

Inherited Retinal Degeneration Caused by Dehydrodolichyl Diphosphate Synthase Mutation–Effect of an ALG6 Modifier Variant


 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Selection of Potential Modifier Variants
2.2. DNA Sequencing Results Shows That Only ALG6 Displays Sequence Variation
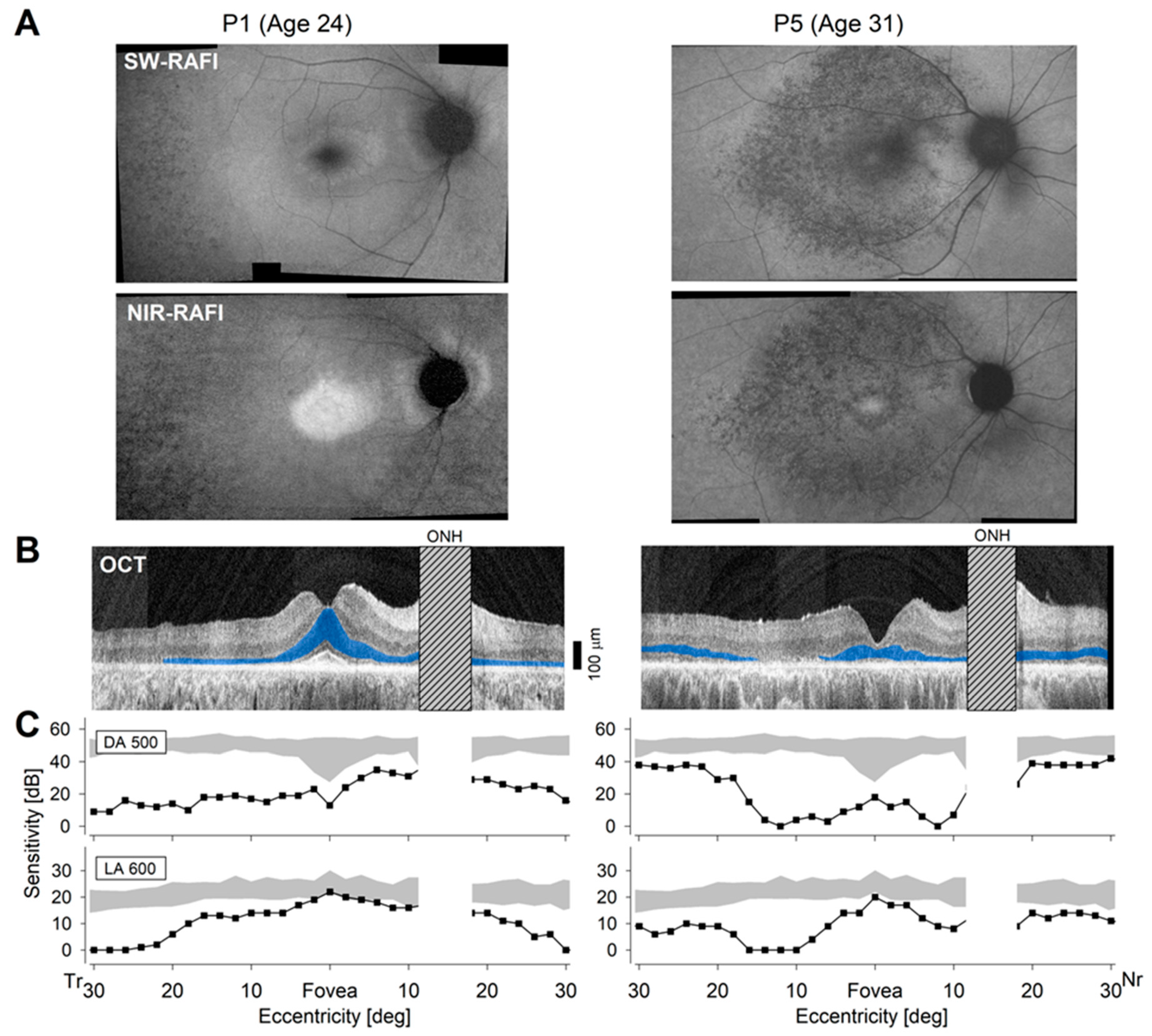
2.3. Differences in Severity of Disease in Patients
3. Discussion
4. Materials and Methods
4.1. Patient DNA Procurement
4.2. Clinical Assessments
4.3. PCR Amplification and DNA Sequencing
4.4. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Varki, A. Biological roles of oligosaccharides: All of the theories are correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhang, H. Glycoproteomics and clinical applications. Proteom. Clin. Appl. 2010, 4, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Schjoldager, K.T.; Narimatsu, Y.; Joshi, H.J.; Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 729–749. [Google Scholar] [CrossRef]
- Aebi, M. N-linked protein glycosylation in the ER. Biochim. Biophys. Acta 2013, 1833, 2430–2437. [Google Scholar] [CrossRef] [PubMed]
- Esmail, S.; Manolson, M.F. Advances in understanding N-glycosylation structure, function, and regulation in health and disease. Eur. J. Cell Biol. 2021, 100, 151186. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J. Congenital disorders of glycosylation. Handb. Clin. Neurol. 2013, 113, 1737–1743. [Google Scholar] [CrossRef]
- Ondruskova, N.; Cechova, A.; Hansikova, H.; Honzik, T.; Jaeken, J. Congenital disorders of glycosylation: Still “hot” in 2020. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129751. [Google Scholar] [CrossRef]
- Verheijen, J.; Tahata, S.; Kozicz, T.; Witters, P.; Morava, E. Therapeutic approaches in Congenital Disorders of Glycosylation (CDG) involving N-linked glycosylation: An update. Genet. Med. 2020, 22, 268–279. [Google Scholar] [CrossRef]
- Jaeken, J. Congenital disorders of glycosylation: A multi-genetic disease family with multiple subcellular locations. J. Mother. Child. 2020, 24, 14–20. [Google Scholar] [CrossRef]
- Carroll, K.K.; Guthrie, N.; Ravi, K. Dolichol: Function, metabolism, and accumulation in human tissues. Biochem. Cell Biol. 1992, 70, 382–384. [Google Scholar] [CrossRef]
- Schwarz, R.T.; Datema, R. Inhibition of the dolichol pathway of protein glycosylation. Methods Enzymol. 1982, 83, 432–443. [Google Scholar] [CrossRef]
- Kean, E.L. The dolichol pathway in the retina and its involvement in the glycosylation of rhodopsin. Biochim. Biophys. Acta 1999, 1473, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Zelinger, L.; Banin, E.; Obolensky, A.; Mizrahi-Meissonnier, L.; Beryozkin, A.; Bandah-Rozenfeld, D.; Frenkel, S.; Ben-Yosef, T.; Merin, S.; Schwartz, S.B.; et al. A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in Ashkenazi Jews. Am. J. Hum. Genet. 2011, 88, 207–215. [Google Scholar] [CrossRef]
