

Could Targeting NPM1c+ Misfolding Be a Promising Strategy for Combating Acute Myeloid Leukemia?



Abstract
:1. Introduction
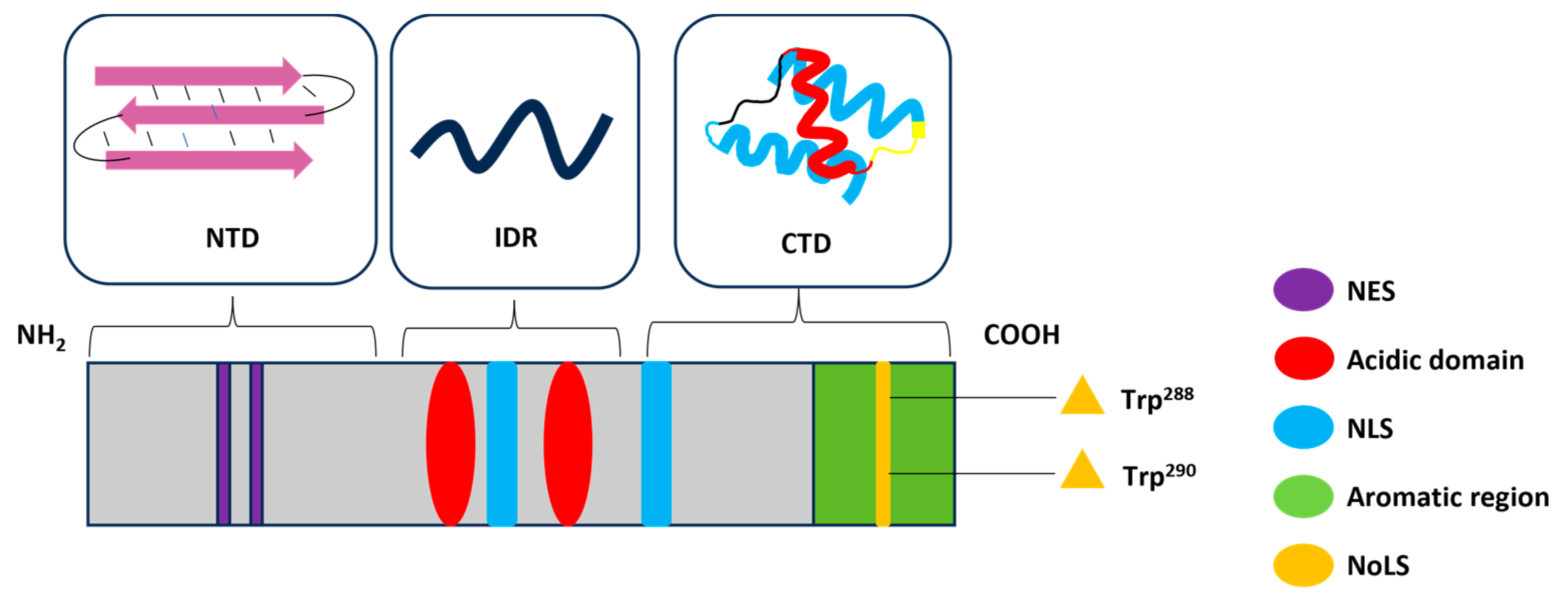
2. Structure and Functions of NPM1
3. NPM1 Is the Most Commonly Mutated Gene in Adult AML
3.1. Common NPM1 Mutations and Structural Consequences
3.2. Rare Mutations of Exon 5 and Fusion Transcripts of NPM1c+
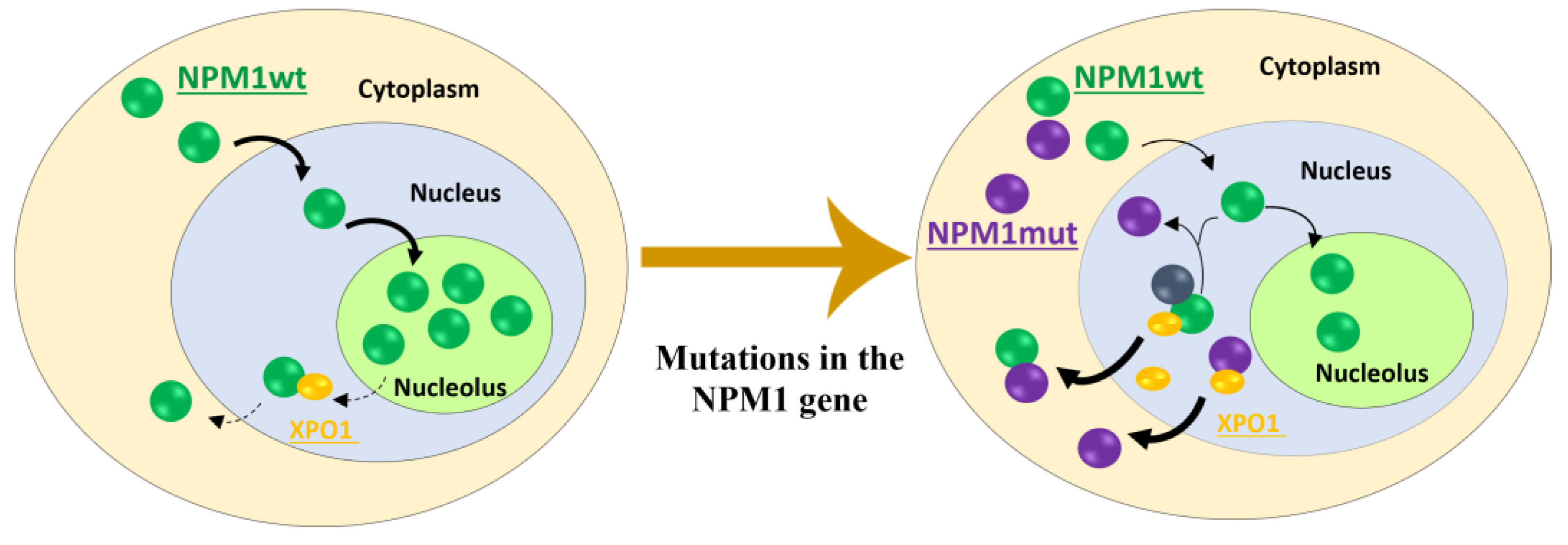
3.3. Consequences of Cytoplasmic Mislocation of NPM1c+
4. NPM1c+ Mutations Govern the Amyloidogenicity of the CTD
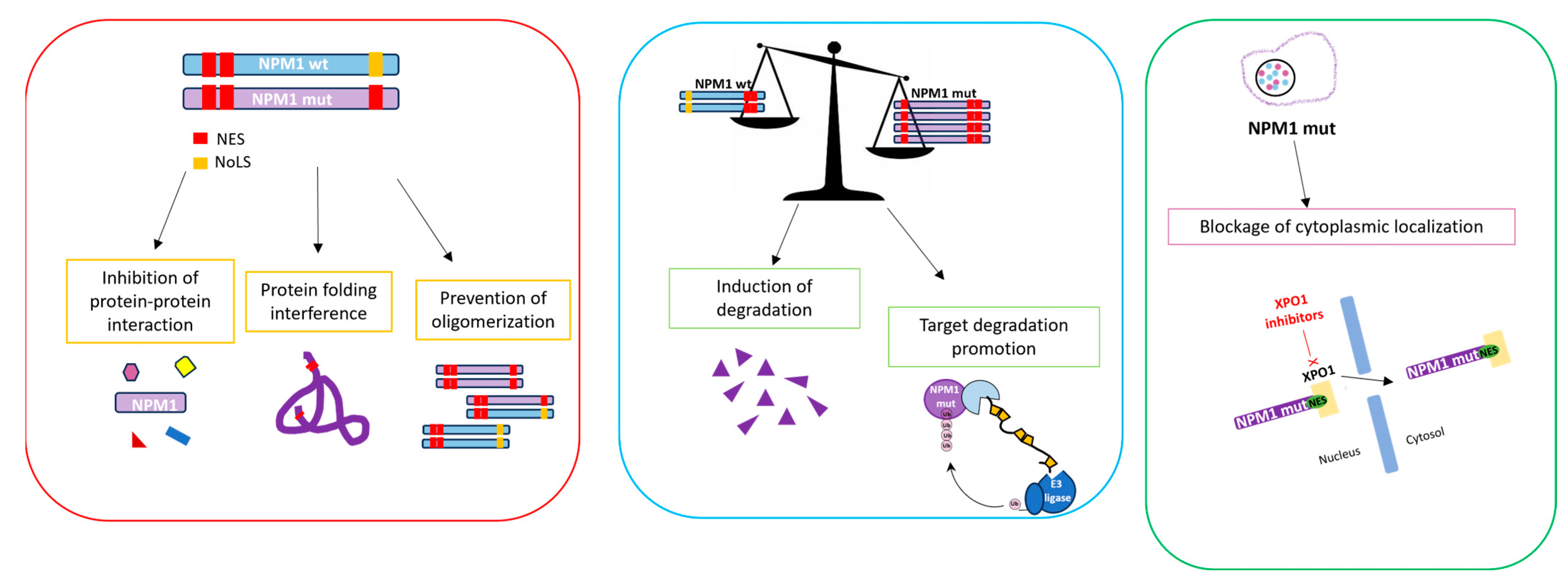
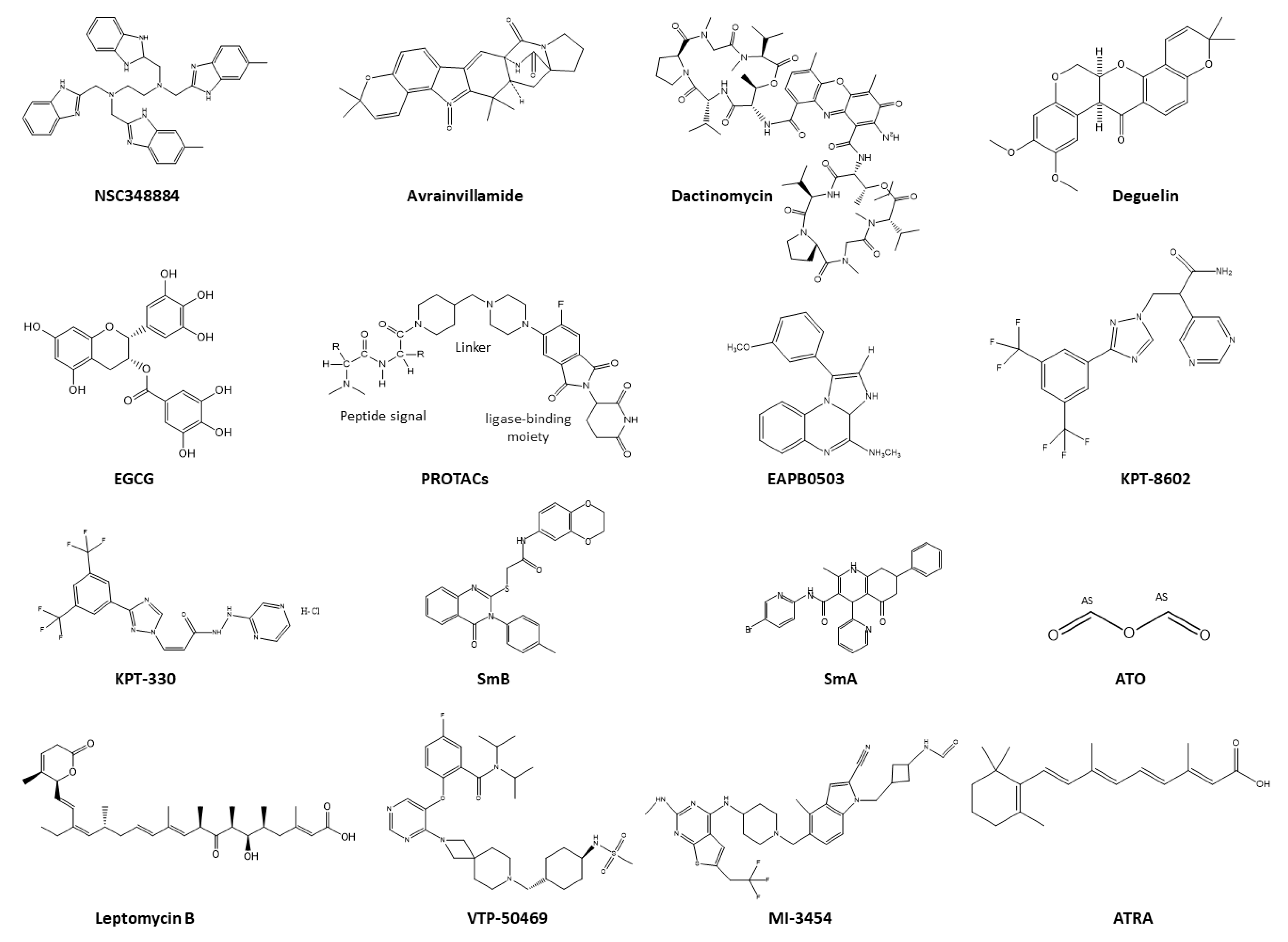
5. Therapeutic Strategies Targeting NPM1c+

5.1. Therapeutics Targeting NPM1 Protein–Protein Interactions
5.2. Therapeutics Targeting the Nucleolus of NPM1c+
5.3. Therapeutics Targeting NPM1c+ Localization
5.4. Therapeutics as Menin Inhibitors
5.5. Therapeutics Targeting Aggregation of NPM1c+
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Straube, J.; Ling, V.Y.; Hill, G.R.; Lane, S.W. The impact of age, NPM1(mut), and FLT3(ITD) allelic ratio in patients with acute myeloid leukemia. Blood 2018, 131, 1148–1153. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Goudswaard, C.S.; Van Putten, W.; Bijl, M.A.; Sanders, M.A.; Hugens, W.; Uitterlinden, A.G.; Erpelinck, C.A.; Delwel, R.; Löwenberg, B. Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): Association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood 2005, 106, 3747–3754. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer Lyon: Lyon, France, 2008; Volume 2, pp. 83–87. [Google Scholar]
- Falini, B.; Gionfriddo, I.; Cecchetti, F.; Ballanti, S.; Pettirossi, V.; Martelli, M.P. Acute myeloid leukemia with mutated nucleophosmin (NPM1): Any hope for a targeted therapy? Blood Rev. 2011, 25, 247–254. [Google Scholar] [PubMed]
- Okuwaki, M.; Matsumoto, K.; Tsujimoto, M.; Nagata, K. Function of nucleophosmin/B23, a nucleolar acidic protein, as a histone chaperone. FEBS Lett. 2001, 506, 272–276. [Google Scholar] [CrossRef]
- Itahana, K.; Bhat, K.P.; Jin, A.; Itahana, Y.; Hawke, D.; Kobayashi, R.; Zhang, Y. Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol. Cell 2003, 12, 1151–1164. [Google Scholar] [CrossRef]
- Grisendi, S.; Bernardi, R.; Rossi, M.; Cheng, K.; Khandker, L.; Manova, K.; Pandolfi, P.P. Role of nucleophosmin in embryonic development and tumorigenesis. Nature 2005, 437, 147–153. [Google Scholar] [CrossRef]
- Raval, A.; Kusler, B.; Pang, W.W.; Weissman, I.L.; Mitchell, B.S.; Park, C.Y. Effect of nucleophosmin1 haploinsufficiency on hematopoietic stem cells. Leukemia 2012, 26, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Sportoletti, P.; Clohessy, J.G.; Silvia, G.; Pandolfi, P.P. The role of nucleophosmin in hematopoietic stem cells and the pathogenesis of myelodysplastic syndrome. Blood 2010, 116, 95. [Google Scholar] [CrossRef]
- Yogev, O.; Saadon, K.; Anzi, S.; Inoue, K.; Shaulian, E. DNA damage–dependent translocation of B23 and p19ARF is regulated by the Jun N-terminal kinase pathway. Cancer Res. 2008, 68, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.K.; Clair, D.K.S. Nucleophosmin blocks mitochondrial localization of p53 and apoptosis. J. Biol. Chem. 2009, 284, 16409–16418. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Alcalay, M.; Pelicci, P. Nucleophosmin and its complex network: A possible therapeutic target in hematological diseases. Oncogene 2011, 30, 2595–2609. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.B., Jr.; Kuchenruether, M.; Dadey, D.Y.; Schwope, R.M.; Grisendi, S.; Townsend, R.R.; Pandolfi, P.P.; Weber, J.D. Nucleophosmin serves as a rate-limiting nuclear export chaperone for the Mammalian ribosome. Mol. Cell. Biol. 2008, 28, 7050–7065. [Google Scholar] [CrossRef]
- Wang, W.; Budhu, A.; Forgues, M.; Wang, X.W. Temporal and spatial control of nucleophosmin by the Ran–Crm1 complex in centrosome duplication. Nat. Cell Biol. 2005, 7, 823–830. [Google Scholar] [CrossRef]
- Lee, H.H.; Kim, H.S.; Kang, J.Y.; Lee, B.I.; Ha, J.Y.; Yoon, H.J.; Lim, S.O.; Jung, G.; Suh, S.W. Crystal structure of human nucleophosmin-core reveals plasticity of the pentamer-pentamer interface. Proteins 2007, 69, 672–678. [Google Scholar] [CrossRef]
