combination: Role of hippocampal BDNF, synapsin, and mTOR. Psychopharmacology 2013, 230, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Horgan, C.P.; McCaffrey, W.M. The dynamic Rab11-FIPs. Biochem. Soc. Trans. 2009, 37, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- da Silva, M.E.; Adrian, M.; Schätzle, P.; Lipka, J.; Watanabe, T.; Cho, S.; Futai, K.; Wierenga, C.J.; Kapitein, L.C.; Hoogenraad, C.C. Positioning of AMPA Receptor-Containing Endosomes Regulates Synapse Architecture. Cell Rep. 2015, 13, 933–943. [Google Scholar]
- Zhang, J.; Su, G.; Wu, Q.; Liu, J.; Tian, Y.; Liu, X.; Zhou, J.; Gao, J.; Chen, W.; Chen, D.; et al. Rab11-mediated recycling endosome role in nervous system development and neurodegenerative diseases. Int. J. Neurosci. 2021, 131, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Bhuin, T.; Roy, J.K. Rab11 in Disease Progression. Int. J. Mol. Cell Med. 2015, 4, 1–8. [Google Scholar]
- Bacaj, T.; Ahmad, M.; Jurado, S.; Malenka, R.C.; Südhof, T.C. Synaptic Function of Rab11Fip5: Selective Requirement for Hippocampal Long-Term Depression. J. Neurosci. 2015, 35, 7460–7474. [Google Scholar] [CrossRef]
- Royo, M.; Gutiérrez, Y.; Fernández-Monreal, M.; Gutiérrez-Eisman, S.; Jiménez, R.; Jurado, S.; Esteban, J.A. A retention-release mechanism based on Rab11-FIP2 for AMPA receptor synaptic delivery during long-term potentiation. J. Cell Sci. 2019, 132, jcs234237. [Google Scholar] [CrossRef]
- Pylypenko, O.; Attanda, W.; Gauquelin, C.; Lahmani, M.; Coulibaly, D.; Baron, B.; Hoos, S.; Titus, M.A.; England, P.; Houdusse, A.M. Structural basis of myosin V Rab GTPase-dependent cargo recognition. Proc. Natl. Acad. Sci. USA 2013, 110, 20443–20448. [Google Scholar] [CrossRef]
- Ji, H.H.; Yao, L.L.; Liu, C.; Li, X.D. Regulation of Myosin-5b by Rab11a and the Rab11 family interacting protein 2. Biosci. Rep. 2019, 39, BSR20181252. [Google Scholar] [CrossRef] [PubMed]
- Schafer, J.C.; Baetz, N.W.; Lapierre, L.A.; McRae, R.E.; Roland, J.T.; Goldenring, J.R. Rab11-FIP2 Interaction with MYO5B Regulates Movement of Rab11a-Containing Recycling Vesicles. Traffic 2014, 15, 292–308. [Google Scholar] [CrossRef] [PubMed]
- Provance, D.W.; Addison, E.J.; Wood, P.R.; Chen, D.Z.; Silan, C.M.; Mercer, J.A. Myosin-Vb functions as a dynamic tether for peripheral endocytic compartments during transferrin trafficking. BMC Cell Biol. 2008, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Pick, J.E.; Ziff, E.B. Regulation of AMPA receptor trafficking and exit from the endoplasmic reticulum. Mol. Cell. Neurosci. 2018, 91, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Park, M. AMPA Receptor Trafficking for Postsynaptic Potentiation. Front. Cell. Neurosci. 2018, 12, 361. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Alonso, J.; Nicoll, R.A. AMPA receptor trafficking and LTP: Carboxy-termini, amino-termini and TARPs. Neuropharmacology 2021, 197, 108710. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, E.; von Engelhardt, J. AMPA receptor complex constituents: Control of receptor assembly, membrane trafficking and subcellular localization. Mol. Cell Neurosci. 2018, 91, 67–75. [Google Scholar] [CrossRef]
- Uchida, S.; Shumyatsky, G.P. Deceivingly dynamic: Learning-dependent changes in stathmin and microtubules. Neurobiol. Learn. Mem. 2015, 124, 52–61. [Google Scholar] [CrossRef]
- Ives, J.H.; Fung, S.; Tiwari, P.; Payne, H.L.; Thompson, C.L. Microtubule-associated protein light chain 2 is a stargazin-AMPA receptor complex-interacting protein in vivo. J. Biol. Chem. 2004, 279, 31002–31009. [Google Scholar] [CrossRef]
- Palenzuela, R.; Gutiérrez, Y.; Draffin, J.E.; Lario, A.; Benoist, M.; Esteban, J.A. MAP1B Light Chain Modulates Synaptic Transmission via AMPA Receptor Intracellular Trapping. J. Neurosci. 2017, 37, 9945–9963. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Noiges, R.; Descovich, L.; Fischer, I.; Albrecht, D.E.; Nothias, F.; Froehner, S.C.; Propst, F. The light chains of microtubule-associated proteins MAP1A and MAP1B interact with alpha1-syntrophin in the central and peripheral nervous system. PLoS ONE 2012, 7, e49722. [Google Scholar] [CrossRef] [PubMed]
- Hammarback, J.A.; Obar, R.A.; Hughes, S.M.; Vallee, R.B. MAP1B is encoded as a polyprotein that is processed to form a complex N-terminal microtubule-binding domain. Neuron 1991, 7, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Takei, Y.; Kikkawa, Y.S.; Atapour, N.; Hensch, T.K.; Hirokawa, N. Defects in Synaptic Plasticity, Reduced NMDA-Receptor Transport, and Instability of Postsynaptic Density Proteins in Mice Lacking Microtubule-Associated Protein 1A. J. Neurosci. 2015, 35, 15539–15554. [Google Scholar] [CrossRef] [PubMed]
- Leenders, A.M.; Lin, L.; Huang, L.D.; Gerwin, C.; Lu, P.H.; Sheng, Z.H. The role of MAP1A light chain 2 in synaptic surface retention of Cav2.2 channels in hippocampal neurons. J. Neurosci. 2008, 28, 11333–11346. [Google Scholar] [CrossRef] [PubMed]
- Hanley, J.G. Actin-dependent mechanisms in AMPA receptor trafficking. Front. Cell. Neurosci. 2014, 8, 381. [Google Scholar] [CrossRef] [PubMed]
- Schulz, T.W.; Nakagawa, T.; Licznerski, P.; Pawlak, V.; Kolleker, A.; Rozov, A.; Kim, J.; Dittgen, T.; Köhr, G.; Sheng, M.; et al. Actin/alpha-actinin-dependent transport of AMPA receptors in dendritic spines: Role of the PDZ-LIM protein RIL. J. Neurosci. 2004, 24, 8584–8594. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Sun, W.; Kujala, P.; Lotfi, Y.; Vida, T.A.; Bean, A.J.; Chiu, T.T.; Patel, N.; Shaw, A.E.; Bamburg, J.R.; et al. CART: An Hrs/actinin-4/BERP/myosin V protein complex required for efficient receptor recycling. Mol. Biol. Cell 2005, 16, 2470–2482. [Google Scholar] [CrossRef]
- Hámor, P.U.; Schwendt, M. Metabotropic Glutamate Receptor Trafficking and its Role in Drug-Induced Neurobehavioral Plasticity. Brain Plast. 2021, 7, 61–76. [Google Scholar] [CrossRef]