- Zuchner, S.; Dallman, J.; Wen, R.; Beecham, G.; Naj, A.; Farooq, A.; Kohli, M.A.; Whitehead, P.L.; Hulme, W.; Konidari, I.; et al. Whole-exome sequencing links a variant in DHDDS to retinitis pigmentosa. Am. J. Hum. Genet. 2011, 88, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.L.; Zuchner, S.L.; Dallman, J.; Wen, R.; Alfonso, E.C.; Vance, J.M.; Pericak-Vance, M.A. Mutation K42E in dehydrodolichol diphosphate synthase (DHDDS) causes recessive retinitis pigmentosa. Adv. Exp. Med. Biol. 2014, 801, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Daiger, S.P.; Rossiter, B.J.F.; Greenberg, J.; Christoffels, A.; Hide, W. Data services and software for identifying genes and mutations causing retinal degeneration. Investig. Ophthalmol. Vis. Sci. 1998, 39, S295. [Google Scholar]
- Hanany, M.; Rivolta, C.; Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 2710–2716. [Google Scholar] [CrossRef]
- Pizzo, L.; Jensen, M.; Polyak, A.; Rosenfeld, J.A.; Mannik, K.; Krishnan, A.; McCready, E.; Pichon, O.; Le Caignec, C.; Van Dijck, A.; et al. Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet. Med. 2019, 21, 816–825. [Google Scholar] [CrossRef]
- Rahit, K.; Tarailo-Graovac, M. Genetic Modifiers and Rare Mendelian Disease. Genes 2020, 11, 239. [Google Scholar] [CrossRef]
- Haider, N.B.; Ikeda, A.; Naggert, J.K.; Nishina, P.M. Genetic modifiers of vision and hearing. Hum. Mol. Genet. 2002, 11, 1195–1206. [Google Scholar] [CrossRef]
- Pacione, L.R.; Szego, M.J.; Ikeda, S.; Nishina, P.M.; McInnes, R.R. Progress toward understanding the genetic and biochemical mechanisms of inherited photoreceptor degenerations. Annu. Rev. Neurosci. 2003, 26, 657–700. [Google Scholar] [CrossRef] [PubMed]
- Gifford, C.A.; Ranade, S.S.; Samarakoon, R.; Salunga, H.T.; de Soysa, T.Y.; Huang, Y.; Zhou, P.; Elfenbein, A.; Wyman, S.K.; Bui, Y.K.; et al. Oligogenic inheritance of a human heart disease involving a genetic modifier. Science 2019, 364, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Bis-Brewer, D.M.; Fazal, S.; Zuchner, S. Genetic modifiers and non-Mendelian aspects of CMT. Brain Res. 2020, 1726, 146459. [Google Scholar] [CrossRef]
- Leclere, J.C.; Le Gac, M.S.; Le Marechal, C.; Ferec, C.; Marianowski, R. GJB2 mutations: Genotypic and phenotypic correlation in a cohort of 690 hearing-impaired patients, toward a new mutation? Int. J. Pediatr. Otorhinolaryngol. 2017, 102, 80–85. [Google Scholar] [CrossRef]
