
Lysine Methylation-Dependent Proteolysis by the Malignant Brain Tumor (MBT) Domain Proteins

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Non-Histone Proteins Are Targeted for Proteolysis
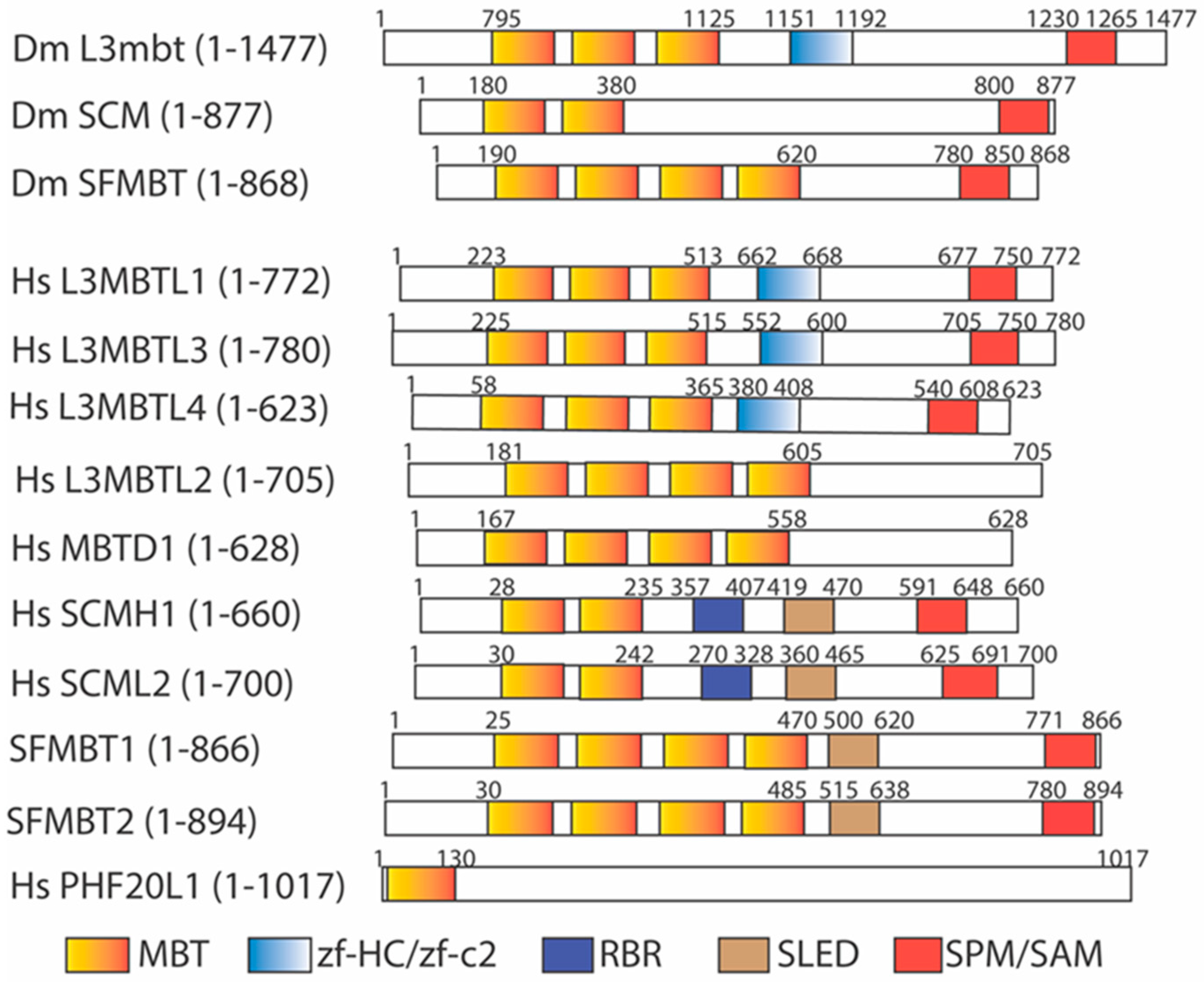
3. The Malignant Brain Tumor (MBT) Domain Serves as a Methyl Lysine Recognition Domain
4. The Methylated DNMT1 as a Substrate for L3MBTL3 and the CRL4DCAF5 Ubiquitin E3 Ligase Complex
5. E2F1 as a Substrate for L3MBTL3 and the CRL4DCAF5 Ubiquitin E3 Ligase Complex
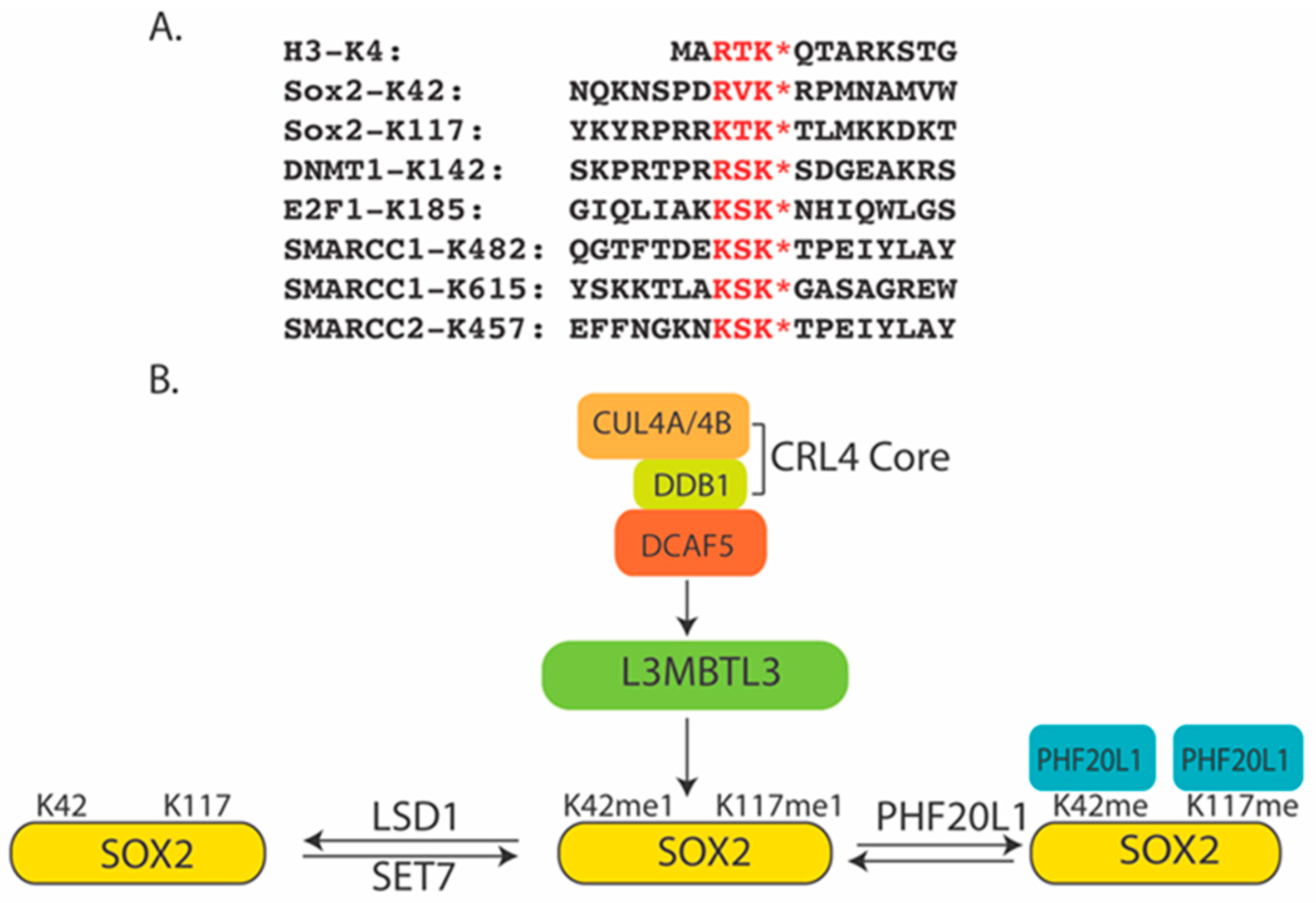
6. SOX2 as a Substrate for L3MBTL3 and the CRL4DCAF5 Ubiquitin E3 Ligase Complex
7. SMARCC1 and SMARCC2 as a Substrate for L3MBTL3 and the CRL4DCAF5 Ubiquitin E3 Ligase Complex
8. L3MBTL3 Inhibitor UNC1215 Serves as a PROTAC Molecule
9. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jambhekar, A.; Dhall, A.; Shi, Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell. Biol. 2019, 20, 625–641. [Google Scholar] [CrossRef]
- Zhang, X.; Wen, H.; Shi, X. Lysine methylation: Beyond histones. Acta Biochim. Biophys. Sin. 2012, 44, 14–27. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010, 11, 285–296. [Google Scholar] [CrossRef]
- Nady, N.; Krichevsky, L.; Zhong, N.; Duan, S.; Tempel, W.; Amaya, M.F.; Ravichandran, M.; Arrowsmith, C.H. Histone recognition by human malignant brain tumor domains. J. Mol. Biol. 2012, 423, 702–718. [Google Scholar] [CrossRef]
- Lu, R.; Wang, G.G. Tudor: A versatile family of histone methylation ‘readers’. Trends Biochem. Sci. 2013, 38, 546–555. [Google Scholar] [CrossRef]
- Klose, R.J.; Zhang, Y. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell. Biol. 2007, 8, 307–318. [Google Scholar] [CrossRef]
- Hamamoto, R.; Saloura, V.; Nakamura, Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat. Rev. Cancer. 2015, 15, 110–124. [Google Scholar] [CrossRef]
- Dhayalan, A.; Kudithipudi, S.; Rathert, P.; Jeltsch, A. Specificity analysis-based identification of new methylation targets of the SET7/9 protein lysine methyltransferase. Chem. Biol. 2011, 18, 111–120. [Google Scholar] [CrossRef]
- Kim, S.K.; Lee, H.; Han, K.; Kim, S.C.; Choi, Y.; Park, S.W.; Bak, G.; Lee, Y.; Choi, J.K.; Kim, T.K.; et al. SET7/9 methylation of the pluripotency factor LIN28A is a nucleolar localization mechanism that blocks let-7 biogenesis in human ESCs. Cell Stem Cell 2014, 15, 735–749. [Google Scholar] [CrossRef]
- Fu, L.; Wu, H.; Cheng, S.Y.; Gao, D.; Zhang, L.; Zhao, Y. Set7 mediated Gli3 methylation plays a positive role in the activation of Sonic Hedgehog pathway in mammals. eLife 2016, 5, e15690. [Google Scholar] [CrossRef]
- Chuikov, S.; Kurash, J.K.; Wilson, J.R.; Xiao, B.; Justin, N.; Ivanov, G.S.; McKinney, K.; Tempst, P.; Prives, C.; Gamblin, S.J.; et al. Regulation of p53 activity through lysine methylation. Nature 2004, 432, 353–360. [Google Scholar] [CrossRef]
- Calnan, D.R.; Webb, A.E.; White, J.L.; Stowe, T.R.; Goswami, T.; Shi, X.; Espejo, A.; Bedford, M.T.; Gozani, O.; Gygi, S.P.; et al. Methylation by Set9 modulates FoxO3 stability and transcriptional activity. Aging 2012, 4, 462–479. [Google Scholar] [CrossRef]
- Kontaki, H.; Talianidis, I. Lysine methylation regulates E2F1-induced cell death. Mol. Cell 2010, 39, 152–160. [Google Scholar] [CrossRef]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef]
- Yang, X.D.; Huang, B.; Li, M.; Lamb, A.; Kelleher, N.L.; Chen, L.F. Negative regulation of NF-kappaB action by Set9-mediated lysine methylation of the RelA subunit. EMBO J. 2009, 28, 1055–1066. [Google Scholar] [CrossRef]
- Esteve, P.O.; Chin, H.G.; Benner, J.; Feehery, G.R.; Samaranayake, M.; Horwitz, G.A.; Jacobsen, S.E.; Pradhan, S. Regulation of DNMT1 stability through SET7-mediated lysine methylation in mammalian cells. Proc. Natl. Acad. Sci. USA 2009, 106, 5076–5081. [Google Scholar] [CrossRef]
- Sharif, J.; Muto, M.; Takebayashi, S.; Suetake, I.; Iwamatsu, A.; Endo, T.A.; Shinga, J.; Mizutani-Koseki, Y.; Toyoda, T.; Okamura, K.; et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 2007, 450, 908–912. [Google Scholar] [CrossRef]