- Correia, S.S.; Bassani, S.; Brown, T.C.; Lisé, M.F.; Backos, D.S.; El-Husseini, A.; Passafaro, M.; Esteban, J.A. Motor protein–dependent transport of AMPA receptors into spines during long-term potentiation. Nat. Neurosci. 2008, 11, 457–466. [Google Scholar] [CrossRef]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef]
- Takenawa, T.; Suetsugu, S. The WASP–WAVE protein network: Connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2007, 8, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Wood, C.L.; Patton, A.P.; Jaafari, N.; Henley, J.M.; Mellor, J.R.; Hanley, J.G. PICK1 inhibition of the Arp2/3 complex controls dendritic spine size and synaptic plasticity. EMBO J. 2011, 30, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Matt, L.; Kim, K.; Hergarden, A.C.; Patriarchi, T.; Malik, Z.A.; Park, D.K.; Chowdhury, D.; Buonarati, O.R.; Henderson, P.B.; Saraç, Ç.G.; et al. alpha-Actinin Anchors PSD-95 at Postsynaptic Sites. Neuron 2018, 97, 1094–1109.e9. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Shi, R.; Hwang, H.; Han, K.S.; Wong, M.H.; Ren, X.; Lewis, L.D.; Brown, E.N.; Xu, W. SAP102 regulates synaptic AMPAR function through a CNIH-2-dependent mechanism. J. Neurophysiol. 2018, 120, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Nissen, K.B.; Haugaard-Kedström, L.M.; Wilbek, T.S.; Nielsen, L.S.; Åberg, E.; Kristensen, A.S.; Bach, A.; Jemth, P.; Strømgaard, K. Targeting protein-protein interactions with trimeric ligands: High affinity inhibitors of the MAGUK protein family. PLoS ONE 2015, 10, e0117668. [Google Scholar] [CrossRef] [PubMed]
- Toto, A.; Pedersen, S.W.; Karlsson, O.A.; Moran, G.E.; Andersson, E.; Chi, C.N.; Strømgaard, K.; Gianni, S.; Jemth, P. Ligand binding to the PDZ domains of postsynaptic density protein 95. Protein Eng. Des. Sel. 2016, 29, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.N.; Bach, A.; Strømgaard, K.; Gianni, S.; Jemth, P. Ligand binding by PDZ domains. Biofactors 2012, 38, 338–348. [Google Scholar] [CrossRef]
- Won, S. and K.W. Roche, Regulation of glutamate receptors by striatal-enriched tyrosine phosphatase 61 (STEP61). J. Physiol. 2021, 599, 443–451. [Google Scholar] [CrossRef]
- Shaikh, S.A.; Dolino, D.M.; Lee, G.; Chatterjee, S.; MacLean, D.M.; Flatebo, C.; Landes, C.F.; Jayaraman, V. Stargazin Modulation of AMPA Receptors. Cell Rep. 2016, 17, 328–335. [Google Scholar] [CrossRef]
- Stein, E.L.A.; Chetkovich, D.M. Regulation of stargazin synaptic trafficking by C-terminal PDZ ligand phosphorylation in bidirectional synaptic plasticity. J. Neurochem. 2010, 113, 42–53. [Google Scholar] [CrossRef]
- Tomita, S.; Adesnik, H.; Sekiguchi, M.; Zhang, W.; Wada, K.; Howe, J.R.; Nicoll, R.A.; Bredt, D.S. Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature 2005, 435, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Tomita, S.; Fukata, M.; Nicoll, R.A.; Bredt, D.S. Dynamic interaction of stargazin-like TARPs with cycling AMPA receptors at synapses. Science 2004, 303, 1508–1511. [Google Scholar] [CrossRef] [PubMed]
- Béïque, J.C.; Lin, D.T.; Kang, M.G.; Aizawa, H.; Takamiya, K.; Huganir, R.L. Synapse-specific regulation of AMPA receptor function by PSD-95. Proc. Natl. Acad. Sci. USA 2006, 103, 19535–19540. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Stodieck, S.K.; Goetze, B.; Cui, L.; Wong, M.H.; Wenzel, C.; Hosang, L.; Dong, Y.; Löwel, S.; Schlüter, O.M. Progressive maturation of silent synapses governs the duration of a critical period. Proc. Natl. Acad. Sci. USA 2015, 112, E3131–E3140. [Google Scholar] [CrossRef] [PubMed]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; VanLeeuwen, J.E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Greger, I.H.; Watson, J.F.; Cull-Candy, S.G. Structural and Functional Architecture of AMPA-Type Glutamate Receptors and Their Auxiliary Proteins. Neuron 2017, 94, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.A.; Elias, G.M.; Elias, L.A.B.; Swat, W.; Nicoll, R.A. The role of SAP97 in synaptic glutamate receptor dynamics. Proc. Natl. Acad. Sci. USA 2010, 107, 3805–3810. [Google Scholar] [CrossRef] [PubMed]
- Nikandrova, Y.A.; Jiao, Y.; Baucum, A.J.; Tavalin, S.J.; Colbran, R.J. Ca2+/calmodulin-dependent protein kinase II binds to and phosphorylates a specific SAP97 splice variant to disrupt association with AKAP79/150 and modulate alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-type glutamate receptor (AMPAR) activity. J. Biol. Chem. 2010, 285, 923–934. [Google Scholar] [CrossRef]
- Anggono, V.; Huganir, R.L. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr. Opin. Neurobiol. 2012, 22, 461–469. [Google Scholar] [CrossRef]
- Cai, C.; Li, H.; Rivera, C.; Keinänen, K. Interaction between SAP97 and PSD-95, two maguk proteins involved in synaptic trafficking of AMPA receptors. J. Biol. Chem. 2006, 281, 4267–4273. [Google Scholar] [CrossRef]
- Vinade, L.; Chang, M.; Schlief, M.L.; Petersen, J.D.; Reese, T.S.; Tao-Cheng, J.H.; Dosemeci, A. Affinity purification of PSD-95-containing postsynaptic complexes. J. Neurochem. 2003, 87, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Fukata, Y.; Tzingounis, A.V.; Trinidad, J.C.; Fukata, M.; Burlingame, A.L.; Nicoll, R.A.; Bredt, D.S. Molecular constituents of neuronal AMPA receptors. J. Cell Biol. 2005, 169, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Rumbaugh, G.; Sia, G.M.; Garner, C.C.; Huganir, R.L. Synapse-associated protein-97 isoform-specific regulation of surface AMPA receptors and synaptic function in cultured neurons. J. Neurosci. 2003, 23, 4567–4576. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, O.M.; Xu, W.; Malenka, R.C. Alternative N-terminal domains of PSD-95 and SAP97 govern activity-dependent regulation of synaptic AMPA receptor function. Neuron 2006, 51, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Schapitz, I.U.; Behrend, B.; Pechmann, Y.; Lappe-Siefke, C.; Kneussel, S.J.; Wallace, K.E.; Stempel, A.V.; Buck, F.; Grant, S.G.N.; Schweizer, M.; et al. Neuroligin 1 Is Dynamically Exchanged at Postsynaptic Sites. J. Neurosci. 2010, 30, 12733–12744. [Google Scholar] [CrossRef] [PubMed]