- Danciger, M.; Matthes, M.T.; Yasamura, D.; Akhmedov, N.B.; Rickabaugh, T.; Gentleman, S.; Redmond, T.M.; La Vail, M.M.; Farber, D.B. A QTL on distal chromosome 3 that influences the severity of light-induced damage to mouse photoreceptors. Mamm. Genome 2000, 11, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, A.; Reme, C.E.; Williams, T.P.; Hafezi, F.; Grimm, C. The Rpe65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration. J. Neurosci. 2001, 21, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Schneider, N.; Sundaresan, Y.; Gopalakrishnan, P.; Beryozkin, A.; Hanany, M.; Levanon, E.Y.; Banin, E.; Ben-Aroya, S.; Sharon, D. Inherited retinal diseases: Linking genes, disease-causing variants, and relevant therapeutic modalities. Prog. Retin. Eye Res. 2022, 89, 101029. [Google Scholar] [CrossRef]
- Rao, K.N.; Zhang, W.; Li, L.; Ronquillo, C.; Baehr, W.; Khanna, H. Ciliopathy-associated protein CEP290 modifies the severity of retinal degeneration due to loss of RPGR. Hum. Mol. Genet. 2016, 25, 2005–2012. [Google Scholar] [CrossRef]
- Rose, A.M.; Shah, A.Z.; Venturini, G.; Krishna, A.; Chakravarti, A.; Rivolta, C.; Bhattacharya, S.S. Transcriptional regulation of PRPF31 gene expression by MSR1 repeat elements causes incomplete penetrance in retinitis pigmentosa. Sci. Rep. 2016, 6, 19450. [Google Scholar] [CrossRef]
- Vollrath, D.; Yasumura, D.; Benchorin, G.; Matthes, M.T.; Feng, W.; Nguyen, N.M.; Sedano, C.D.; Calton, M.A.; LaVail, M.M. Tyro3 Modulates Mertk-Associated Retinal Degeneration. PLoS Genet. 2015, 11, e1005723. [Google Scholar] [CrossRef]
- Fahim, A.T.; Bowne, S.J.; Sullivan, L.S.; Webb, K.D.; Williams, J.T.; Wheaton, D.K.; Birch, D.G.; Daiger, S.P. Allelic heterogeneity and genetic modifier loci contribute to clinical variation in males with X-linked retinitis pigmentosa due to RPGR mutations. PLoS ONE 2011, 6, e23021. [Google Scholar] [CrossRef]
- Appelbaum, T.; Murgiano, L.; Becker, D.; Santana, E.; Aguirre, G.D. Candidate Genetic Modifiers for RPGR Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2020, 61, 20. [Google Scholar] [CrossRef]
- Ripolles-Garcia, A.; Murgiano, L.; Ziolkowska, N.; Marinho, F.P.; Roszak, K.; Iffrig, S.; Aguirre, G.D.; Miyadera, K. Natural disease history of a canine model of oligogenic RPGRIP1-cone-rod dystrophy establishes variable effects of previously and newly mapped modifier loci. Hum. Mol. Genet. 2023, 32, 2139–2151. [Google Scholar] [CrossRef] [PubMed]
- Sabry, S.; Vuillaumier-Barrot, S.; Mintet, E.; Fasseu, M.; Valayannopoulos, V.; Heron, D.; Dorison, N.; Mignot, C.; Seta, N.; Chantret, I.; et al. A case of fatal Type I congenital disorders of glycosylation (CDG I) associated with low dehydrodolichol diphosphate synthase (DHDDS) activity. Orphanet J. Rare Dis. 2016, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- Imbach, T.; Grunewald, S.; Schenk, B.; Burda, P.; Schollen, E.; Wevers, R.A.; Jaeken, J.; de Klerk, J.B.; Berger, E.G.; Matthijs, G.; et al. Multi-allelic origin of congenital disorder of glycosylation (CDG)-Ic. Hum. Genet. 