- Qin, W.; Leonhardt, H.; Pichler, G. Regulation of DNA methyltransferase 1 by interactions and modifications. Nucleus 2011, 2, 392–402. [Google Scholar] [CrossRef]
- Rose, N.R.; Klose, R.J. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta 2014, 1839, 1362–1372. [Google Scholar] [CrossRef]
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [Google Scholar] [CrossRef]
- Schoofs, T.; Berdel, W.E.; Muller-Tidow, C. Origins of aberrant DNA methylation in acute myeloid leukemia. Leukemia 2014, 28, 1–14. [Google Scholar] [CrossRef]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef]
- Kooistra, S.M.; Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell. Biol. 2012, 13, 297–311. [Google Scholar] [CrossRef]
- Wang, J.; Hevi, S.; Kurash, J.K.; Lei, H.; Gay, F.; Bajko, J.; Su, H.; Sun, W.; Chang, H.; Xu, G.; et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 2009, 41, 125–129. [Google Scholar] [CrossRef]
- O’Connell, B.C.; Harper, J.W. Ubiquitin proteasome system (UPS): What can chromatin do for you? Curr. Opin. Cell Biol. 2007, 19, 206–214. [Google Scholar] [CrossRef]
- Petroski, M.D.; Deshaies, R.J. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell. Biol. 2005, 6, 9–20. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Du, Z.; Song, J.; Wang, Y.; Zhao, Y.; Guda, K.; Yang, S.; Kao, H.Y.; Xu, Y.; Willis, J.; Markowitz, S.D.; et al. DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci. Signal. 2010, 3, ra80. [Google Scholar] [CrossRef] [PubMed]
- Driessens, G.; Blanpain, C. Long live sox2: Sox2 lasts a lifetime. Cell Stem Cell 2011, 9, 283–284. [Google Scholar] [CrossRef]
- Yamanaka, S. A fresh look at iPS cells. Cell 2009, 137, 13–17. [Google Scholar] [CrossRef]
- Hagey, D.W.; Muhr, J. Sox2 acts in a dose-dependent fashion to regulate proliferation of cortical progenitors. Cell Rep. 2014, 9, 1908–1920. [Google Scholar] [CrossRef]
- Taranova, O.V.; Magness, S.T.; Fagan, B.M.; Wu, Y.; Surzenko, N.; Hutton, S.R.; Pevny, L.H. SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev. 2006, 20, 1187–1202. [Google Scholar] [CrossRef]
- Sarkar, A.; Hochedlinger, K. The sox family of transcription factors: Versatile regulators of stem and progenitor cell fate. Cell Stem Cell 2013, 12, 15–30. [Google Scholar] [CrossRef]
- Wu, J.; Izpisua Belmonte, J.C. The Molecular Harbingers of Early Mammalian Embryo Patterning. Cell 2016, 165, 13–15. [Google Scholar] [CrossRef]
- Goolam, M.; Scialdone, A.; Graham, S.J.; Macaulay, I.C.; Jedrusik, A.; Hupalowska, A.; Voet, T.; Marioni, J.C.; Zernicka-Goetz, M. Heterogeneity in Oct4 and Sox2 Targets Biases Cell Fate in 4-Cell Mouse Embryos. Cell 2016, 165, 61–74. [Google Scholar] [CrossRef] [PubMed]
- White, M.D.; Angiolini, J.F.; Alvarez, Y.D.; Kaur, G.; Zhao, Z.W.; Mocskos, E.; Bruno, L.; Bissiere, S.; Levi, V.; Plachta, N. Long-Lived Binding of Sox2 to DNA Predicts Cell Fate in the Four-Cell Mouse Embryo. Cell 2016, 165, 75–87. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Hirano, K.; Nagata, S.; Tada, T. Sox2 expression effects on direct reprogramming efficiency as determined by alternative somatic cell fate. Stem Cell Res. 2011, 6, 177–186. [Google Scholar] [CrossRef]
- Rizzino, A.; Wuebben, E.L. Sox2/Oct4: A delicately balanced partnership in pluripotent stem cells and embryogenesis. Biochim. Biophys. Acta 2016, 1859, 780–791. [Google Scholar] [CrossRef]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef]
- Young, R.A. Control of the embryonic stem cell state. Cell 2011, 144, 940–954. [Google Scholar] [CrossRef]
- Arnold, K.; Sarkar, A.; Yram, M.A.; Polo, J.M.; Bronson, R.; Sengupta, S.; Seandel, M.; Geijsen, N.; Hochedlinger, K. Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 2011, 9, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Williamson, K.A.; FitzPatrick, D.R. SOX2-Related Eye Disorders; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; GeneReviews((R)): Seattle, WA, USA, 1993. [Google Scholar]