- Fantini, D.; Vascotto, C.; Marasco, D.; D’ambrosio, C.; Romanello, M.; Vitagliano, L.; Pedone, C.; Poletto, M.; Cesaratto, L.; Quadrifoglio, F. Critical lysine residues within the overlooked N-terminal domain of human APE1 regulate its biological functions. Nucleic Acids Res. 2010, 38, 8239–8256. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Grace, C.R.; Buljan, M.; Yun, M.-K.; Pytel, N.J.; Satumba, J.; Nourse, A.; Park, C.-G.; Madan Babu, M.; White, S.W. Structural polymorphism in the N-terminal oligomerization domain of NPM1. Proc. Natl. Acad. Sci. 2014, 111, 4466–4471. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Cika, J.A.; Guy, C.S.; Ban, D.; Banerjee, P.R.; Stanley, C.B.; Nourse, A.; Deniz, A.A.; Kriwacki, R.W. Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. eLife 2016, 5, e13571. [Google Scholar] [PubMed]
- Bolli, N.; Nicoletti, I.; De Marco, M.F.; Bigerna, B.; Pucciarini, A.; Mannucci, R.; Martelli, M.P.; Liso, A.; Mecucci, C.; Fabbiano, F. Born to be exported: COOH-terminal nuclear export signals of different strength ensure cytoplasmic accumulation of nucleophosmin leukemic mutants. Cancer Res. 2007, 67, 6230–6237. [Google Scholar] [PubMed]
- Hisaoka, M.; Nagata, K.; Okuwaki, M. Intrinsically disordered regions of nucleophosmin/B23 regulate its RNA binding activity through their inter- and intra-molecular association. Nucleic Acids Res. 2014, 42, 1180–1195. [Google Scholar] [PubMed]
- Gadad, S.S.; Senapati, P.; Syed, S.H.; Rajan, R.E.; Shandilya, J.; Swaminathan, V.; Chatterjee, S.; Colombo, E.; Dimitrov, S.; Pelicci, P.G.; et al. The multifunctional protein nucleophosmin (NPM1) is a human linker histone H1 chaperone. Biochemistry 2011, 50, 2780–2789. [Google Scholar] [CrossRef]
- Swaminathan, V.; Kishore, A.H.; Febitha, K.K.; Kundu, T.K. Human histone chaperone nucleophosmin enhances acetylation-dependent chromatin transcription. Mol. Cell. Biol. 2005, 25, 7534–7545. [Google Scholar] [CrossRef] [PubMed]
- Federici, L.; Falini, B. Nucleophosmin mutations in acute myeloid leukemia: A tale of protein unfolding and mislocalization. Protein Sci. 2013, 22, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Mitrea, D.M.; Cika, J.A.; Stanley, C.B.; Nourse, A.; Onuchic, P.L.; Banerjee, P.R.; Phillips, A.H.; Park, C.G.; Deniz, A.A.; Kriwacki, R.W. Self-interaction of NPM1 modulates multiple mechanisms of liquid-liquid phase separation. Nat. Commun. 2018, 9, 842. [Google Scholar]
- Mitrea, D.M.; Kriwacki, R.W. Phase separation in biology; Functional organization of a higher order. Cell Commun. Signal. 2016, 14, 1. [Google Scholar]
- Mitrea, D.M.; Kriwacki, R.W. On the relationship status for Arf and NPM1—It’s complicated. FEBS J. 2018, 285, 828–831. [Google Scholar] [CrossRef]
- Hingorani, K.; Szebeni, A.; Olson, M.O. Mapping the functional domains of nucleolar protein B23. J. Biol. Chem. 2000, 275, 24451–24457. [Google Scholar] [CrossRef]
- Grummitt, C.G.; Townsley, F.M.; Johnson, C.M.; Warren, A.J.; Bycroft, M. Structural consequences of nucleophosmin mutations in acute myeloid leukemia. J. Biol. Chem. 2008, 283, 23326–23332. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Ohkubo, T.; Furuichi, Y.; Umekawa, H. Tryptophans 286 and 288 in the C-terminal region of protein B23.1 are important for its nucleolar localization. Biosci. Biotechnol. Biochem. 2002, 66, 2239–2242. [Google Scholar] [CrossRef] [PubMed]
- Burra, S.; Marasco, D.; Malfatti, M.C.; Antoniali, G.; Virgilio, A.; Esposito, V.; Demple, B.; Galeone, A.; Tell, G. Human AP-endonuclease (Ape1) activity on telomeric G4 structures is modulated by acetylatable lysine residues in the N-terminal sequence. DNA Repair 2019, 73, 129–143. [Google Scholar] [PubMed]
- Falini, B.; Tiacci, E.; Martelli, M.P.; Ascani, S.; Pileri, S.A. New classification of acute myeloid leukemia and precursor-related neoplasms: Changes and unsolved issues. Discov. Med. 2010, 10, 281–292. [Google Scholar] [PubMed]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Döhner, K.; Schlenk, R.F.; Habdank, M.; Scholl, C.; Rücker, F.G.; Corbacioglu, A.; Bullinger, L.; Fröhling, S.; Döhner, H.; for the AML Study Group (AMLSG). Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: Interaction with other gene mutations. Blood 2005, 106, 3740–3746. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, S.; Schoch, C.; Kern, W.; Mecucci, C.; Tschulik, C.; Martelli, M.F.; Haferlach, T.; Hiddemann, W.; Falini, B. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood 2005, 106, 3733–3739. [Google Scholar] [CrossRef]