2000, 106, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Westphal, V.; Kjaergaard, S.; Schollen, E.; Martens, K.; Grunewald, S.; Schwartz, M.; Matthijs, G.; Freeze, H.H. A frequent mild mutation in ALG6 may exacerbate the clinical severity of patients with congenital disorder of glycosylation Ia (CDG-Ia) caused by phosphomannomutase deficiency. Hum. Mol. Genet. 2002, 11, 599–604. [Google Scholar] [CrossRef]
- Bloch, J.S.; Pesciullesi, G.; Boilevin, J.; Nosol, K.; Irobalieva, R.N.; Darbre, T.; Aebi, M.; Kossiakoff, A.A.; Reymond, J.L.; Locher, K.P. Structure and mechanism of the ER-based glucosyltransferase ALG6. Nature 2020, 579, 443–447. [Google Scholar] [CrossRef]
- Yamagata, T.; Tsuru, T.; Momoi, M.Y.; Suwa, K.; Nozaki, Y.; Mukasa, T.; Ohashi, H.; Fukushima, Y.; Momoi, T. Genome organization of human 48-kDa oligosaccharyltransferase (DDOST). Genomics 1997, 45, 535–540. [Google Scholar] [CrossRef]
- Schenk, B.; Imbach, T.; Frank, C.G.; Grubenmann, C.E.; Raymond, G.V.; Hurvitz, H.; Korn-Lubetzki, I.; Revel-Vik, S.; Raas-Rotschild, A.; Luder, A.S.; et al. MPDU1 mutations underlie a novel human congenital disorder of glycosylation, designated type If. J. Clin. Investig. 2001, 108, 1687–1695. [Google Scholar] [CrossRef]
- Anand, M.; Rush, J.S.; Ray, S.; Doucey, M.A.; Weik, J.; Ware, F.E.; Hofsteenge, J.; Waechter, C.J.; Lehrman, M.A. Requirement of the Lec35 gene for all known classes of monosaccharide-P-dolichol-dependent glycosyltransferase reactions in mammals. Mol. Biol. Cell 2001, 12, 487–501. [Google Scholar] [CrossRef]
- Yue, X.; Tiwari, N.; Zhu, L.; Ngo, H.D.T.; Lim, J.M.; Gim, B.; Jing, S.; Wang, Y.; Qian, Y.; Lee, I. Tankyrase-1-mediated degradation of Golgin45 regulates glycosyltransferase trafficking and protein glycosylation in Rab2-GTP-dependent manner. Commun. Biol. 2021, 4, 1370. [Google Scholar] [CrossRef] [PubMed]
- Sauna, Z.E.; Kimchi-Sarfaty, C. Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 2011, 12, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Hunt, R.C.; Simhadri, V.L.; Iandoli, M.; Sauna, Z.E.; Kimchi-Sarfaty, C. Exposing synonymous mutations. Trends Genet. 2014, 30, 308–321. [Google Scholar] [CrossRef]
- Rauscher, R.; Ignatova, Z. Timing during translation matters: Synonymous mutations in human pathologies influence protein folding and function. Biochem. Soc. Trans. 2018, 46, 937–944. [Google Scholar] [CrossRef]
- Westphal, V.; Schottstädt, C.; Marquardt, T.; Freeze, H.H. Analysis of multiple mutations in the hALG6 gene in a patient with congenital disorder of glycosylation Ic. Mol. Genet. Metab. 2000, 70, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Matsui, R.; Cideciyan, A.V.; Schwartz, S.B.; Sumaroka, A.; Roman, A.J.; Swider, M.; Huang, W.C.; Sheplock, R.; Jacobson, S.G. Molecular heterogeneity within the clinical diagnosis of pericentral retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6007–6018. [Google Scholar] [CrossRef] [PubMed]