- Williamson, K.A.; Hever, A.M.; Rainger, J.; Rogers, R.C.; Magee, A.; Fiedler, Z.; Keng, W.T.; Sharkey, F.H.; McGill, N.; Hill, C.J.; et al. Mutations in SOX2 cause anophthalmia-esophageal-genital (AEG) syndrome. Hum. Mol. Genet. 2006, 15, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Fantes, J.; Ragge, N.K.; Lynch, S.A.; McGill, N.I.; Collin, J.R.; Howard-Peebles, P.N.; Hayward, C.; Vivian, A.J.; Williamson, K.; van Heyningen, V.; et al. Mutations in SOX2 cause anophthalmia. Nat. Genet. 2003, 33, 461–463. [Google Scholar] [CrossRef]
- Bani-Yaghoub, M.; Tremblay, R.G.; Lei, J.X.; Zhang, D.; Zurakowski, B.; Sandhu, J.K.; Smith, B.; Ribecco-Lutkiewicz, M.; Kennedy, J.; Walker, P.R.; et al. Role of Sox2 in the development of the mouse neocortex. Dev. Biol. 2006, 295, 52–66. [Google Scholar] [CrossRef]
- Bass, A.J.; Watanabe, H.; Mermel, C.H.; Yu, S.; Perner, S.; Verhaak, R.G.; Kim, S.Y.; Wardwell, L.; Tamayo, P.; Gat-Viks, I.; et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat. Genet. 2009, 41, 1238–1242. [Google Scholar] [CrossRef]
- Hussenet, T.; Dali, S.; Exinger, J.; Monga, B.; Jost, B.; Dembele, D.; Martinet, N.; Thibault, C.; Huelsken, J.; Brambilla, E.; et al. SOX2 is an oncogene activated by recurrent 3q26.3 amplifications in human lung squamous cell carcinomas. PLoS ONE 2010, 5, e8960. [Google Scholar] [CrossRef]
- Hussenet, T.; du Manoir, S. SOX2 in squamous cell carcinoma: Amplifying a pleiotropic oncogene along carcinogenesis. Cell Cycle 2010, 9, 1480–1486. [Google Scholar] [CrossRef] [PubMed]
- Maier, S.; Wilbertz, T.; Braun, M.; Scheble, V.; Reischl, M.; Mikut, R.; Menon, R.; Nikolov, P.; Petersen, K.; Beschorner, C.; et al. SOX2 amplification is a common event in squamous cell carcinomas of different organ sites. Hum. Pathol. 2011, 42, 1078–1088. [Google Scholar] [CrossRef]
- Weina, K.; Utikal, J. SOX2 and cancer: Current research and its implications in the clinic. Clin. Transl. Med. 2014, 3, 19. [Google Scholar] [CrossRef]
- Rudin, C.M.; Durinck, S.; Stawiski, E.W.; Poirier, J.T.; Modrusan, Z.; Shames, D.S.; Bergbower, E.A.; Guan, Y.; Shin, J.; Guillory, J.; et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat. Genet. 2012, 44, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.M.; Diez-Valle, R.; Manterola, L.; Rubio, A.; Liu, D.; Cortes-Santiago, N.; Urquiza, L.; Jauregi, P.; Lopez de Munain, A.; Sampron, N.; et al. Genetic and epigenetic modifications of Sox2 contribute to the invasive phenotype of malignant gliomas. PLoS ONE 2011, 6, e26740. [Google Scholar] [CrossRef] [PubMed]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Sholl, L.M.; Barletta, J.A.; Yeap, B.Y.; Chirieac, L.R.; Hornick, J.L. Sox2 protein expression is an independent poor prognostic indicator in stage I lung adenocarcinoma. Am. J. Surg. Pathol. 2010, 34, 1193–1198. [Google Scholar] [CrossRef]
- Sholl, L.M.; Long, K.B.; Hornick, J.L. Sox2 expression in pulmonary non-small cell and neuroendocrine carcinomas. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Maihle, N.J.; Huang, Y. Pluripotency factors Lin28 and Oct4 identify a sub-population of stem cell-like cells in ovarian cancer. Oncogene 2010, 29, 2153–2159. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Li, N.; Liang, S.; Huang, Q.; Coukos, G.; Zhang, L. Identification of microRNAs regulating reprogramming factor LIN28 in embryonic stem cells and cancer cells. J. Biol. Chem. 2010, 285, 41961–41971. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Bahr, F.; Horst, D.; Kriegl, L.; Engel, J.; Luque, R.M.; Gerhard, M.; Kirchner, T.; Jung, A. SOX2 expression correlates with lymph-node metastases and distant spread in right-sided colon cancer. BMC Cancer 2011, 11, 518. [Google Scholar] [CrossRef]
- Kim, J.B.; Sebastiano, V.; Wu, G.; Arauzo-Bravo, M.J.; Sasse, P.; Gentile, L.; Ko, K.; Ruau, D.; Ehrich, M.; van den Boom, D.; et al. Oct4-induced pluripotency in adult neural stem cells. Cell 2009, 136, 411–419. [Google Scholar] [CrossRef]