- Choquet, D.; Opazo, P. The role of AMPAR lateral diffusion in memory. Semin. Cell Dev. Biol. 2022, 125, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Levy, J.M.; Hou, A.; Winters, C.; Azzam, R.; Sousa, A.A.; Leapman, R.D.; Nicoll, R.A.; Reese, T.S. PSD-95 family MAGUKs are essential for anchoring AMPA and NMDA receptor complexes at the postsynaptic density. Proc. Natl. Acad. Sci. USA 2015, 112, E6983–E6992. [Google Scholar] [CrossRef]
- Man, H.Y.; Sekine-Aizawa, Y.; Huganir, R.L. Regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc. Natl. Acad. Sci. USA 2007, 104, 3579–3584. [Google Scholar] [CrossRef]
- Amer, Y.O.; Hebert-Chatelain, E. Mitochondrial cAMP-PKA signaling: What do we really know? Biochim. Biophys. Acta (BBA) Bioenerg. 2018, 1859, 868–877. [Google Scholar] [CrossRef]
- Park, P.; Kang, H.; Sanderson, T.M.; Bortolotto, Z.A.; Georgiou, J.; Zhuo, M.; Kaang, B.K.; Collingridge, G.L. The Role of Calcium-Permeable AMPARs in Long-Term Potentiation at Principal Neurons in the Rodent Hippocampus. Front. Synaptic Neurosci. 2018, 10, 42. [Google Scholar] [CrossRef]
- Park, P.; Sanderson, T.M.; Amici, M.; Choi, S.L.; Bortolotto, Z.A.; Zhuo, M.; Kaang, B.K.; Collingridge, G.L. Calcium-Permeable AMPA Receptors Mediate the Induction of the Protein Kinase A-Dependent Component of Long-Term Potentiation in the Hippocampus. J. Neurosci. 2016, 36, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, J.L.; Freund, R.K.; Gorski, J.A.; Dellacqua, M.L. β-Amyloid disruption of LTP/LTD balance is mediated by AKAP150-anchored PKA and Calcineurin regulation of Ca2+-permeable AMPA receptors. Cell Rep. 2021, 37, 109786. [Google Scholar] [CrossRef] [PubMed]
- Plant, K.; Pelkey, K.A.; Bortolotto, Z.A.; Morita, D.; Terashima, A.; McBain, C.J.; Collingridge, G.L.; Isaac, J.T.R. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 2006, 9, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Suppiramaniam, V.; Wooten, M.W. Posttranslational Modifications and Receptor-Associated Proteins in AMPA Receptor Trafficking and Synaptic Plasticity. Neurosignals 2006, 15, 266–282. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Ji, J.; Zhao, X.; Lei, Y.; Li, H.; Hao, Y.; Zhang, S.; Zhang, J.; Liu, C.; Nie, J.; et al. The Role of PKC in Regulating NMDARs in Aluminum-Induced Learning and Memory Impairment in Rats. Neurotox. Res. 2021, 39, 2042–2055. [Google Scholar] [CrossRef] [PubMed]
- Boehm, J.; Kang, M.G.; Johnson, R.C.; Esteban, J.; Huganir, R.L.; Malinow, R. Synaptic Incorporation of AMPA Receptors during LTP Is Controlled by a PKC Phosphorylation Site on GluR1. Neuron 2006, 51, 213–225. [Google Scholar] [CrossRef]
- Boehm, J.; Kang, M.-G.; Johnson, R.C.; Esteban, J.; Huganir, R.L.; Malinow, R. Spinal PKC alpha inhibition and gene-silencing for pain relief: AMPAR trafficking at the synapses between primary afferents and sensory interneurons. Sci. Rep. 2018, 8, 10285. [Google Scholar]
- Kopach, O.; Viatchenko-Karpinski, V.; Atianjoh, F.E.; Belan, P.; Tao, Y.X.; Voitenko, N. PKCalpha is required for inflammation-induced trafficking of extrasynaptic AMPA receptors in tonically firing lamina II dorsal horn neurons during the maintenance of persistent inflammatory pain. J. Pain 2013, 14, 182–192. [Google Scholar] [CrossRef]
- Terashima, A.; Cotton, L.; Dev, K.K.; Meyer, G.; Zaman, S.; Duprat, F.; Henley, J.M.; Collingridge, G.L.; Isaac, J.T.R. Regulation of synaptic strength and AMPA receptor subunit composition by PICK1. J. Neurosci. 2004, 24, 5381–5390. [Google Scholar] [CrossRef]
- Yao, Y.; Kelly, M.T.; Sajikumar, S.; Serrano, P.; Tian, D.; Bergold, P.J.; Frey, J.U.; Sacktor, T.C. PKM zeta maintains late long-term potentiation by N-ethylmaleimide-sensitive factor/GluR2-dependent trafficking of postsynaptic AMPA receptors. J. Neurosci. 2008, 28, 7820–7827. [Google Scholar] [CrossRef]
- Kelly, M.T.; Yao, Y.; Sondhi, R.; Sacktor, T.C. Actin polymerization regulates the synthesis of PKMζ in LTP. Neuropharmacology 2007, 52, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Faux, M.C.; Rollins, E.N.; Edwards, A.S.; Langeberg, L.K.; Newton, A.C.; Scott, J.D. Mechanism of A-kinase-anchoring protein 79 (AKAP79) and protein kinase C interaction. Biochem. J. 1999, 343 Pt 2, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Nauert, J.; Klauck, T.M.; Langeberg, L.K.; Scott, J.D. Gravin, an autoantigen recognized by serum from myasthenia gravis patients, is a kinase scaffold protein. Curr. Biol. 1997, 7, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Kuntziger, T.; Rogne, M.; Folstad, R.L.S.; Collas, P. Association of PP1 with its regulatory subunit AKAP149 is regulated by serine phosphorylation flanking the RVXF motif of AKAP149. Biochemistry 2006, 45, 5868–5877. [Google Scholar] [CrossRef] [PubMed]
- Klauck, T.M.; Faux, M.C.; Labudda, K.; Langeberg, L.K.; Jaken, S.; Scott, J.D. Coordination of three signaling enzymes by AKAP79, a mammalian scaffold protein. Science 1996, 271, 1589–1592. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.L.; Chiu, S.L.; Zhu, Q.; Huganir, R.L. GRIP1 regulates synaptic plasticity and learning and memory. Proc. Natl. Acad. Sci. USA 2020, 117, 25085–25091. [Google Scholar] [CrossRef] [PubMed]
- Cisani, F.; Olivero, G.; Usai, C.; Van Camp, G.; Maccari, S.; Morley-Fletcher, S.; Pittaluga, A.M. Antibodies Against the NH2-Terminus of the GluA Subunits Affect the AMPA-Evoked Releasing Activity: The Role of Complement. Front. Immunol. 2021, 12, 586521. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Ziff, E.B. PICK1 Interacts with ABP/GRIP to Regulate AMPA Receptor Trafficking. Neuron 2005, 47, 407–421. [Google Scholar] [CrossRef]