- Venturini, G.; Koskiniemi-Kuendig, H.; Harper, S.; Berson, E.L.; Rivolta, C. Two specific mutations are prevalent causes of recessive retinitis pigmentosa in North American patients of Jewish ancestry. Genet. Med. 2015, 17, 285–290. [Google Scholar] [CrossRef]
- Biswas, P.; Duncan, J.L.; Maranhao, B.; Kozak, I.; Branham, K.; Gabriel, L.; Lin, J.H.; Barteselli, G.; Navani, M.; Suk, J.; et al. Genetic analysis of 10 pedigrees with inherited retinal degeneration by exome sequencing and phenotype-genotype association. Physiol. Genom. 2017, 49, 216–229. [Google Scholar] [CrossRef]
- Hariri, A.H.; Gui, W.; Datoo O’Keefe, G.A.; Ip, M.S.; Sadda, S.R.; Gorin, M.B. Ultra-Widefield Fundus Autofluorescence Imaging of Patients with Retinitis Pigmentosa: A Standardized Grading System in Different Genotypes. Ophthalmol. Retin. 2018, 2, 735–745. [Google Scholar] [CrossRef]
- Kimchi, A.; Khateb, S.; Wen, R.; Guan, Z.; Obolensky, A.; Beryozkin, A.; Kurtzman, S.; Blumenfeld, A.; Pras, E.; Jacobson, S.G.; et al. Nonsyndromic Retinitis Pigmentosa in the Ashkenazi Jewish Population: Genetic and Clinical Aspects. Ophthalmology 2018, 125, 725–734. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Swider, M.; Aleman, T.S.; Sumaroka, A.; Schwartz, S.B.; Roman, M.I.; Milam, A.H.; Bennett, J.; Stone, E.M.; Jacobson, S.G. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4739–4746. [Google Scholar] [CrossRef]
- Aleman, T.S.; Uyhazi, K.E.; Serrano, L.W.; Vasireddy, V.; Bowman, S.J.; Ammar, M.J.; Pearson, D.J.; Maguire, A.M.; Bennett, J. RDH12 Mutations Cause a Severe Retinal Degeneration With Relatively Spared Rod Function. Investig. Ophthalmol. Vis. Sci. 2018, 59, 5225–5236. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Jacobson, S.G.; Sumaroka, A.; Swider, M.; Krishnan, A.K.; Sheplock, R.; Garafalo, A.V.; Guziewicz, K.E.; Aguirre, G.D.; Beltran, W.A.; et al. Photoreceptor function and structure in retinal degenerations caused by biallelic BEST1 mutations. Vis. Res. 2023, 203, 108157. [Google Scholar] [CrossRef]
- Burgunder, J.M. Mechanisms underlying phenotypic variation in neurogenetic disorders. Nat. Rev. Neurol. 2023, 19, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Vuillaumier-Barrot, S.; Le Bizec, C.; Durand, G.; Seta, N. The T911C (F304S) substitution in the human ALG6 gene is a common polymorphism and not a causal mutation of CDG-Ic. J. Hum. Genet. 2001, 46, 547–548. [Google Scholar] [CrossRef] [PubMed]