- Alpermann, T.; Schnittger, S.; Eder, C.; Dicker, F.; Meggendorfer, M.; Kern, W.; Schmid, C.; Aul, C.; Staib, P.; Wendtner, C.-M. Molecular subtypes of NPM1 mutations have different clinical profiles, specific patterns of accompanying molecular mutations and varying outcomes in intermediate risk acute myeloid leukemia. Haematologica 2016, 101, e55. [Google Scholar] [CrossRef]
- Alpermann, T.; Haferlach, C.; Dicker, F.; Eder, C.; Kohlmann, A.; Kern, W.; Haferlach, T.; Schnittger, S. Evaluation of different NPM1 mutations in AML patients according to clinical, cytogenetic and molecular features and impact on outcome. Blood 2013, 122, 51. [Google Scholar] [CrossRef]
- Falini, B.; Bolli, N.; Shan, J.; Martelli, M.P.; Liso, A.; Pucciarini, A.; Bigerna, B.; Pasqualucci, L.; Mannucci, R.; Rosati, R.; et al. Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML. Blood 2006, 107, 4514–4523. [Google Scholar] [CrossRef]
- Nakagawa, M. Nucleophosmin in acute myelogenous leukemia. N. Engl. J. Med. 2005, 352, 1819–1820. [Google Scholar] [PubMed]
- Falini, B.; Brunetti, L.; Sportoletti, P.; Martelli, M.P. NPM1-mutated acute myeloid leukemia: From bench to bedside. Blood J. Am. Soc. Hematol. 2020, 136, 1707–1721. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.-H.; Ramabadran, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B. Mutant NPM1 maintains the leukemic state through HOX expression. Cancer Cell 2018, 34, 499–512.e499. [Google Scholar] [CrossRef] [PubMed]
- Martelli, M.P.; Rossi, R.; Venanzi, A.; Meggendorfer, M.; Perriello, V.M.; Martino, G.; Spinelli, O.; Ciurnelli, R.; Varasano, E.; Brunetti, L. Novel NPM1 exon 5 mutations and gene fusions leading to aberrant cytoplasmic nucleophosmin in AML. Blood J. Am. Soc. Hematol. 2021, 138, 2696–2701. [Google Scholar] [CrossRef] [PubMed]
- Martelli, M.P.; Rossi, R.; Varasano, E.; Specchia, G.; Di Raimondo, F.; Avvisati, G.; Tiacci, E.; Falzetti, F.; Sportoletti, P.; Falini, B. Identification and characterization of novel rare nucleophosmin (NPM1) gene mutations in acute myeloid leukemia (AML) by a combinatorial approach of immunohistochemistry and molecular analyses. Blood 2016, 128, 1717. [Google Scholar] [CrossRef]
- Yao, Y.; Lin, X.; Wang, C.; Gu, Y.; Jin, J.; Zhu, Y.; Wang, H. Identification of a novel NPM1 mutation in acute myeloid leukemia. Exp. Hematol. Oncol. 2023, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Colombo, F.L.C.; Mecucci, C.; Falini, B.; Pelicci, P.G.; Meani, N.; Diverio, D.; Bernard, L.; Tizzoni, L.; Volorio, S.; Luzi, L. Acute myeloid leukemia bearing cytoplasmic nucleophosmin (NPMc+ AML) shows a distinct gene expression profile characterized by up-regulation of genes involved in stem-cell maintenance. Blood 2005, 106, 899–902. [Google Scholar]
- Ozyerli-Goknar, E.; Nizamuddin, S.; Timmers, H.M. A box of chemistry to inhibit the MEN1 tumor suppressor gene promoting leukemia. ChemMedChem 2021, 16, 1391–1402. [Google Scholar] [CrossRef]
- Uddin, M.H.; Aboukameel, A.; Khan, H.; Bannoura, S.; Deol, A.; Yang, J.; Dyson, G.; Azmi, A.; Polin, L.; Maciejewski, J.P. The Clinical Menin Inhibitor Ziftomenib and the Nuclear Export Inhibitor Selinexor Synergistically Inhibit the Growth of MLL-r AML. Blood 2023, 142, 4168. [Google Scholar] [CrossRef]
- Bain, B.J.; Heller, M.; Toma, S.; Pavlu, J. The cytological features of NPM1-mutated acute myeloid leukemia. Am. J. Hematol. 2015, 90, 560. [Google Scholar] [CrossRef]
- Weinberg, O.K.; Hasserjian, R.P.; Baraban, E.; Ok, C.Y.; Geyer, J.T.; Philip, J.K.; Kurzer, J.H.; Rogers, H.J.; Nardi, V.; Stone, R.M. Clinical, immunophenotypic, and genomic findings of acute undifferentiated leukemia and comparison to acute myeloid leukemia with minimal differentiation: A study from the bone marrow pathology group. Mod. Pathol. 2019, 32, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Bain, B.J.; Béné, M.C. Morphological and immunophenotypic clues to the WHO categories of acute myeloid leukaemia. Acta Haematol. 