- Tan, H.L.; Queenan, B.N.; Huganir, R.L. GRIP1 is required for homeostatic regulation of AMPAR trafficking. Proc. Natl. Acad. Sci. USA 2015, 112, 10026–10031. [Google Scholar] [CrossRef]
- Diakowski, W.; Grzybek, M.; Sikorski, A.F. Protein 4.1, a component of the erythrocyte membrane skeleton and its related homologue proteins forming the protein 4.1/FERM superfamily. Folia Histochem. Cytobiol. 2006, 44, 231–248. [Google Scholar]
- Baines, A.J.; Lu, H.C.; Bennett, P.M. The Protein 4.1 family: Hub proteins in animals for organizing membrane proteins. Biochim. Biophys. Acta (BBA) Biomembr. 2014, 1838, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.T.; Makino, Y.; Sharma, K.; Hayashi, T.; Neve, R.; Takamiya, K.; Huganir, R.L. Regulation of AMPA receptor extrasynaptic insertion by 4.1N, phosphorylation and palmitoylation. Nat. Neurosci. 2009, 12, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Fukatsu, K.; Bannai, H.; Inoue, T.; Mikoshiba, K. 4.1N binding regions of inositol 1,4,5-trisphosphate receptor type 1. Biochem. Biophys. Res. Commun. 2006, 342, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Baines, A.J. A FERM-adjacent (FA) region defines a subset of the 4.1 superfamily and is a potential regulator of FERM domain function. BMC Genom. 2006, 7, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Jurado, S.; Goswami, D.; Zhang, Y.; Molina, A.J.M.; Südhof, T.C.; Malenka, R.C. LTP requires a unique postsynaptic SNARE fusion machinery. Neuron 2013, 77, 542–558. [Google Scholar] [CrossRef] [PubMed]
- Rizo, J. Molecular Mechanisms Underlying Neurotransmitter Release. Annu. Rev. Biophys. 2022, 51, 377–408. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T.C.; Rothman, J.E. Membrane fusion: Grappling with SNARE and SM proteins. Science 2009, 323, 474–477. [Google Scholar] [CrossRef]
- Peters, J.J.; Leitz, J.; Oses-Prieto, J.A.; Burlingame, A.L.; Brunger, A.T. Molecular Characterization of AMPA-Receptor-Containing Vesicles. Front. Mol. Neurosci. 2021, 14, 754631. [Google Scholar] [CrossRef]
- Koike, S.; Jahn, R. SNARE proteins: Zip codes in vesicle targeting? Biochem. J. 2022, 479, 273–288. [Google Scholar] [CrossRef]
- Xie, M.J.; Iwata, K.; Ishikawa, Y.; Nomura, Y.; Tani, T.; Murata, K.; Fukazawa, Y.; Matsuzaki, H. Autistic-Like Behavior and Impairment of Serotonin Transporter and AMPA Receptor Trafficking in N-Ethylmaleimide Sensitive Factor Gene-Deficient Mice. Front. Genet. 2021, 12, 748627. [Google Scholar] [CrossRef]
- Migues, P.V.; Hardt, O.; Finnie, P.; Wang, Y.W.; Nader, K. The maintenance of long-term memory in the hippocampus depends on the interaction between N-ethylmaleimide-sensitive factor and GluA2. Hippocampus 2014, 24, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Zhou, Z.; Cai, S.; Xie, W.; Jia, Z. Hippocampal Long-Term Depression in the Presence of Calcium-Permeable AMPA Receptors. Front. Synaptic Neurosci. 2018, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Beretta, F.; Sala, C.; Saglietti, L.; Hirling, H.; Sheng, M.; Passafaro, M. NSF interaction is important for direct insertion of GluR2 at synaptic sites. Mol. Cell. Neurosci. 2005, 28, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Umanah, G.K.; Ghasemi, M.; Yin, X.; Chang, M.; Kim, J.W.; Zhang, J.; Ma, E.; Scarffe, L.A.; Lee, Y.I.; Chen, R.; et al. AMPA Receptor Surface Expression Is Regulated by S-Nitrosylation of Thorase and Transnitrosylation of NSF. Cell Rep. 2020, 33, 108329. [Google Scholar] [CrossRef] [PubMed]
- Moore, F.B.; Baleja, J.D. Molecular remodeling mechanisms of the neural somatodendritic compartment. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2012, 1823, 1720–1730. [Google Scholar] [CrossRef] [PubMed]
- Kastning, K.; Kukhtina, V.; Kittler, J.T.; Chen, G.; Pechstein, A.; Enders, S.; Lee, S.H.; Sheng, M.; Yan, Z.; Haucke, V. Molecular determinants for the interaction between AMPA receptors and the clathrin adaptor complex AP-2. Proc. Natl. Acad. Sci. USA 2007, 104, 2991–2996. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Liu, L.; Wang, Y.T.; Sheng, M. Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron 2002, 36, 661–674. [Google Scholar] [CrossRef]
- DaSilva, L.L.P.; Wall, M.J.; de Almeida, L.P.; Wauters, S.C.; Januário, Y.C.; Müller, J.; Corrêa, S.A.L. Activity-Regulated Cytoskeleton-Associated Protein Controls AMPAR Endocytosis through a Direct Interaction with Clathrin-Adaptor Protein 2. eNeuro 2016, 3. [Google Scholar] [CrossRef]
- Hosokawa, T.; Liu, P.W.; Cai, Q.; Ferreira, J.S.; Levet, F.; Butler, C.; Sibarita, J.B.; Choquet, D.; Groc, L.; Hosy, E.; et al. CaMKII activation persistently segregates postsynaptic proteins via liquid phase separation. Nat. Neurosci. 2021, 24, 777–785. [Google Scholar] [CrossRef]
- Hausser, A.; Schlett, K. Coordination of AMPA receptor trafficking by Rab GTPases. Small GTPases 2017, 10, 419–432. [Google Scholar] [CrossRef]
- Rudolf, R.; Bittins, C.M.; Gerdes, H.H. The role of myosin V in exocytosis and synaptic plasticity. J. Neurochem. 2010, 116, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Opazo, P.; Labrecque, S.; Tigaret, C.M.; Frouin, A.; Wiseman, P.W.; De Koninck, P.; Choquet, D. CaMKII Triggers the Diffusional Trapping of Surface AMPARs through Phosphorylation of Stargazin. Neuron 2010, 67, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Jurado, S. The dendritic SNARE fusion machinery involved in AMPARs insertion during long-term potentiation. Front. Cell. Neurosci. 2014, 8, 407. [Google Scholar] [CrossRef] [PubMed]
- Chazeau, A.; Giannone, G. Organization and dynamics of the actin cytoskeleton during dendritic spine morphological remodeling. Cell. Mol. Life Sci. 2016, 73, 3053–3073. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, G.T.; Chamberlain, S.E.L.; Jaafari, N.; Turvey, M.; Mellor, J.R.; Hanley, J.G. Cortactin regulates endo-lysosomal sorting of AMPARs via direct interaction with GluA2 subunit. Sci. Rep. 2018, 8, 4155. [Google Scholar] [CrossRef] [PubMed]