- Leis, O.; Eguiara, A.; Lopez-Arribillaga, E.; Alberdi, M.J.; Hernandez-Garcia, S.; Elorriaga, K.; Pandiella, A.; Rezola, R.; Martin, A.G. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene 2012, 31, 1354–1365. [Google Scholar] [CrossRef]
- Matsuoka, J.; Yashiro, M.; Sakurai, K.; Kubo, N.; Tanaka, H.; Muguruma, K.; Sawada, T.; Ohira, M.; Hirakawa, K. Role of the stemness factors sox2, oct3/4, and nanog in gastric carcinoma. J. Surg. Res. 2012, 174, 130–135. [Google Scholar] [CrossRef]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef]
- Tsukamoto, T.; Mizoshita, T.; Mihara, M.; Tanaka, H.; Takenaka, Y.; Yamamura, Y.; Nakamura, S.; Ushijima, T.; Tatematsu, M. Sox2 expression in human stomach adenocarcinomas with gastric and gastric-and-intestinal-mixed phenotypes. Histopathology 2005, 46, 649–658. [Google Scholar] [CrossRef]
- Boumahdi, S.; Driessens, G.; Lapouge, G.; Rorive, S.; Nassar, D.; Le Mercier, M.; Delatte, B.; Caauwe, A.; Lenglez, S.; Nkusi, E.; et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 2014, 511, 246–250. [Google Scholar] [CrossRef]
- Vanner, R.J.; Remke, M.; Gallo, M.; Selvadurai, H.J.; Coutinho, F.; Lee, L.; Kushida, M.; Head, R.; Morrissy, S.; Zhu, X.; et al. Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell 2014, 26, 33–47. [Google Scholar] [CrossRef]
- Fang, L.; Zhang, L.; Wei, W.; Jin, X.; Wang, P.; Tong, Y.; Li, J.; Du, J.X.; Wong, J. A methylation-phosphorylation switch determines Sox2 stability and function in ESC maintenance or differentiation. Mol. Cell 2014, 55, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Wismar, J.; Loffler, T.; Habtemichael, N.; Vef, O.; Geissen, M.; Zirwes, R.; Altmeyer, W.; Sass, H.; Gateff, E. The Drosophila melanogaster tumor suppressor gene lethal(3)malignant brain tumor encodes a proline-rich protein with a novel zinc finger. Mech. Dev. 1995, 53, 141–154. [Google Scholar] [CrossRef]
- Gateff, E.; Loffler, T.; Wismar, J. A temperature-sensitive brain tumor suppressor mutation of Drosophila melanogaster: Developmental studies and molecular localization of the gene. Mech. Dev. 1993, 41, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.; Ponting, C.P.; Hofmann, K.; Bork, P. SAM as a protein interaction domain involved in developmental regulation. Protein Sci. 1997, 6, 249–253. [Google Scholar] [CrossRef]
- Bornemann, D.; Miller, E.; Simon, J. The Drosophila Polycomb group gene Sex comb on midleg (Scm) encodes a zinc finger protein with similarity to polyhomeotic protein. Development 1996, 122, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Breen, T.R.; Duncan, I.M. Maternal expression of genes that regulate the bithorax complex of Drosophila melanogaster. Dev. Biol. 1986, 118, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, T.; Papp, B.; Fischle, W.; Kocher, T.; Schelder, M.; Fritsch, C.; Wild, B.; Wilm, M.; Muller, J. A Polycomb group protein complex with sequence-specific DNA-binding and selective methyl-lysine-binding activities. Genes Dev. 2006, 20, 1110–1122. [Google Scholar] [CrossRef]
- Koga, H.; Matsui, S.; Hirota, T.; Takebayashi, S.; Okumura, K.; Saya, H. A human homolog of Drosophila lethal(3)malignant brain tumor (l(3)mbt) protein associates with condensed mitotic chromosomes. Oncogene 1999, 18, 3799–3809. [Google Scholar] [CrossRef] [PubMed]
- Wismar, J. Molecular characterization of h-l(3)mbt-like: A new member of the human mbt family. FEBS Lett. 2001, 507, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Miyazaki, T. Impaired maturation of myeloid progenitors in mice lacking novel Polycomb group protein MBT-1. EMBO J. 2005, 24, 1863–1873. [Google Scholar] [CrossRef]
- Berger, J.; Kurahashi, H.; Takihara, Y.; Shimada, K.; Brock, H.W.; Randazzo, F. The human homolog of Sex comb on midleg (SCMH1) maps to chromosome 1p34. Gene 1999, 237, 185–191. [Google Scholar] [CrossRef]
- Montini, E.; Buchner, G.; Spalluto, C.; Andolfi, G.; Caruso, A.; den Dunnen, J.T.; Trump, D.; Rocchi, M.; Ballabio, A.; Franco, B. Identification of SCML2, a second human gene homologous to the Drosophila sex comb on midleg (Scm): A new gene cluster on Xp22. Genomics 1999, 58, 65–72. [Google Scholar] [CrossRef]