- Goreta, S.S.; Dabelic, S.; Pavlinic, D.; Lauc, G.; Dumic, J. Frequency Determination of α-1,3 Glucosyltransferase p.Y131H and p.F304S Polymorphisms in the Croatian Population Revealed Five Novel Single Nucleotide Polymorphisms in the hALG6 Gene. Genet. Test. Mol. Biomark. 2012, 16, 50–53. [Google Scholar] [CrossRef]
- Wang, N.; Li, S.T.; Xiang, M.H.; Gao, X.D. Alg mannosyltransferases: From functional and structural analyses to the lipid-linked oligosaccharide pathway reconstitution. Biochim. Biophys. Acta Gen. Subj. 2022, 1866, 130112. [Google Scholar] [CrossRef]
- Kornfeld, R.; Kornfeld, S. Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 1985, 54, 631–664. [Google Scholar] [CrossRef]
- Elbein, A.D. Inhibitors of the biosynthesis and processing of N-linked oligosaccharides. CRC Crit. Rev. Biochem. 1984, 16, 21–49. [Google Scholar] [CrossRef]
- Yan, A.; Lennarz, W.J. Unraveling the mechanism of protein N-glycosylation. J. Biol. Chem. 2005, 280, 3121–3124. [Google Scholar] [CrossRef]
- Shrimal, S.; Gilmore, R. Oligosaccharyltransferase structures provide novel insight into the mechanism of asparagine-linked glycosylation in prokaryotic and eukaryotic cells. Glycobiology 2019, 29, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Haeuptle, M.A.; Hennet, T. Congenital disorders of glycosylation: An update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum. Mutat. 2009, 30, 1628–1641. [Google Scholar] [CrossRef]
- Zahn-Zabal, M.; Michel, P.A.; Gateau, A.; Nikitin, F.; Schaeffer, M.; Audot, E.; Gaudet, P.; Duek, P.D.; Teixeira, D.; Rech de Laval, V.; et al. The neXtProt knowledgebase in 2020: Data, tools and usability improvements. Nucleic Acids Res. 2020, 48, D328–D334. [Google Scholar] [CrossRef] [PubMed]
- Dercksen, M.; Crutchley, A.C.; Honey, E.M.; Lippert, M.M.; Matthijs, G.; Mienie, L.J.; Schuman, H.C.; Vorster, B.C.; Jaeken, J. ALG6-CDG in South Africa: Genotype-Phenotype Description of Five Novel Patients. JIMD Rep. 2013, 8, 17–23. [Google Scholar] [CrossRef]
- Willemse, S.W.; van Es, M.A. Susceptibility and disease modifier genes in amyotrophic lateral sclerosis: From genetic associations to therapeutic implications. Curr. Opin. Neurol. 2023, 36, 365–370. [Google Scholar] [CrossRef]
- Mésinèle, J.; Ruffin, M.; Guillot, L.; Corvol, H. Modifier Factors of Cystic Fibrosis Phenotypes: A Focus on Modifier Genes. Int. J. Mol. Sci. 2022, 23, 4205. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Yagasaki, K.; Feuer, W.J.; Roman, A.J. Interocular asymmetry of visual function in heterozygotes of X-linked retinitis pigmentosa. Exp. Eye Res. 1989, 48, 679–691. [Google Scholar] [CrossRef]
- Jacobson, S.G.; Cideciyan, A.V.; Kemp, C.M.; Sheffield, V.C.; Stone, E.M. Photoreceptor function in heterozygotes with insertion or deletion mutations in the RDS gene. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1662–1674. [Google Scholar]