2019, 141, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Redner, R.L.; Rush, E.A.; Faas, S.; Rudert, W.A.; Corey, S.J. The t(5;17) variant of acute promyelocytic leukemia expresses a nucleophosmin-retinoic acid receptor fusion. Blood 1996, 87, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.; Kirstein, M.; Valentine, M.; Dittmer, K.; Shapiro, D.; Look, A.; Saltman, D. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1995, 267, 316–317. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Nicoletti, I.; Martelli, M.F.; Mecucci, C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): Biologic and clinical features. Blood 2007, 109, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Motea, E.A.; Berdis, A.J. Terminal deoxynucleotidyl transferase: The story of a misguided DNA polymerase. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2010, 1804, 1151–1166. [Google Scholar] [CrossRef]
- Cheng, K.; Sportoletti, P.; Ito, K.; Clohessy, J.G.; Teruya-Feldstein, J.; Kutok, J.L.; Pandolfi, P.P. The cytoplasmic NPM mutant induces myeloproliferation in a transgenic mouse model. Blood 2010, 115, 3341–3345. [Google Scholar] [CrossRef]
- Zarka, J.; Short, N.J.; Kanagal-Shamanna, R.; Issa, G.C. Nucleophosmin 1 mutations in acute myeloid leukemia. Genes 2020, 11, 649. [Google Scholar] [CrossRef]
- Spencer, D.H.; Young, M.A.; Lamprecht, T.L.; Helton, N.M.; Fulton, R.; O’Laughlin, M.; Fronick, C.; Magrini, V.; Demeter, R.T.; Miller, C.A. Epigenomic analysis of the HOX gene loci reveals mechanisms that may control canonical expression patterns in AML and normal hematopoietic cells. Leukemia 2015, 29, 1279–1289. [Google Scholar] [CrossRef]
- Pianigiani, G.; Betti, C.; Bigerna, B.; Rossi, R.; Brunetti, L. PU. 1 subcellular localization in acute myeloid leukaemia with mutated NPM1. Br. J. Haematol. 2020, 188, 184–187. [Google Scholar] [CrossRef]
- Dawson, M.; Gudgin, E.; Horton, S.; Giotopoulos, G.; Meduri, E.; Robson, S.; Cannizzaro, E.; Osaki, H.; Wiese, M.; Putwain, S. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia 2014, 28, 311–320. [Google Scholar] [CrossRef]
- Marasco, D.; Ruggiero, A.; Vascotto, C.; Poletto, M.; Scognamiglio, P.L.; Tell, G.; Vitagliano, L. Role of mutual interactions in the chemical and thermal stability of nucleophosmin NPM1 domains. Biochem. Biophys. Res. Commun. 2013, 430, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Di Natale, C.; La Manna, S.; De Benedictis, I.; Brandi, P.; Marasco, D. Perspectives in peptide-based vaccination strategies for syndrome coronavirus 2 pandemic. Front. Pharmacol. 2020, 11, 578382. [Google Scholar] [CrossRef] [PubMed]
- Di Natale, C.; Scognamiglio, P.L.; Cascella, R.; Cecchi, C.; Russo, A.; Leone, M.; Penco, A.; Relini, A.; Federici, L.; Di Matteo, A. Nucleophosmin contains amyloidogenic regions that are able to form toxic aggregates under physiological conditions. FASEB J. 2015, 29, 3689–3701. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Diaferia, C.; La Manna, S.; Giannini, C.; Sibillano, T.; Accardo, A.; Morelli, G.; Novellino, E.; Marasco, D. Insights into amyloid-like aggregation of H2 region of the C-terminal domain of nucleophosmin. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2017, 1865, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Scognamiglio, P.L.; Di Natale, C.; Leone, M.; Cascella, R.; Cecchi, C.; Lirussi, L.; Antoniali, G.; Riccardi, D.; Morelli, G.; Tell, G. Destabilisation, aggregation, toxicity and cytosolic mislocalisation of nucleophosmin regions associated with acute myeloid leukemia. Oncotarget. 2016, 7, 59129. [Google Scholar] [CrossRef] [PubMed]
- Scognamiglio, P.L.; Di Natale, C.; Leone, M.; Poletto, M.; Vitagliano, L.; Tell, G.; Marasco, D. G-quadruplex DNA recognition by nucleophosmin: New insights from protein dissection. Biochim. Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 2050–2059. [Google Scholar] [CrossRef]
- De Santis, A.; La Manna, S.; Krauss, I.R.; Malfitano, A.M.; Novellino, E.; Federici, L.; De Cola, A.; Di Matteo, A.; D’Errico, G.; Marasco, D. Nucleophosmin-1 regions associated with acute myeloid leukemia interact differently with lipid membranes. Biochim. Biophys. Acta (BBA) Gen. Subj. 2018, 1862, 967–978. [Google Scholar] [CrossRef]
- Di Natale, C.; Florio, D.; Di Somma, S.; Di Matteo, A.; Federici, L.; Netti, P.A.; Morelli, G.; Malfitano, A.M.; Marasco, D. Proteostasis unbalance of nucleophosmin 1 in Acute Myeloid Leukemia: An aggregomic perspective. Int. J. Biol. Macromol. 2020, 164, 3501–3507. [Google Scholar] [CrossRef]