- Usui, H.; Ichikawa, T.; Kobayashi, K.; Kumanishi, T. Cloning of a novel murine gene Sfmbt, Scm-related gene containing four mbt domains, structurally belonging to the Polycomb group of genes. Gene 2000, 248, 127–135. [Google Scholar] [CrossRef]
- Kuzmin, A.; Han, Z.; Golding, M.C.; Mann, M.R.; Latham, K.E.; Varmuza, S. The PcG gene Sfmbt2 is paternally expressed in extraembryonic tissues. Gene Expr. Patterns 2008, 8, 107–116. [Google Scholar] [CrossRef]
- Honda, H.; Takubo, K.; Oda, H.; Kosaki, K.; Tazaki, T.; Yamasaki, N.; Miyazaki, K.; Moore, K.A.; Honda, Z.; Suda, T.; et al. Hemp, an mbt domain-containing protein, plays essential roles in hematopoietic stem cell function and skeletal formation. Proc. Natl. Acad. Sci. USA 2011, 108, 2468–2473. [Google Scholar] [CrossRef]
- Addou-Klouche, L.; Adelaide, J.; Finetti, P.; Cervera, N.; Ferrari, A.; Bekhouche, I.; Sircoulomb, F.; Sotiriou, C.; Viens, P.; Moulessehoul, S.; et al. Loss, mutation and deregulation of L3MBTL4 in breast cancers. Mol. Cancer 2010, 9, 213. [Google Scholar] [CrossRef]
- Boccuni, P.; MacGrogan, D.; Scandura, J.M.; Nimer, S.D. The human L(3)MBT polycomb group protein is a transcriptional repressor and interacts physically and functionally with TEL (ETV6). J. Biol. Chem. 2003, 278, 15412–15420. [Google Scholar] [CrossRef]
- Liu, X.; Hu, C.; Bao, M.; Li, J.; Liu, X.; Tan, X.; Zhou, Y.; Chen, Y.; Wu, S.; Chen, S.; et al. Genome Wide Association Study Identifies L3MBTL4 as a Novel Susceptibility Gene for Hypertension. Sci. Rep. 2016, 6, 30811. [Google Scholar] [CrossRef]
- Lin, S.; Shen, H.; Li, J.L.; Tang, S.; Gu, Y.; Chen, Z.; Hu, C.; Rice, J.C.; Lu, J.; Wu, L. Proteomic and functional analyses reveal the role of chromatin reader SFMBT1 in regulating epigenetic silencing and the myogenic gene program. J. Biol. Chem. 2013, 288, 6238–6247. [Google Scholar] [CrossRef]
- Mendjan, S.; Taipale, M.; Kind, J.; Holz, H.; Gebhardt, P.; Schelder, M.; Vermeulen, M.; Buscaino, A.; Duncan, K.; Mueller, J.; et al. Nuclear pore components are involved in the transcriptional regulation of dosage compensation in Drosophila. Mol. Cell 2006, 21, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Jin, J.; Swanson, S.K.; Cole, M.D.; Choi, S.H.; Florens, L.; Washburn, M.P.; Conaway, J.W.; Conaway, R.C. Subunit composition and substrate specificity of a MOF-containing histone acetyltransferase distinct from the male-specific lethal (MSL) complex. J. Biol. Chem. 2010, 285, 4268–4272. [Google Scholar] [CrossRef]
- Esteve, P.O.; Terragni, J.; Deepti, K.; Chin, H.G.; Dai, N.; Espejo, A.; Correa, I.R., Jr.; Bedford, M.T.; Pradhan, S. Methyllysine reader plant homeodomain (PHD) finger protein 20-like 1 (PHF20L1) antagonizes DNA (cytosine-5) methyltransferase 1 (DNMT1) proteasomal degradation. J. Biol. Chem. 2014, 289, 8277–8287. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Daniel, J.; Espejo, A.; Lake, A.; Krishna, M.; Xia, L.; Zhang, Y.; Bedford, M.T. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006, 7, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Maurer-Stroh, S.; Dickens, N.J.; Hughes-Davies, L.; Kouzarides, T.; Eisenhaber, F.; Ponting, C.P. The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem. Sci. 2003, 28, 69–74. [Google Scholar] [CrossRef]
- Wang, W.K.; Tereshko, V.; Boccuni, P.; MacGrogan, D.; Nimer, S.D.; Patel, D.J. Malignant brain tumor repeats: A three-leaved propeller architecture with ligand/peptide binding pockets. Structure 2003, 11, 775–789. [Google Scholar] [CrossRef]
- Min, J.; Allali-Hassani, A.; Nady, N.; Qi, C.; Ouyang, H.; Liu, Y.; MacKenzie, F.; Vedadi, M.; Arrowsmith, C.H. L3MBTL1 recognition of mono- and dimethylated histones. Nat. Struct. Mol. Biol. 2007, 14, 1229–1230. [Google Scholar] [CrossRef]
- Li, H.; Fischle, W.; Wang, W.; Duncan, E.M.; Liang, L.; Murakami-Ishibe, S.; Allis, C.D.; Patel, D.J. Structural basis for lower lysine methylation state-specific readout by MBT repeats of L3MBTL1 and an engineered PHD finger. Mol. Cell 2007, 28, 677–691. [Google Scholar] [CrossRef]