- Jacobson, S.G.; Voigt, W.J.; Parel, J.M.; Apathy, P.P.; Nghiem-Phu, L.; Myers, S.W.; Patella, V.M. Automated light- and dark-adapted perimetry for evaluating retinitis pigmentosa. Ophthalmology 1986, 93, 1604–1611. [Google Scholar] [CrossRef]
- Roman, A.J.; Schwartz, S.B.; Aleman, T.S.; Cideciyan, A.V.; Chico, J.D.; Windsor, E.A.M.; Gardner, L.M.; Ying, G.S.; Smilko, E.E.; Maguire, M.G.; et al. Quantifying rod photoreceptor-mediated vision in retinal degenerations: Dark-adapted thresholds as outcome measures. Exp. Eye Res. 2005, 80, 259–272. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Krishnan, A.K.; Roman, A.J.; Sumaroka, A.; Swider, M.; Jacobson, S.G. Measures of function and structure to determine phenotypic features, natural history, and treatment outcomes in inherited retinal diseases. Annu. Rev. Vis. Sci. 2021, 7, 747–772. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Cideciyan, A.V.; Aleman, T.S.; Pianta, M.J.; Sumaroka, A.; Schwartz, S.B.; Smilko, E.E.; Milam, A.H.; Sheffield, V.C.; Stone, E.M. Crumbs homolog 1 (CRB1) mutations result in a thick human retina with abnormal lamination. Hum. Mol. Genet. 2003, 12, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Hufnagel, R.B.; Carroll, J.; Sumaroka, A.; Luo, X.; Schwartz, S.B.; Dubra, A.; Land, M.; Michaelides, M.; Gardner, J.C.; et al. Human cone visual pigment deletions spare sufficient photoreceptors to warrant gene therapy. Hum. Gene Ther. 2013, 24, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Swider, M.; Aleman, T.S.; Roman, M.I.; Sumaroka, A.; Schwartz, S.B.; Stone, E.M.; Jacobson, S.G. Reduced-illuminance autofluorescence imaging in ABCA4-associated retinal degenerations. J. Opt. Soc. Am. A 2007, 24, 1457. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Ramachandra Rao, S.; Fliesler, S.J.; Kotla, P.; Nguyen, M.N.; Pittler, S.J. Lack of Overt Retinal Degeneration in a K42E Dhdds Knock-In Mouse Model of RP59. Cells 2020, 9, 896. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene | Location | Protein | Mutation Type | Reference |
|---|---|---|---|---|
| ALG6 | c.911 T→C | F304S | Missense | [34,35,45] |
| ALG8 | c.1068C→G | P356 = ** | Point (synonymous) | [34] |
| DDOST | c.679A→G | I227V | Missense | [34] |
| MPDU1 | c.393C→T | V131 = ** | Point (synonymous) | [34] |
| TNKS | c.1945G→A | D649N | Missense | [34] |
| Patient | ALG6 | ALG8 | DDOST | MPDU1 | TNKS | DHDDS |
|---|---|---|---|---|---|---|
| P1 | −/− | −/− | +/+ | −/− | −/− | +/+ |
| P2 | −/− | −/− | +/+ | −/− | −/− | +/+ |
| P3 | −/− | −/− | +/+ | −/− | −/− | +/+ |
| P4 | +/− | −/− | +/+ | −/− | −/− | +/+ |
| P5 | +/− | −/− | +/+ | −/− | −/− | +/+ |
| P6 | +/− | −/− | +/+ | −/− | −/− | +/+ |
| P7 | −/− | −/− | +/+ | −/− | −/− | +/+ |