- Di Natale, C.; La Manna, S.; Malfitano, A.M.; Di Somma, S.; Florio, D.; Scognamiglio, P.L.; Novellino, E.; Netti, P.A.; Marasco, D. Structural insights into amyloid structures of the C-terminal region of nucleophosmin 1 in type A mutation of acute myeloid leukemia. Biochim. Biophys Acta Proteins Proteom 2019, 1867, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Florio, D.; Cuomo, M.; Iacobucci, I.; Ferraro, G.; Mansour, A.M.; Monti, M.; Merlino, A.; Marasco, D. Modulation of amyloidogenic peptide aggregation by photoactivatable CO-releasing ruthenium (II) complexes. Pharmaceuticals 2020, 13, 171. [Google Scholar] [CrossRef] [PubMed]
- La Manna, S.; Florio, D.; Di Natale, C.; Napolitano, F.; Malfitano, A.M.; Netti, P.A.; De Benedictis, I.; Marasco, D. Conformational consequences of NPM1 rare mutations: An aggregation perspective in Acute Myeloid Leukemia. Bioorganic Chem. 2021, 113, 104997. [Google Scholar] [CrossRef] [PubMed]
- La Manna, S.; Florio, D.; Di Natale, C.; Scognamiglio, P.L.; Sibillano, T.; Netti, P.A.; Giannini, C.; Marasco, D. Type F mutation of nucleophosmin 1 Acute Myeloid Leukemia: A tale of disorder and aggregation. Int. J. Biol. Macromol. 2021, 188, 207–214. [Google Scholar] [CrossRef] [PubMed]
- La Manna, S.; Roviello, V.; Scognamiglio, P.L.; Diaferia, C.; Giannini, C.; Sibillano, T.; Morelli, G.; Novellino, E.; Marasco, D. Amyloid fibers deriving from the aromatic core of C-terminal domain of nucleophosmin 1. Int. J. Biol. Macromol. 2019, 122, 517–525. [Google Scholar] [CrossRef]
- La Manna, S.; Scognamiglio, P.L.; Roviello, V.; Borbone, F.; Florio, D.; Di Natale, C.; Bigi, A.; Cecchi, C.; Cascella, R.; Giannini, C. The acute myeloid leukemia-associated Nucleophosmin 1 gene mutations dictate amyloidogenicity of the C-terminal domain. FEBS J. 2019, 286, 2311–2328. [Google Scholar] [CrossRef]
- La Manna, S.; Florio, D.; Di Natale, C.; Lagreca, E.; Sibillano, T.; Giannini, C.; Marasco, D. Type C mutation of nucleophosmin 1 acute myeloid leukemia: Consequences of intrinsic disorder. Biochim. Biophys. Acta (BBA) Gen. Subj. 2022, 1866, 130173. [Google Scholar] [CrossRef]
- Almeida, Z.L.; Brito, R.M. Structure and aggregation mechanisms in amyloids. Molecules 2020, 25, 1195. [Google Scholar] [CrossRef]
- Florio, D.; La Manna, S.; Di Natale, C.; Leone, M.; Mercurio, F.A.; Napolitano, F.; Malfitano, A.M.; Marasco, D. Insights into Network of Hot Spots of Aggregation in Nucleophosmin 1. Int. J. Mol. Sci. 2022, 23, 14704. [Google Scholar] [CrossRef]
- Wang, R.; Xu, P.; Chang, L.-L.; Zhang, S.-Z.; Zhu, H.-H. Targeted therapy in NPM1-mutated AML: Knowns and unknowns. Front. Oncol. 2022, 12, 972606. [Google Scholar] [CrossRef]
- Ranieri, R.; Pianigiani, G.; Sciabolacci, S.; Perriello, V.M.; Marra, A.; Cardinali, V.; Pierangeli, S.; Milano, F.; Gionfriddo, I.; Brunetti, L.; et al. Current status and future perspectives in targeted therapy of NPM1-mutated AML. Leukemia 2022, 36, 2351–2367. [Google Scholar] [CrossRef]
- Falini, B.; Brunetti, L.; Martelli, M.P. Dactinomycin in NPM1-mutated acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1180–1182. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Shakalya, K.; Stejskal, A.; Goldman, A.; Beeck, S.; Cooke, L.; Mahadevan, D. NSC348884, a nucleophosmin inhibitor disrupts oligomer formation and induces apoptosis in human cancer cells. Oncogene 2008, 27, 4210–4220. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.F.; Han, L.L.; Ma, Y.; Wang, Y.X.; Zhang, F. The effect of small molecule inhibitor NSC348884 on nucleophosmin 1-mutated acute myeloid leukemia cells. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 9145–9151. [Google Scholar] [PubMed]