- Wennagel, D.; Braz, B.Y.; Capizzi, M.; Barnat, M.; Humbert, S. Huntingtin coordinates dendritic spine morphology and function through cofilin-mediated control of the actin cytoskeleton. Cell Rep. 2022, 40, 111261. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, L.; Farley, M.M.; Waxham, M.N. Calcium-calmodulin-dependent protein kinase II isoforms differentially impact the dynamics and structure of the actin cytoskeleton. Biochemistry 2013, 52, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Borovac, J.; Bosch, M.; Okamoto, K. Regulation of actin dynamics during structural plasticity of dendritic spines: Signaling messengers and actin-binding proteins. Mol. Cell. Neurosci. 2018, 91, 122–130. [Google Scholar] [CrossRef]
- Perez-Alvarez, A.; Yin, S.; Schulze, C.; Hammer, J.A.; Wagner, W.; Oertner, T.G. Endoplasmic reticulum visits highly active spines and prevents runaway potentiation of synapses. Nat. Commun. 2020, 11, 5083. [Google Scholar] [CrossRef]
- Wu, Y.; Whiteus, C.; Xu, C.S.; Hayworth, K.J.; Weinberg, R.J.; Hess, H.F.; De Camilli, P. Contacts between the endoplasmic reticulum and other membranes in neurons. Proc. Natl. Acad. Sci. USA 2017, 114, E4859–E4867. [Google Scholar] [CrossRef]
- Harris, K.M. Structural LTP: From synaptogenesis to regulated synapse enlargement and clustering. Curr. Opin. Neurobiol. 2020, 63, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Konietzny, A.; Grendel, J.; Kadek, A.; Bucher, M.; Han, Y.; Hertrich, N.; Dekkers, D.H.W.; Demmers, J.A.A.; Grünewald, K.; Uetrecht, C.; et al. Caldendrin and myosin V regulate synaptic spine apparatus localization via ER stabilization in dendritic spines. EMBO J. 2022, 41, e106523. [Google Scholar] [CrossRef] [PubMed]
- Jungenitz, T.; Beining, M.; Radic, T.; Deller, T.; Cuntz, H.; Jedlicka, P.; Schwarzacher, S.W. Structural homo- and heterosynaptic plasticity in mature and adult newborn rat hippocampal granule cells. Proc. Natl. Acad. Sci. USA 2018, 115, E4670–E4679. [Google Scholar] [CrossRef] [PubMed]
- Saneyoshi, T.; Hayashi, Y. The Ca2+ and Rho GTPase signaling pathways underlying activity-dependent actin remodeling at dendritic spines. Cytoskeleton 2012, 69, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Jędrzejewska-Szmek, J.; Blackwell, K.T. From membrane receptors to protein synthesis and actin cytoskeleton: Mechanisms underlying long lasting forms of synaptic plasticity. Semin. Cell Dev. Biol. 2019, 95, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Barnat, M.; Le Friec, J.; Benstaali, C.; Humbert, S. Huntingtin-Mediated Multipolar-Bipolar Transition of Newborn Cortical Neurons Is Critical for Their Postnatal Neuronal Morphology. Neuron 2016, 93, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Richards, P.; Didszun, C.; Campesan, S.; Simpson, A.; Horley, B.; Young, K.W.; Glynn, P.; Cain, K.; Kyriacou, C.P.; Giorgini, F.; et al. Dendritic spine loss and neurodegeneration is rescued by Rab11 in models of Huntington’s disease. Cell Death Differ. 2011, 18, 191–200. [Google Scholar] [CrossRef]
- Arbo, B.D.; Cechinel, L.R.; Palazzo, R.P.; Siqueira, I.R. Endosomal dysfunction impacts extracellular vesicle release: Central role in Abeta pathology. Ageing Res. Rev. 2020, 58, 101006. [Google Scholar] [CrossRef]
- Schürmann, B.; Bermingham, D.P.; Kopeikina, K.J.; Myczek, K.; Yoon, S.; Horan, K.E.; Kelly, C.J.; Martin-De-Saavedra, M.D.; Forrest, M.P.; Fawcett-Patel, J.M.; et al. A novel role for the late-onset Alzheimer’s disease (LOAD)-associated protein Bin1 in regulating postsynaptic trafficking and glutamatergic signaling. Mol. Psychiatry 2019, 25, 2000–2016. [Google Scholar] [CrossRef]
- Achzet, L.M.; Davison, C.J.; Shea, M.; Sturgeon, I.; Jackson, D.A. Oxidative Stress Underlies the Ischemia/Reperfusion-Induced Internalization and Degradation of AMPA Receptors. Int. J. Mol. Sci. 2021, 22, 717. [Google Scholar] [CrossRef]
- Inoshita, T.; Arano, T.; Hosaka, Y.; Meng, H.; Umezaki, Y.; Kosugi, S.; Morimoto, T.; Koike, M.; Chang, H.Y.; Imai, Y.; et al. Vps35 in cooperation with LRRK2 regulates synaptic vesicle endocytosis through the endosomal pathway in Drosophila. Hum. Mol. Genet. 2017, 26, 2933–2948. [Google Scholar] [CrossRef] [PubMed]
- Opazo, P.; da Silva, S.V.; Carta, M.; Breillat, C.; Coultrap, S.J.; Grillo-Bosch, D.; Sainlos, M.; Coussen, F.; Bayer, K.U.; Mulle, C.; et al. CaMKII Metaplasticity Drives Abeta Oligomer-Mediated Synaptotoxicity. Cell Rep. 2018, 23, 3137–3145. [Google Scholar] [CrossRef] [PubMed]
- Küry, S.; van Woerden, G.M.; Besnard, T.; Onori, M.P.; Latypova, X.; Towne, M.C.; Cho, M.T.; Prescott, T.E.; Ploeg, M.A.; Sanders, S.; et al. De Novo Mutations in Protein Kinase Genes CAMK2A and CAMK2B Cause Intellectual Disability. Am. J. Hum. Genet. 2017, 101, 768–788. [Google Scholar] [CrossRef] [PubMed]
- Savas, J.N.; Wang, Y.Z.; DeNardo, L.A.; Martinez-Bartolome, S.; McClatchy, D.B.; Hark, T.J.; Shanks, N.F.; Cozzolino, K.A.; Lavallée-Adam, M.; Smukowski, S.N.; et al. Amyloid Accumulation Drives Proteome-wide Alterations in Mouse Models of Alzheimer’s Disease-like Pathology. Cell Rep. 2017, 21, 2614–2627. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, G.L.; Inácio, A.S.; Beltrão, N.; Barreto, C.A.V.; Rodrigues, M.V.; Rondão, T.; Macedo, R.; Gouveia, R.P.; Edfawy, M.; Guedes, J.; et al. Aberrant hippocampal transmission and behavior in mice with a stargazin mutation linked to intellectual disability. Mol. Psychiatry 2022, 27, 2457–2469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, C.; Vincent, J.; Zala, D.; Benstaali, C.; Sainlos, M.; Grillo-Bosch, D.; Daburon, S.; Coussen, F.; Cho, Y.; et al. Modulation of AMPA receptor surface diffusion restores hippocampal plasticity and memory in Huntington’s disease models. Nat. Commun. 2018, 9, 4272. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Su, C.W.; Xiong, S.; Li, T.; Liang, H.Y.; Lin, Y.H.; Chang, L.; Wu, H.Y.; Li, F.; Zhu, D.Y.; et al. Enhanced AMPAR-dependent synaptic transmission by S-nitrosylation in the vmPFC contributes to chronic inflammatory pain-induced persistent anxiety in mice. Acta Pharmacol. Sin. 2022, 44, 954–968. [Google Scholar] [CrossRef] [PubMed]