| P8 | −/− | −/− | +/+ | −/− | −/− | +/+ |
| P9 | +/− | −/− | +/+ | −/− | −/− | +/+ |
| P10 | −/− | −/− | +/+ | −/− | −/− | +/+ |
| P11 | +/− | −/− | +/+ | −/− | −/− | +/+ |
| C1 | −/− | −/− | +/+ | −/− | −/− | −/− |
| C2 | +/+ | −/− | +/+ | −/− | −/− | −/− |
| C3 | +/− | −/− | +/+ | −/− | −/− | −/− |
| Patient | Alternate ID † | Gender | Diagnosis ** |
|---|---|---|---|
| P1 | CHRD5308 | M | RP |
| P2 | CHRD4047 | M | RP |
| P3 | CHRD3323 | M | RP |
| P4 | CHRD0262 | M | RP |
| P5 | * | F | RP |
| P6 | CHRD5151 | M | RP |
| P7 | CHRD0677 | M | RP |
| P8 | CHRD3458 | M | RP |
| P9 | MOL0884-2 | F | CRD |
| P10 | MOL0884-1 | F | CRD |
| P11 | MOL0884-4 | M | CRD |
| C1 | * | F | control |
| C2 | * | F | control |
| C3 | * | F | control |
| Gene | Forward Primer | Reverse Primer | Size (nt) |
|---|---|---|---|
| ALG6 | 5′-TCTAGTAGCTTCCTGCTCCCT | 5′-ATCCTTTGGAAGAGGGCTGAA | 575 |
| ALG8 | 5′-GCTGTCTTTCAGAGATGATGCAA | 5′-GCCACCCAAACATAAAGGAGC | 198 |
| DDOST | 5′-GTGGCCGATCCTGATAACCC | 5′-CCAGCAATGAGGAGGGTGTT | 373 |
| MPDU1 | 5′-CTGCTTCCTGGTCATGCACT | 5′-GGGTGACTACAGTCAAGGGC | 240 |
| TNKS | 5′-TTGTGTGGCTTCCCTAGGTTTG | 5′-CTTCACAGTTTCCAAGTCTCCA | 276 |
| DHDDS | 5′-TCACCTTGGGGTGTAGTGTCT | 5′-AACACTCTCCAACCACAGCAA | 291 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monson, E.; Cideciyan, A.V.; Roman, A.J.; Sumaroka, A.; Swider, M.; Wu, V.; Viarbitskaya, I.; Jacobson, S.G.; Fliesler, S.J.; Pittler, S.J. Inherited Retinal Degeneration Caused by Dehydrodolichyl Diphosphate Synthase Mutation–Effect of an ALG6 Modifier Variant. Int. J. Mol. Sci. 2024, 25, 1004. https://doi.org/10.3390/ijms25021004
Monson E, Cideciyan AV, Roman AJ, Sumaroka A, Swider M, Wu V, Viarbitskaya I, Jacobson SG, Fliesler SJ, Pittler SJ. Inherited Retinal Degeneration Caused by Dehydrodolichyl Diphosphate Synthase Mutation–Effect of an ALG6 Modifier Variant. International Journal of Molecular Sciences. 2024; 25(2):1004. https://doi.org/10.3390/ijms25021004
Chicago/Turabian StyleMonson, Elisha, Artur V. Cideciyan, Alejandro J. Roman, Alexander Sumaroka, Malgorzata Swider, Vivian Wu, Iryna Viarbitskaya, Samuel G. Jacobson, Steven J. Fliesler, and Steven J. Pittler. 2024. "Inherited Retinal Degeneration Caused by Dehydrodolichyl Diphosphate Synthase Mutation–Effect of an ALG6 Modifier Variant" International Journal of Molecular Sciences 25, no. 2: 1004. https://doi.org/10.3390/ijms25021004
APA StyleMonson, E., Cideciyan, A. V., Roman, A. J., Sumaroka, A., Swider, M., Wu, V., Viarbitskaya, I., Jacobson, S. G., Fliesler, S. J., & Pittler, S. J. (2024). Inherited Retinal Degeneration Caused by Dehydrodolichyl Diphosphate Synthase Mutation–Effect of an ALG6 Modifier Variant. International Journal of Molecular Sciences, 25(2), 1004. https://doi.org/10.3390/ijms25021004