- Šašinková, M.; Heřman, P.; Holoubek, A.; Strachotová, D.; Otevřelová, P.; Grebeňová, D.; Kuželová, K.; Brodská, B. NSC348884 cytotoxicity is not mediated by inhibition of nucleophosmin oligomerization. Sci. Rep. 2021, 11, 1084. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, H.; Chan, K.-P.; Andresen, V.; Hanley, M.L.; Gjertsen, B.T.; Myers, A.G. Interactions of the natural product (+)-avrainvillamide with nucleophosmin and exportin-1 mediate the cellular localization of nucleophosmin and its AML-associated mutants. ACS Chem. Biol. 2015, 10, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Andresen, V.; Erikstein, B.S.; Mukherjee, H.; Sulen, A.; Popa, M.; Sørnes, S.; Reikvam, H.; Chan, K.-P.; Hovland, R.; McCormack, E. Anti-proliferative activity of the NPM1 interacting natural product avrainvillamide in acute myeloid leukemia. Cell Death Dis. 2016, 7, e2497. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, U. Actinomycin. Chemistry and mechanism of action. Chem. Rev. 1974, 74, 625–652. [Google Scholar] [CrossRef]
- Gionfriddo, I.; Brunetti, L.; Mezzasoma, F.; Milano, F.; Cardinali, V.; Ranieri, R.; Venanzi, A.; Pierangeli, S.; Vetro, C.; Spinozzi, G. Dactinomycin induces complete remission associated with nucleolar stress response in relapsed/refractory NPM1-mutated AML. Leukemia 2021, 35, 2552–2562. [Google Scholar] [CrossRef]
- Martelli, M.P.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Pierangeli, S.; Mulas, F.; Pacini, R.; Tabarrini, A.; Pettirossi, V.; Rossi, R. Arsenic trioxide and all-trans retinoic acid target NPM1 mutant oncoprotein levels and induce apoptosis in NPM1-mutated AML cells. Blood J. Am. Soc. Hematol. 2015, 125, 3455–3465. [Google Scholar] [CrossRef]
- Wu, H.C.; Rerolle, D.; Berthier, C.; Hleihel, R.; Sakamoto, T.; Quentin, S.; Benhenda, S.; Morganti, C.; Wu, C.; Conte, L.; et al. Actinomycin D Targets NPM1c-Primed Mitochondria to Restore PML-Driven Senescence in AML Therapy. Cancer Discov. 2021, 11, 3198–3213. [Google Scholar] [CrossRef]
- Davison, K.; Mann, K.K.; Miller, W.H., Jr. Arsenic trioxide: Mechanisms of action. Semin. Hematol. 2002, 39, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Siddikuzzaman, N.; Guruvayoorappan, C.; Berlin Grace, V.M. All trans retinoic acid and cancer. Immunopharmacol. Immunotoxicol. 2010, 33, 241–249. [Google Scholar] [CrossRef] [PubMed]
- El Hajj, H.; Dassouki, Z.; Berthier, C.; Raffoux, E.; Ades, L.; Legrand, O.; Hleihel, R.; Sahin, U.; Tawil, N.; Salameh, A.; et al. Retinoic acid and arsenic trioxide trigger degradation of mutated NPM1, resulting in apoptosis of AML cells. Blood 2015, 125, 3447–3454. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Y. Molecular mechanisms of deguelin-induced apoptosis in transformed human bronchial epithelial cells. Biochem. Pharmacol. 2004, 68, 1119–1124. [Google Scholar] [CrossRef] [PubMed]
- Nagle, D.G.; Ferreira, D.; Zhou, Y.-D. Epigallocatechin-3-gallate (EGCG): Chemical and biomedical perspectives. Phytochemistry 2006, 67, 1849–1855. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Wen, L.; He, J.; Wang, Y.; Zhao, F.; Zhao, J.; Zhao, Z.; Cui, G.; Chen, Y. Deguelin, a selective silencer of the NPM1 mutant, potentiates apoptosis and induces differentiation in AML cells carrying the NPM1 mutation. Ann. Hematol. 2015, 94, 201–210. [Google Scholar] [CrossRef]
- Chi, H.T.; Ly, B.T.; Vu, H.A.; Sato, Y.; Dung, P.C.; Xinh, P.T. Down-regulated expression of NPM1 in IMS-M2 cell line by (−)-epigallocatechin-3-gallate. Asian Pac. J. Trop. Biomed. 2014, 4, 570–574. [Google Scholar] [CrossRef]
- Lafaille, F.; Solassol, I.; Enjalbal, C.; Bertrand, B.; Doulain, P.-E.; Vappiani, J.; Bonnet, P.-A.; Deleuze-Masquéfa, C.; Bressolle, F.M. Structural characterization of in vitro metabolites of the new anticancer agent EAPB0503 by liquid chromatography–tandem mass spectrometry. J. Pharm. Biomed. Anal. 2014, 88, 429–440. [Google Scholar] [CrossRef]
- Skayneh, H.; Jishi, B.; Hleihel, R.; Hamie, M.; El Hajj, R.; Deleuze-Masquefa, C.; Bonnet, P.-A.; El Sabban, M.; El Hajj, H. EAPB0503, an imidazoquinoxaline derivative modulates SENP3/ARF mediated SUMOylation, and induces NPM1c degradation in NPM1 mutant AML. Int. J. Mol. Sci. 2022, 23, 3421. [Google Scholar] [CrossRef]
- Nabbouh, A.I.; Hleihel, R.S.; Saliba, J.L.; Karam, M.M.; Hamie, M.H.; Wu, H.C.J.; Berthier, C.P.; Tawil, N.M.; Bonnet, P.A.A.; Deleuze-Masquefa, C. Imidazoquinoxaline derivative EAPB0503: A promising drug targeting mutant nucleophosmin 1 in acute myeloid leukemia. Cancer 2017, 123, 1662–1673. [Google Scholar] [CrossRef]