- Silberberg, G.; Levit, A.; Collier, D.; Clair, D.S.; Munro, J.; Kerwin, R.W.; Tondo, L.; Floris, G.; Breen, G.; Navon, R. Stargazin involvement with bipolar disorder and response to lithium treatment. Pharmacogenet. Genom. 2008, 18, 403–412. [Google Scholar] [CrossRef]
- Tomita, S.; Byrd, R.K.; Rouach, N.; Bellone, C.; Venegas, A.; O’Brien, J.L.; Kim, K.S.; Olsen, O.; Nicoll, R.A.; Bredt, D.S. AMPA receptors and stargazin-like transmembrane AMPA receptor-regulatory proteins mediate hippocampal kainate neurotoxicity. Proc. Natl. Acad. Sci. USA 2007, 104, 18784–18788. [Google Scholar] [CrossRef]
- Dixon, R.M.; Mellor, J.R.; Hanley, J.G. PICK1-mediated glutamate receptor subunit 2 (GluR2) trafficking contributes to cell death in oxygen/glucose-deprived hippocampal neurons. J. Biol. Chem. 2009, 284, 14230–14235. [Google Scholar] [CrossRef]
- Koszegi, Z.; Fiuza, M.; Hanley, J.G. Endocytosis and lysosomal degradation of GluA2/3 AMPARs in response to oxygen/glucose deprivation in hippocampal but not cortical neurons. Sci. Rep. 2017, 7, 12318. [Google Scholar] [CrossRef] [PubMed]
- Atianjoh, F.E.; Yaster, M.; Zhao, X.; Takamiya, K.; Xia, J.; Gauda, E.B.; Huganir, R.L.; Tao, Y.X. Spinal cord protein interacting with C kinase 1 is required for the maintenance of complete Freund’s adjuvant-induced inflammatory pain but not for incision-induced post-operative pain. Pain 2010, 151, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Mignogna, M.L.; Giannandrea, M.; Gurgone, A.; Fanelli, F.; Raimondi, F.; Mapelli, L.; Bassani, S.; Fang, H.; Van Anken, E.; Alessio, M.; et al. The intellectual disability protein RAB39B selectively regulates GluA2 trafficking to determine synaptic AMPAR composition. Nat. Commun. 2015, 6, 6504. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Xie, C.; McCormack, S.G.; Chiang, H.C.; Michalak, M.K.; Lin, X.; Chandran, J.; Shim, H.; Shimoji, M.; Cookson, M.R.; et al. Amyotrophic lateral sclerosis 2-deficiency leads to neuronal degeneration in amyotrophic lateral sclerosis through altered AMPA receptor trafficking. J. Neurosci. 2006, 26, 11798–11806. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Buniel, M.C.; Chang, L.; Fernandez, S.J.; Gong, Y.; Viola, K.L.; Lambert, M.P.; Velasco, P.T.; Bigio, E.H.; Finch, C.E.; et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J. Neurosci. 2004, 24, 10191–10200. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Feyder, M.; Karlsson, R.M.; Mathur, P.; Lyman, M.; Bock, R.; Momenan, R.; Munasinghe, J.; Scattoni, M.L.; Ihne, J.; Camp, M.; et al. Association of mouse Dlg4 (PSD-95) gene deletion and human Dlg4 gene variation with phenotypes relevant to autism spectrum disorders and Williams’ syndrome. Am. J. Psychiatry 2010, 167, 1508–1517. [Google Scholar] [CrossRef]
- Mandal, M.; Wei, J.; Zhong, P.; Cheng, J.; Duffney, L.J.; Liu, W.; Yuen, E.Y.; Twelvetrees, A.E.; Li, S.; Li, X.J.; et al. Impaired alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor trafficking and function by mutant huntingtin. J. Biol. Chem. 2011, 286, 33719–33728. [Google Scholar] [CrossRef]
- Wang, G.; Li, S.; Gilbert, J.; Gritton, H.J.; Wang, Z.; Li, Z.; Han, X.; Selkoe, D.J.; Man, H.Y. Crucial Roles for SIRT2 and AMPA Receptor Acetylation in Synaptic Plasticity and Memory. Cell Rep. 2017, 20, 1335–1347. [Google Scholar] [CrossRef]
- Diering, G.H.; Gustina, A.S.; Huganir, R.L. PKA-GluA1 coupling via AKAP5 controls AMPA receptor phosphorylation and cell-surface targeting during bidirectional homeostatic plasticity. Neuron 2014, 84, 790–805. [Google Scholar] [CrossRef]
- Purkey, A.M.; Dell’Acqua, M.L. Phosphorylation-Dependent Regulation of Ca2+-Permeable AMPA Receptors During Hippocampal Synaptic Plasticity. Front. Synaptic Neurosci. 2020, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Wickens, M.M.; Kirkland, J.M.; Knouse, M.C.; McGrath, A.G.; Briand, L.A. Sex-specific role for prefrontal cortical protein interacting with C kinase 1 in cue-induced cocaine seeking. Addict. Biol. 2021, 26, e13051. [Google Scholar] [CrossRef] [PubMed]
- Lussier, M.P.; Gu, X.; Lu, W.; Roche, K.W. Casein kinase 2 phosphorylates GluA1 and regulates its surface expression. Eur. J. Neurosci. 2014, 39, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, T.; Mitsushima, D.; Kaneko, R.; Hayashi, Y. Stoichiometry and phosphoisotypes of hippocampal AMPA-type glutamate receptor phosphorylation. Neuron 2014, 85, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.C.; Derkach, V.A.; Guire, E.S.; Soderling, T.R. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J. Biol. Chem. 2006, 281, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, L.A.; Hall, B.J.; Patrick, G.N. Activity-Dependent Ubiquitination of GluA1 Mediates a Distinct AMPA Receptor Endocytosis and Sorting Pathway. J. Neurosci. 2010, 30, 16718–16729. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Hou, Q.; Jarzylo, L.; Amato, S.; Gilbert, J.; Shang, F.; Man, H.Y. Nedd4-mediated AMPA receptor ubiquitination regulates receptor turnover and trafficking. J. Neurochem. 2011, 119, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Lee, K.Y.; Jewett, K.A.; Man, H.Y.; Chung, H.J.; Tsai, N.P. Epilepsy-associated gene Nedd4-2 mediates neuronal activity and seizure susceptibility through AMPA receptors. PLOS Genet. 2017, 13, e1006634. [Google Scholar] [CrossRef]