- Trojer, P.; Li, G.; Sims, R.J., 3rd; Vaquero, A.; Kalakonda, N.; Boccuni, P.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Nimer, S.D.; et al. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell 2007, 129, 915–928. [Google Scholar] [CrossRef]
- Huang, J.; Sengupta, R.; Espejo, A.B.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T.; et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007, 449, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Kachirskaia, I.; Yamaguchi, H.; West, L.E.; Wen, H.; Wang, E.W.; Dutta, S.; Appella, E.; Gozani, O. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell 2007, 27, 636–646. [Google Scholar] [CrossRef] [PubMed]
- West, L.E.; Roy, S.; Lachmi-Weiner, K.; Hayashi, R.; Shi, X.; Appella, E.; Kutateladze, T.G.; Gozani, O. The MBT repeats of L3MBTL1 link SET8-mediated p53 methylation at lysine 382 to target gene repression. J. Biol. Chem. 2010, 285, 37725–37732. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Nady, N.; Qi, C.; Allali-Hassani, A.; Zhu, H.; Pan, P.; Adams-Cioaba, M.A.; Amaya, M.F.; Dong, A.; Vedadi, M.; et al. Methylation-state-specific recognition of histones by the MBT repeat protein L3MBTL2. Nucleic Acids Res. 2009, 37, 2204–2210. [Google Scholar] [CrossRef]
- Eryilmaz, J.; Pan, P.; Amaya, M.F.; Allali-Hassani, A.; Dong, A.; Adams-Cioaba, M.A.; Mackenzie, F.; Vedadi, M.; Min, J. Structural studies of a four-MBT repeat protein MBTD1. PLoS ONE 2009, 4, e7274. [Google Scholar] [CrossRef]
- Northcott, P.A.; Nakahara, Y.; Wu, X.; Feuk, L.; Ellison, D.W.; Croul, S.; Mack, S.; Kongkham, P.N.; Peacock, J.; Dubuc, A.; et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 2009, 41, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Trojer, P.; Cao, A.R.; Gao, Z.; Li, Y.; Zhang, J.; Xu, X.; Li, G.; Losson, R.; Erdjument-Bromage, H.; Tempst, P.; et al. L3MBTL2 protein acts in concert with PcG protein-mediated monoubiquitination of H2A to establish a repressive chromatin structure. Mol. Cell 2011, 42, 438–450. [Google Scholar] [CrossRef]
- Stielow, B.; Finkernagel, F.; Stiewe, T.; Nist, A.; Suske, G. MGA, L3MBTL2 and E2F6 determine genomic binding of the non-canonical Polycomb repressive complex PRC1.6. PLoS Genet. 2018, 14, e1007193. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, J.; Bonasio, R.; Strino, F.; Sawai, A.; Parisi, F.; Kluger, Y.; Reinberg, D. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 2012, 45, 344–356. [Google Scholar] [CrossRef]
- Qin, J.; Whyte, W.A.; Anderssen, E.; Apostolou, E.; Chen, H.H.; Akbarian, S.; Bronson, R.T.; Hochedlinger, K.; Ramaswamy, S.; Young, R.A.; et al. The polycomb group protein L3mbtl2 assembles an atypical PRC1-family complex that is essential in pluripotent stem cells and early development. Cell Stem Cell 2012, 11, 319–332. [Google Scholar] [CrossRef]
- Grimm, C.; de Ayala Alonso, A.G.; Rybin, V.; Steuerwald, U.; Ly-Hartig, N.; Fischle, W.; Muller, J.; Muller, C.W. Structural and functional analyses of methyl-lysine binding by the malignant brain tumour repeat protein Sex comb on midleg. EMBO Rep. 2007, 8, 1031–1037. [Google Scholar] [CrossRef]
- Takada, Y.; Isono, K.; Shinga, J.; Turner, J.M.; Kitamura, H.; Ohara, O.; Watanabe, G.; Singh, P.B.; Kamijo, T.; Jenuwein, T.; et al. Mammalian Polycomb Scmh1 mediates exclusion of Polycomb complexes from the XY body in the pachytene spermatocytes. Development 2007, 134, 579–590. [Google Scholar] [CrossRef]
- Hasegawa, K.; Sin, H.S.; Maezawa, S.; Broering, T.J.; Kartashov, A.V.; Alavattam, K.G.; Ichijima, Y.; Zhang, F.; Bacon, W.C.; Greis, K.D.; et al. SCML2 establishes the male germline epigenome through regulation of histone H2A ubiquitination. Dev. Cell 2015, 32, 574–588. [Google Scholar] [CrossRef]