- Li, K.; Crews, C.M. PROTACs: Past, present and future. Chem. Soc. Rev. 2022, 51, 5214–5236. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Hu, W.; Zhang, S.; He, L.; Hu, C.; Yang, S. Proteolysis-targeting chimeras: A promising technique in cancer therapy for gaining insights into tumor development. Cancer Lett. 2022, 539, 215716. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Khan, S.; Huo, Z.; Lv, D.; Zhang, X.; Liu, X.; Yuan, Y.; Hromas, R.; Xu, M.; Zheng, G. Proteolysis targeting chimeras (PROTACs) are emerging therapeutics for hematologic malignancies. J. Hematol. Oncol. 2020, 13, 103. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Pradeep, S.; Wu, S.Y.; Rupaimoole, R.; Zand, B.; Wen, Y.; Gharpure, K.M.; Nagaraja, A.S.; Hu, W.; Cho, M.S. XPO1/CRM1 inhibition causes antitumor effects by mitochondrial accumulation of eIF5A. Clin. Cancer Res. 2015, 21, 3286–3297. [Google Scholar] [CrossRef] [PubMed]
- Kudo, N.; Wolff, B.; Sekimoto, T.; Schreiner, E.P.; Yoneda, Y.; Yanagida, M.; Horinouchi, S.; Yoshida, M. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp. Cell Res. 1998, 242, 540–547. [Google Scholar] [CrossRef]
- Sun, Q.; Chen, X.; Zhou, Q.; Burstein, E.; Yang, S.; Jia, D. Inhibiting cancer cell hallmark features through nuclear export inhibition. Signal Transduct. Target. Ther. 2016, 1, 16010. [Google Scholar] [CrossRef]
- Garzon, R.; Savona, M.; Baz, R.; Andreeff, M.; Gabrail, N.; Gutierrez, M.; Savoie, L.; Mau-Sorensen, P.M.; Wagner-Johnston, N.; Yee, K. A phase 1 clinical trial of single-agent selinexor in acute myeloid leukemia. Blood J. Am. Soc. Hematol. 2017, 129, 3165–3174. [Google Scholar] [CrossRef]
- Etchin, J.; Berezovskaya, A.; Conway, A.S.; Galinsky, I.A.; Stone, R.M.; Baloglu, E.; Senapedis, W.; Landesman, Y.; Kauffman, M.; Shacham, S. KPT-8602, a second-generation inhibitor of XPO1-mediated nuclear export, is well tolerated and highly active against AML blasts and leukemia-initiating cells. Leukemia 2017, 31, 143–150. [Google Scholar] [CrossRef]
- Fischer, M.A.; Friedlander, S.Y.; Arrate, M.P.; Chang, H.; Gorska, A.E.; Fuller, L.D.; Ramsey, H.E.; Kashyap, T.; Argueta, C.; Debler, S. Venetoclax response is enhanced by selective inhibitor of nuclear export compounds in hematologic malignancies. Blood Adv. 2020, 4, 586–598. [Google Scholar] [CrossRef]
- Matkar, S.; Thiel, A.; Hua, X. Menin: A scaffold protein that controls gene expression and cell signaling. Trends Biochem. Sci. 2013, 38, 394–402. [Google Scholar] [CrossRef]
- Argiropoulos, B.; Humphries, R. Hox genes in hematopoiesis and leukemogenesis. Oncogene 2007, 26, 6766–6776. [Google Scholar] [CrossRef] [PubMed]
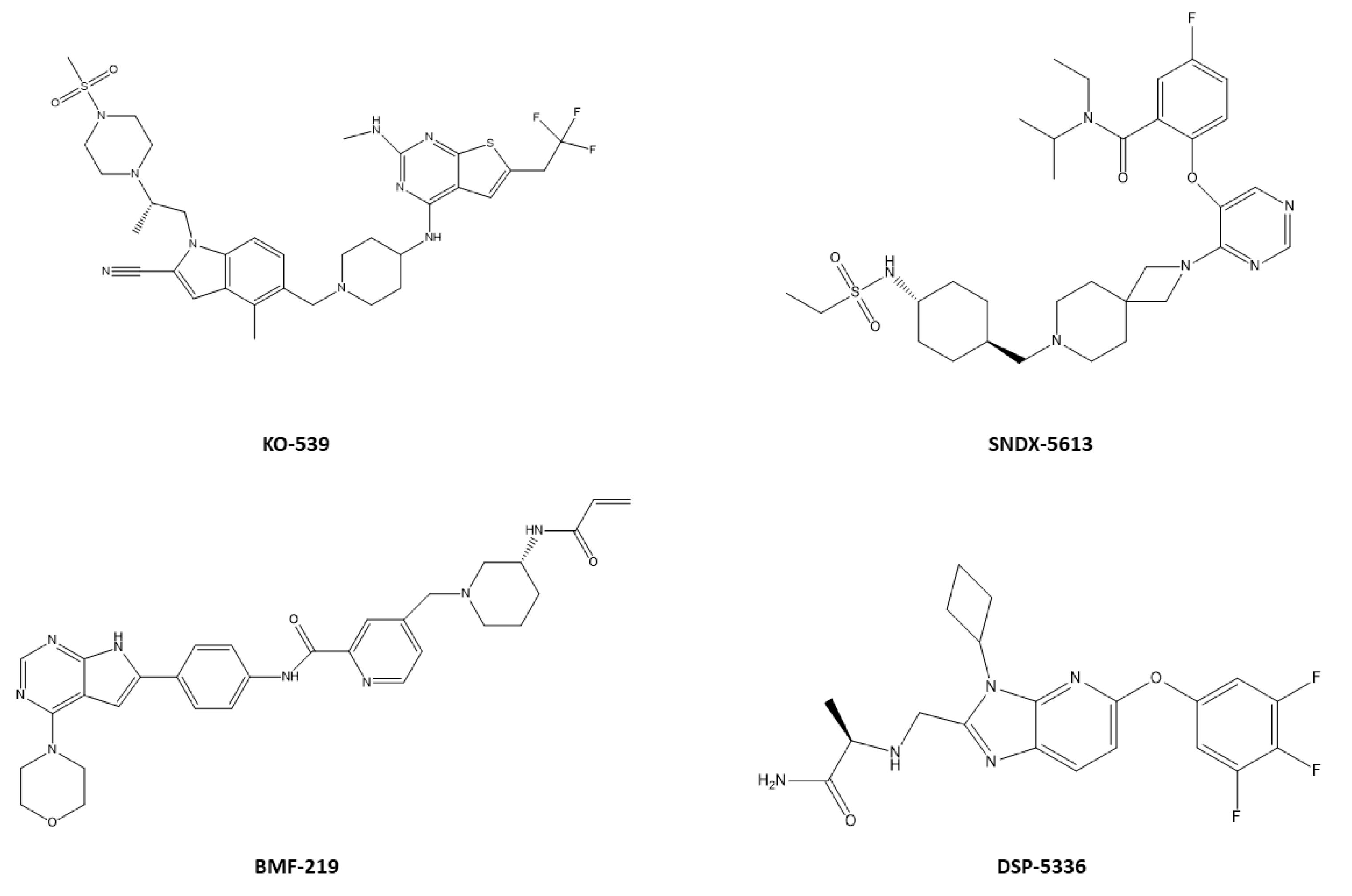
- Klossowski, S.; Miao, H.; Kempinska, K.; Wu, T.; Purohit, T.; Kim, E.; Linhares, B.M.; Chen, D.; Jih, G.; Perkey, E. Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J. Clin. Investig. 2020, 130, 981–997. [Google Scholar] [CrossRef] [PubMed]
- Uckelmann, H.J.; Kim, S.M.; Wong, E.M.; Hatton, C.; Giovinazzo, H.; Gadrey, J.Y.; Krivtsov, A.V.; Rücker, F.G.; Döhner, K.; McGeehan, G.M. Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science 2020, 367, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, M.; Bourgeois, W.; Armstrong, S.A.; Wang, E.S. Menin inhibitors in acute myeloid leukemia—What does the future hold? Cancer J. 2022, 28, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Evans, K.; Gadrey, J.Y.; Eschle, B.K.; Hatton, C.; Uckelmann, H.J.; Ross, K.N.; Perner, F.; Olsen, S.N.; Pritchard, T. A menin-MLL inhibitor induces specific chromatin changes and eradicates disease in models of MLL-rearranged leukemia. Cancer Cell 2019, 36, 660–673.e611. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.C.; Querolle, O.; Dai, X.; Thuring, J.W.; Verhulst, T.; Marien, A.; Goffin, D.; Cai, W.; Keersmaekers, V.; Eyassu, F. Pharmacological characterization of JNJ-75276617, a menin-KMT2A inhibitor, as targeted treatment for KMT2A-altered and NPM1-mutant acute leukemia. Blood 2022, 140, 5928–5929. [Google Scholar] [CrossRef]
- Somanath, P.; Lu, D.; Law, B.; Archer, T.C.; Cacovean, A.; Palmer, J.T.; Kinoshita, T.; Butler, T. Novel Irreversible Menin Inhibitor, BMF-219, Shows Potent Single Agent Activity in Clinically Relevant DLBCL Cells. Blood 2021, 138, 4318. [Google Scholar] [CrossRef]
- Ravandi-Kashani, F.; Kishtagari, A.; Carraway, H.; Schiller, G.; Curran, E.; Yadav, B.; Cacovean, A.; Morris, S.; Butler, T.; Lancet, J. P587: COVALENT-101: A phase 1 study of BMF-219, a novel oral irreversible menin inhibitor, in patients with relapsed/refractory acute leukemia, diffuse large B-cell lymphoma, and multiple myeloma. HemaSphere 2022, 6, 486–487. [Google Scholar] [CrossRef]