- Fu, A.K.; Hung, K.W.; Fu, W.Y.; Shen, C.; Chen, Y.; Xia, J.; Lai, K.O.; Ip, N.Y. APC(Cdh1) mediates EphA4-dependent downregulation of AMPA receptors in homeostatic plasticity. Nat. Neurosci. 2011, 14, 181–263. [Google Scholar] [CrossRef]
- Lussier, M.P.; Herring, B.E.; Nasu-Nishimura, Y.; Neutzner, A.; Karbowski, M.; Youle, R.J.; Nicoll, R.A.; Roche, K.W. Ubiquitin ligase RNF167 regulates AMPA receptor-mediated synaptic transmission. Proc. Natl. Acad. Sci. USA 2012, 109, 19426–19431. [Google Scholar] [CrossRef]
- Goo, M.S.; Scudder, S.L.; Patrick, G.N. Ubiquitin-dependent trafficking and turnover of ionotropic glutamate receptors. Front. Mol. Neurosci. 2015, 8, 60. [Google Scholar] [CrossRef] [PubMed]
- Widagdo, J.; Kerk, J.W.; Guntupalli, S.; Huganir, R.L.; Anggono, V. Subunit-Specific Augmentation of AMPA Receptor Ubiquitination by Phorbol Ester. Cell. Mol. Neurobiol. 2020, 40, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- George, A.J.; Hoffiz, Y.C.; Charles, A.J.; Zhu, Y.; Mabb, A.M. A Comprehensive Atlas of E3 Ubiquitin Ligase Mutations in Neurological Disorders. Front. Genet. 2018, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Wan, L.P.; Li, Y.; He, C.H.; Song, N.N.; Zhao, S.; Wang, H.; Ding, Y.Q.; Mao, B.; Sheng, N. RNF220 is an E3 ubiquitin ligase for AMPA receptors to regulate synaptic transmission. Sci. Adv. 2022, 8, eabq4736. [Google Scholar] [CrossRef] [PubMed]
- Widagdo, J.; Chai, Y.J.; Ridder, M.C.; Chau, Y.Q.; Johnson, R.C.; Sah, P.; Huganir, R.L.; Anggono, V. Activity-Dependent Ubiquitination of GluA1 and GluA2 Regulates AMPA Receptor Intracellular Sorting and Degradation. Cell Rep. 2015, 10, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Lee, D.S.; Kim, M.J.; Kang, T.C. PLPP/CIN-mediated NEDD4-2 S448 dephosphorylation regulates neuronal excitability via GluA1 ubiquitination. Cell Death Dis. 2019, 10, 545. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Khatri, N.; Hou, Q.; Gilbert, J.; Wang, G.; Man, H.Y. The deubiquitinating enzyme USP46 regulates AMPA receptor ubiquitination and trafficking. J. Neurochem. 2015, 134, 1067–1080. [Google Scholar] [CrossRef]
- Widagdo, J.; Guntupalli, S.; Jang, S.E.; Anggono, V. Regulation of AMPA Receptor Trafficking by Protein Ubiquitination. Front. Mol. Neurosci. 2017, 10, 347. [Google Scholar] [CrossRef]
- Scudder, S.L.; Goo, M.S.; Cartier, A.E.; Molteni, A.; Schwarz, L.A.; Wright, R.; Patrick, G.N. Synaptic strength is bidirectionally controlled by opposing activity-dependent regulation of Nedd4-1 and USP8. J. Neurosci. 2014, 34, 16637–16649. [Google Scholar] [CrossRef]
- Henley, J.M.; Wilkinson, K.A. Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 2016, 17, 337–350. [Google Scholar] [CrossRef]
- Lalanne, T.; Oyrer, J.; Farrant, M.; Sjöström, P.J. Synapse Type-Dependent Expression of Calcium-Permeable AMPA Receptors. Front. Synaptic Neurosci. 2018, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Koster, K.P. AMPAR Palmitoylation Tunes Synaptic Strength: Implications for Synaptic Plasticity and Disease. J. Neurosci. 2019, 39, 5040–5043. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, M.; Fusco, S.; Mainardi, M.; Scala, F.; Natale, F.; Lapenta, R.; Mattera, A.; Rinaudo, M.; Puma, D.D.L.; Ripoli, C.; et al. Brain insulin resistance impairs hippocampal synaptic plasticity and memory by increasing GluA1 palmitoylation through FoxO3a. Nat. Commun. 2017, 8, 2009. [Google Scholar] [CrossRef] [PubMed]
- Casas, M.; Fadó, R.; Domínguez, J.L.; Roig, A.; Kaku, M.; Chohnan, S.; Solé, M.; Unzeta, M.; Miñano-Molina, A.J.; Rodríguez-Álvarez, J.; et al. Sensing of nutrients by CPT1C controls SAC1 activity to regulate AMPA receptor trafficking. J. Cell Biol. 2020, 219, e201912045. [Google Scholar] [CrossRef] [PubMed]
- Gratacos-Batlle, E.; Yefimenko, N.; Cascos-Garcia, H.; Soto, D. AMPAR interacting protein CPT1C enhances surface expression of GluA1-containing receptors. Front. Cell Neurosci. 2014, 8, 469. [Google Scholar] [PubMed]
- Gan, Q.; Salussolia, C.L.; Wollmuth, L.P. Assembly of AMPA receptors: Mechanisms and regulation. J. Physiol. 2014, 593, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Gratacòs-Batlle, E.; Olivella, M.; Sánchez-Fernández, N.; Yefimenko, N.; Miguez-Cabello, F.; Fadó, R.; Casals, N.; Gasull, X.; Ambrosio, S.; Soto, D. Mechanisms of CPT1C-Dependent AMPAR Trafficking Enhancement. Front. Mol. Neurosci. 2018, 11, 275. [Google Scholar] [CrossRef]
- Itoh, M.; Yamashita, M.; Kaneko, M.; Okuno, H.; Abe, M.; Yamazaki, M.; Natsume, R.; Yamada, D.; Kaizuka, T.; Suwa, R.; et al. Deficiency of AMPAR–Palmitoylation Aggravates Seizure Susceptibility. J. Neurosci. 2018, 38, 10220–10235. [Google Scholar] [CrossRef]
- Guntupalli, S.; Jang, S.E.; Zhu, T.; Huganir, R.L.; Widagdo, J.; Anggono, V. GluA1 subunit ubiquitination mediates amyloid-beta-induced loss of surface alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. J. Biol. Chem. 2017, 292, 8186–8194. [Google Scholar] [CrossRef]
- Yang, G.; Xiong, W.; Kojic, L.; Cynader, M.S. Subunit-selective palmitoylation regulates the intracellular trafficking of AMPA receptor. Eur. J. Neurosci. 2009, 30, 35–46. [Google Scholar] [CrossRef]
- Kessels, H.W.; Kopec, C.D.; Klein, M.E.; Malinow, R. Roles of stargazin and phosphorylation in the control of AMPA receptor subcellular distribution. Nat. Neurosci. 2009, 12, 888–896. [Google Scholar] [CrossRef]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef]
- Christensen, N.R.; De Luca, M.; Lever, M.B.; Richner, M.; Hansen, A.B.; Noes-Holt, G.; Jensen, K.L.; Rathje, M.; Jensen, D.B.; Erlendsson, S.; et al. A high-affinity, bivalent PDZ domain inhibitor complexes PICK1 to alleviate neuropathic pain. EMBO Mol. Med. 2020, 12, e11248. [Google Scholar] [CrossRef] [PubMed]