- Leng, F.; Yu, J.; Zhang, C.; Alejo, S.; Hoang, N.; Sun, H.; Lu, F.; Zhang, H. Methylated DNMT1 and E2F1 are targeted for proteolysis by L3MBTL3 and CRL4(DCAF5) ubiquitin ligase. Nat. Commun. 2018, 9, 1641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hoang, N.; Leng, F.; Saxena, L.; Lee, L.; Alejo, S.; Qi, D.; Khal, A.; Sun, H.; Lu, F.; et al. LSD1 demethylase and the methyl-binding protein PHF20L1 prevent SET7 methyltransferase-dependent proteolysis of the stem-cell protein SOX2. J. Biol. Chem. 2018, 293, 3663–3674. [Google Scholar] [CrossRef]
- Zhang, C.; Leng, F.; Saxena, L.; Hoang, N.; Yu, J.; Alejo, S.; Lee, L.; Qi, D.; Lu, F.; Sun, H.; et al. Proteolysis of methylated SOX2 protein is regulated by L3MBTL3 and CRL4(DCAF5) ubiquitin ligase. J. Biol. Chem. 2019, 294, 476–489. [Google Scholar] [CrossRef] [PubMed]
- Esteve, P.O.; Chang, Y.; Samaranayake, M.; Upadhyay, A.K.; Horton, J.R.; Feehery, G.R.; Cheng, X.; Pradhan, S. A methylation and phosphorylation switch between an adjacent lysine and serine determines human DNMT1 stability. Nat. Struct. Mol. Biol. 2011, 18, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bedford, M.T. Titivated for destruction: The methyl degron. Mol. Cell 2012, 48, 487–488. [Google Scholar] [CrossRef]
- Higa, L.A.; Mihaylov, I.S.; Banks, D.P.; Zheng, J.; Zhang, H. Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat. Cell Biol. 2003, 5, 1008–1015. [Google Scholar] [CrossRef]
- Higa, L.A.; Banks, D.; Wu, M.; Kobayashi, R.; Sun, H.; Zhang, H. L2DTL/CDT2 interacts with the CUL4/DDB1 complex and PCNA and regulates CDT1 proteolysis in response to DNA damage. Cell Cycle 2006, 5, 1675–1680. [Google Scholar] [CrossRef]
- Higa, L.A.; Wu, M.; Ye, T.; Kobayashi, R.; Sun, H.; Zhang, H. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat. Cell Biol. 2006, 8, 1277–1283. [Google Scholar] [CrossRef]
- Jin, J.; Arias, E.E.; Chen, J.; Harper, J.W.; Walter, J.C. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 2006, 23, 709–721. [Google Scholar] [CrossRef]
- Higa, L.A.; Zhang, H. Stealing the spotlight: CUL4-DDB1 ubiquitin ligase docks WD40-repeat proteins to destroy. Cell Div. 2007, 2, 5. [Google Scholar] [CrossRef]
- Zhang, H. Regulation of DNA Replication Licensing and Re-Replication by Cdt1. Int. J. Mol. Sci. 2021, 22, 5195. [Google Scholar] [CrossRef] [PubMed]
- Oehl-Jaschkowitz, B.; Vanakker, O.M.; De Paepe, A.; Menten, B.; Martin, T.; Weber, G.; Christmann, A.; Krier, R.; Scheid, S.; McNerlan, S.E.; et al. Deletions in 14q24.1q24.3 are associated with congenital heart defects, brachydactyly, and mild intellectual disability. Am. J. Med. Genet. A 2014, 164, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.P.; Beesley, J.; Amin Al Olama, A.; Michailidou, K.; Tyrer, J.; Kote-Jarai, Z.; Lawrenson, K.; Lindstrom, S.; Ramus, S.J.; Thompson, D.J.; et al. Genome-Wide Meta-Analyses of Breast, Ovarian, and Prostate Cancer Association Studies Identify Multiple New Susceptibility Loci Shared by at Least Two Cancer Types. Cancer Discov. 2016, 6, 1052–1067. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bonasio, R.; Strino, F.; Kluger, Y.; Holloway, J.K.; Modzelewski, A.J.; Cohen, P.E.; Reinberg, D. SFMBT1 functions with LSD1 to regulate expression of canonical histone genes and chromatin-related factors. Genes Dev. 2013, 27, 749–766. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Park, S.S.; Giaimo, B.D.; Hall, D.; Ferrante, F.; Ho, D.M.; Hori, K.; Anhezini, L.; Ertl, I.; Bartkuhn, M.; et al. RBPJ/CBF1 interacts with L3MBTL3/MBT1 to promote repression of Notch signaling via histone demethylase KDM1A/LSD1. EMBO J. 2017, 36, 3232–3249. [Google Scholar] [CrossRef]
- Bonasio, R.; Lecona, E.; Reinberg, D. MBT domain proteins in development and disease. Semin. Cell Dev. Biol. 2010, 21, 221–230. [Google Scholar] [CrossRef]
- Wang, J.; Lu, F.; Ren, Q.; Sun, H.; Xu, Z.; Lan, R.; Liu, Y.; Ward, D.; Quan, J.; Ye, T.; et al. Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res. 2011, 71, 7238–7249. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, F.; Wang, J.; Yin, F.; Xu, Z.; Qi, D.; Wu, X.; Cao, Y.; Liang, W.; Liu, Y.; et al. Pluripotent stem cell protein Sox2 confers sensitivity to LSD1 inhibition in cancer cells. Cell Rep. 2013, 5, 445–457. [Google Scholar] [CrossRef]