- Eguchi, K.; Shimizu, T.; Kato, D.; Furuta, Y.; Kamioka, S.; Ban, H.; Ymamoto, S.; Yokoyama, A.; Kitabayashi, I. Preclinical Evaluation of a Novel Orally Bioavailable Menin-MLL Interaction Inhibitor, DSP-5336, for the Treatment of Acute Leukemia Patients with MLL-Rearrangement or NPM1 Mutation. Blood 2021, 138, 3339. [Google Scholar] [CrossRef]
- Ajmal, M.R. Protein Misfolding and Aggregation in Proteinopathies: Causes, Mechanism and Cellular Response. Diseases 2023, 11, 30. [Google Scholar] [CrossRef]
- Florio, D.; Roviello, V.; La Manna, S.; Napolitano, F.; Maria Malfitano, A.; Marasco, D. Small molecules enhancers of amyloid aggregation of C-terminal domain of Nucleophosmin 1 in acute myeloid leukemia. Bioorg. Chem. 2022, 127, 106001. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Martelli, M.P.; Brunetti, L.; Gjertsen, B.T.; Andresen, V. The NPM1 mutant defines AML irrespective of blast count. Am. J. Hematol. 2023, 98, E187–E189. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Brunetti, L.; Martelli, M.P. How I diagnose and treat NPM1-mutated AML. Blood J. Am. Soc. Hematol. 2021, 137, 589–599. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleotide Sequence | Protein | |
|---|---|---|
| wt NPM1 | GATCTCTG…GCAGT…GGAGGAAGTCTCTTTAAGAAAATAG | 286DLWQWRKSL294 |
| Common mutations | GATCTCTGTCTGGCAGT…GGAGGAAGTCTCTTTAAGAAAATAG | 286DLCLAVEEVSLRK298 |
| GATCTCTGCATGGCAGT…GGAGGAAGTCTCTTTAAGAAAATAG | 286DLCMAVEEVSLRK298 | |
| GATCTCTGCGTGGCAGT…GGAGGAAGTCTCTTTAAGAAAATAG | 286DLCVAVEEVSLRK298 | |
| GATCTCTGCCTGGCAGT…GGAGGAAGTCTCTTTAAGAAAATAG | 286DLCLAVEEVSLRK298 | |
| GATCTCTG…GCAGTCTCTTGCCCAAGTCTCTTTAAGAAAATAG | 286DLWQSLAQVSLRK298 | |
| GATCTCTG…GCAGTCCCTGGAGAAAGTCTCTTTAAGAAAATAG | 286DLWQSLEKVSLRK298 | |
| GATCTCTG…GCAGTCTCTTTCTAAAGTCTCTTTAAGAAAATAG | 286DLWQSLSKVSLRK298 | |
| GATCTCTCCCGGGCAGT…AAGTCTCTTTAAGAAAATAG | 286DLSRAVEEVSLRK298 | |
| GATCTCTG…GCAGTCCCTTTCCAAAGTCTCTTTAAGAAAATAG | 286DLWQSLSKVSLRK298 | |
| GATCTCTGTAGCGCAGT…GGAGGAAGTCTCTTTAAGAAAATAG | 286DLCTAVEEVSLRK298 | |
| GATCTCTGCCACGCAGT…GGAGGAAGTCTCTTTAAGAAAATAG | 286DLCHAVEEVSLRK298 | |
| GATCTCTGGCAGCGTTTCCAGGAAGTCTCTTTAAGAAAATAG | 286DLWQRFQEVSLRK298 | |
| GATCTCTGTACCTTCCT…GGAGGAAGTCTCTTTAAGAAAATAG | 286DLCTFLEEVSLRK298 | |
| GATCTCTG…GCAGAGGATGGAGGAAGTCTCTTTAAGAAAATAG | 286DLWQRMEEVSLRK298 | |
| Nucleotide Change | ||
| Rare mutations | c.864_876delinsTCGGAGTCTCGGCGGAC | 286DLCRSLGGLSLRKA299 |
| c.864_873delinsTCAAGACTTTCTTA | 286DLCQDFLKVSLRKA299 | |
| c.867_875delinsAGATTTCTTAAATC | 286DLWQDFLNRLFKRIVA301 | |
| c.868_876delinsGGGATAGCGATGC | 286DLWQGIAMLSLRKA299 | |
| c.868_876delinsGGGGTGGGGAATC | 286DLWQGVGNLSLRKA299 | |
| c.863_871delinsCGACCCTCCTGGG | 286DLSTLLGEVSLRKA299 |
| In-Frame Insertion/Duplications | Protein | |
|---|---|---|
| Exon 5 Mutations | c408–409 (F,5′-GCGGAGGATGTGAAACTCTTA) | DVKLL136AEDVKLL…286DLWQWRKSL294 |
| c409–410 (F,5′-AATGATCTGTCACTTCTG) | DVKLL137K | |
| c424–425 (F,5′-TTTCTGCCTTAAGTATATCTGGAAAGC) | ISGK141LSALSISGK…286DLWQWRKSL294 | |
| c399–400 (F,5′-CAACTCTTA) and c400–401 (F,5′-GTGGGCTGC) | EEDV134QLLSGLQ…286DLWQWRKSL294 | |
| c406–423 (F,5′-GCCCTGGAACTGGGGAAC) | DVKL135ALELGNLSI…286DLWQWRKSL294 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Florio, D.; Marasco, D. Could Targeting NPM1c+ Misfolding Be a Promising Strategy for Combating Acute Myeloid Leukemia? Int. J. Mol. Sci. 2024, 25, 811. https://doi.org/10.3390/ijms25020811
Florio D, Marasco D. Could Targeting NPM1c+ Misfolding Be a Promising Strategy for Combating Acute Myeloid Leukemia? International Journal of Molecular Sciences. 2024; 25(2):811. https://doi.org/10.3390/ijms25020811
Chicago/Turabian StyleFlorio, Daniele, and Daniela Marasco. 2024. "Could Targeting NPM1c+ Misfolding Be a Promising Strategy for Combating Acute Myeloid Leukemia?" International Journal of Molecular Sciences 25, no. 2: 811. https://doi.org/10.3390/ijms25020811
APA StyleFlorio, D., & Marasco, D. (2024). Could Targeting NPM1c+ Misfolding Be a Promising Strategy for Combating Acute Myeloid Leukemia? International Journal of Molecular Sciences, 25(2), 811. https://doi.org/10.3390/ijms25020811