- Miletic, G.; Hermes, J.L.; Bosscher, G.L.; Meier, B.M.; Miletic, V. Protein kinase C gamma-mediated phosphorylation of GluA1 in the postsynaptic density of spinal dorsal horn neurons accompanies neuropathic pain, and dephosphorylation by calcineurin is associated with prolonged analgesia. Pain 2015, 156, 2514–2520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lei, M.; Wen, Q.; Zhang, D.; Qin, G.; Zhou, J.; Chen, L. Dopamine receptor D2 regulates GLUA1-containing AMPA receptor trafficking and central sensitization through the PI3K signaling pathway in a male rat model of chronic migraine. J. Headache Pain 2022, 23, 98. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yuan, J.; Yu, X.; Liu, X.; Tan, C.; Chen, Y.; Xu, T. Vezatin regulates seizures by controlling AMPAR-mediated synaptic activity. Cell Death Dis. 2021, 12, 936. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, O.; Huo, Y.; Wang, G.; Man, H.Y. Amyloid-beta Induces AMPA Receptor Ubiquitination and Degradation in Primary Neurons and Human Brains of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1789–1801. [Google Scholar] [CrossRef]
- Amin, J.B.; Salussolia, C.L.; Chan, K.; Regan, M.C.; Dai, J.; Zhou, H.X.; Furukawa, H.; Bowen, M.E.; Wollmuth, L.P. Divergent roles of a peripheral transmembrane segment in AMPA and NMDA receptors. J. Gen. Physiol. 2017, 149, 661–680. [Google Scholar] [CrossRef]
- O’Connor, M.; Shentu, Y.P.; Wang, G.; Hu, W.T.; Xu, Z.D.; Wang, X.C.; Liu, R.; Man, H.Y. Acetylation of AMPA Receptors Regulates Receptor Trafficking and Rescues Memory Deficits in Alzheimer’s Disease. iScience 2020, 23, 101465. [Google Scholar] [CrossRef]
- Van Dolah, D.K.; Mao, L.M.; Shaffer, C.; Guo, M.L.; Fibuch, E.E.; Chu, X.P.; Buch, S.; Wang, J.Q. Reversible Palmitoylation Regulates Surface Stability of AMPA Receptors in the Nucleus Accumbens in Response to Cocaine In Vivo. Biol. Psychiatry 2011, 69, 1035–1042. [Google Scholar] [CrossRef]
- Kamalova, A.; Nakagawa, T. AMPA receptor structure and auxiliary subunits. J. Physiol. 2021, 599, 453–469. [Google Scholar] [CrossRef] [PubMed]
- Bowie, D. The many faces of the AMPA-type ionotropic glutamate receptor. Neuropharmacology 2022, 208, 108975. [Google Scholar] [CrossRef] [PubMed]
- Hanley, J.G. Regulation of AMPAR expression by microRNAs. Neuropharmacology 2021, 197, 108723. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Bach, J.R.; Sharma, H.S.; Saberi, H.; Jeon, S.R.; Guo, X.; Shetty, A.; Hawamdeh, Z.; Sharma, A.; von Wild, K.; et al. The 2022 yearbook of Neurorestoratology. J. Neurorestoratol. 2023, 11, 100054. [Google Scholar] [CrossRef]
- Aleksandrova, L.R.; Phillips, A.G. Neuroplasticity as a convergent mechanism of ketamine and classical psychedelics. Trends Pharmacol. Sci. 2021, 42, 929–942. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Interactor | Proposed Function | Interaction Subunits | Citations | |
|---|---|---|---|---|
| Vesicle formation | Rab11 | AMPAR is transported in Rab11-positive recycling endosomes along microtubule tracks within the dendritic shaft | GluA1 | [26] |
| CaMKII | Phosphorylates Stg and GluA1 as well as facilitates the localization and transport of GluA1 | GluA1 | [35] | |
| Vesicle trafficking | MAP | Binds to microtubules and microfilaments, mediating or regulating the interaction between axonal microtubules and actin filaments | [43] | |
| Actin | Promotes receptor localization and provides transport routes and power | GluA1 | [47] | |
| Post-synaptic site of AMPAR | PSD-95 | Induces AMPARs to localize on the PSD | GluA1 | [54,58] |
| SAP-97 | Promotes the expression of GluA1 at the membrane | GluA1 | [70] | |
| PKA | Promotes the expression of GluA1 | GluA1 | [78] | |
| PKC | Reduces the affinity of GRIP1/2 for GluA2 and promotes the internalization of GluA2 | GluA2 | [97] | |
| EPB41L1 | AMPAR is anchored to the PSD by SAP97 and EPB41L1 | GluA1 | [100] | |
| Vesicle fusion | SNARE complex | Mediates vesicle localization and membrane fusion | GluA2 | [106] |
| NSF | Promotes the decomposition of SNARE complexes to accelerate circulation and increase the expression of AMPARs at the membrane | GluA2 | [110] | |
| AP-2 | Induces the internalization of AMPARs | GluA2 | [116] |
| AMPAR PTM | Functional Subunits and Sites | Related Proteins | Diseases | Citations |
|---|---|---|---|---|
| Phosphorylation | GluA2 (S880) | / | AD | [192] |
| GluA2 (S880) | PKCα, PICK1 | Neuropathic Pain | [152,193] | |
| GluA1 (S831) | PKC γ, PI3K/AKT | Neuropathic Pain | [194,195] | |
| GluA1 (S845) | Vezatin, PKA | Epilepsy | [196] | |
| Ubiquitination | GluA1 (S845) | Nedd4, USP46 | AD | [197,198] |
| GluA1 (S845) | Nedd4-2, PLPP/CIN | Epilepsy | [168,176] | |
| Palmitoylation | GluA1(K813, K819, K822, K868) | P300 | AD | [199] |
| Acetylation | GluA1 (C811) | Cys1 | Epilepsy | [188] |
| GluA1, GluA3 | / | Addictive Disorders | [200] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Y.-Y.; Wu, L.-L.; Li, X.-N.; Yuan, Y.-L.; Zhao, W.-W.; Qi, J.-X.; Zhao, X.-Y.; Ward, N.; Wang, J. Molecular Mechanisms of AMPA Receptor Trafficking in the Nervous System. Int. J. Mol. Sci. 2024, 25, 111. https://doi.org/10.3390/ijms25010111
Cao Y-Y, Wu L-L, Li X-N, Yuan Y-L, Zhao W-W, Qi J-X, Zhao X-Y, Ward N, Wang J. Molecular Mechanisms of AMPA Receptor Trafficking in the Nervous System. International Journal of Molecular Sciences. 2024; 25(1):111. https://doi.org/10.3390/ijms25010111
Chicago/Turabian StyleCao, Yi-Yang, Ling-Ling Wu, Xiao-Nan Li, Yu-Lian Yuan, Wan-Wei Zhao, Jing-Xuan Qi, Xu-Yu Zhao, Natalie Ward, and Jiao Wang. 2024. "Molecular Mechanisms of AMPA Receptor Trafficking in the Nervous System" International Journal of Molecular Sciences 25, no. 1: 111. https://doi.org/10.3390/ijms25010111
APA StyleCao, Y. -Y., Wu, L. -L., Li, X. -N., Yuan, Y. -L., Zhao, W. -W., Qi, J. -X., Zhao, X. -Y., Ward, N., & Wang, J. (2024). Molecular Mechanisms of AMPA Receptor Trafficking in the Nervous System. International Journal of Molecular Sciences, 25(1), 111. https://doi.org/10.3390/ijms25010111