- James, L.I.; Barsyte-Lovejoy, D.; Zhong, N.; Krichevsky, L.; Korboukh, V.K.; Herold, J.M.; MacNevin, C.J.; Norris, J.L.; Sagum, C.A.; Tempel, W.; et al. Discovery of a chemical probe for the L3MBTL3 methyllysine reader domain. Nat. Chem. Biol. 2013, 9, 184–191. [Google Scholar] [CrossRef]
- Guo, P.; Hoang, N.; Sanchez, J.; Zhang, E.H.; Rajawasam, K.; Trinidad, K.; Sun, H.; Zhang, H. The assembly of mammalian SWI/SNF chromatin remodeling complexes is regulated by lysine-methylation dependent proteolysis. Nat. Commun. 2022, 13, 6696. [Google Scholar] [CrossRef]
- Ho, L.; Crabtree, G.R. Chromatin remodelling during development. Nature 2010, 463, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Crabtree, G.R. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 2015, 1, e1500447. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Ho, L.; Jothi, R.; Ronan, J.L.; Cui, K.; Zhao, K.; Crabtree, G.R. An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc. Natl. Acad. Sci. USA 2009, 106, 5187–5191. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Ronan, J.L.; Wu, J.; Staahl, B.T.; Chen, L.; Kuo, A.; Lessard, J.; Nesvizhskii, A.I.; Ranish, J.; Crabtree, G.R. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc. Natl. Acad. Sci. USA 2009, 106, 5181–5186. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Wang, Z.; Sharova, L.; Sharov, A.A.; Ling, C.; Piao, Y.; Aiba, K.; Matoba, R.; Wang, W.; Ko, M.S. BAF250B-associated SWI/SNF chromatin-remodeling complex is required to maintain undifferentiated mouse embryonic stem cells. Stem Cells 2008, 26, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Esch, D.; Stehling, M.; Scholer, H.R. BRG1 Is Required to Maintain Pluripotency of Murine Embryonic Stem Cells. BioResearch Open Access 2014, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Graumann, J.; Wu, G.; Arauzo-Bravo, M.J.; Han, D.W.; Greber, B.; Gentile, L.; Mann, M.; Scholer, H.R. Chromatin-Remodeling Components of the BAF Complex Facilitate Reprogramming. Cell 2010, 141, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Jeon, S.; Sohn, D.H.; Lee, C.; Ahn, S.; Kim, W.K.; Chung, H.; Seong, R.H. SRG3, a core component of mouse SWI/SNF complex, is essential for extra-embryonic vascular development. Dev. Biol. 2008, 315, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Bultman, S.; Gebuhr, T.; Yee, D.; La Mantia, C.; Nicholson, J.; Gilliam, A.; Randazzo, F.; Metzger, D.; Chambon, P.; Crabtree, G.; et al. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol. Cell 2000, 6, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Tuoc, T.C.; Boretius, S.; Sansom, S.N.; Pitulescu, M.E.; Frahm, J.; Livesey, F.J.; Stoykova, A. Chromatin regulation by BAF170 controls cerebral cortical size and thickness. Dev. Cell 2013, 25, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sansam, C.G.; Thom, C.S.; Metzger, D.; Evans, J.A.; Nguyen, P.T.; Roberts, C.W. Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res. 2009, 69, 8094–8101. [Google Scholar] [CrossRef]
- Helming, K.C.; Wang, X.; Wilson, B.G.; Vazquez, F.; Haswell, J.R.; Manchester, H.E.; Kim, Y.; Kryukov, G.V.; Ghandi, M.; Aguirre, A.J.; et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med. 2014, 20, 251–254. [Google Scholar] [CrossRef]
- Schick, S.; Rendeiro, A.F.; Runggatscher, K.; Ringler, A.; Boidol, B.; Hinkel, M.; Majek, P.; Vulliard, L.; Penz, T.; Parapatics, K.; et al. Systematic characterization of BAF mutations provides insights into intracomplex synthetic lethalities in human cancers. Nat. Genet. 2019, 51, 1399–1410. [Google Scholar] [CrossRef]
- Narayanan, R.; Pirouz, M.; Kerimoglu, C.; Pham, L.; Wagener, R.J.; Kiszka, K.A.; Rosenbusch, J.; Seong, R.H.; Kessel, M.; Fischer, A.; et al. Loss of BAF (mSWI/SNF) Complexes Causes Global Transcriptional and Chromatin State Changes in Forebrain Development. Cell Rep. 2015, 13, 1842–1854. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Helming, K.C.; Wang, X.; Kim, Y.; Vazquez, F.; Jagani, Z.; Hahn, W.C.; Roberts, C.W. Residual complexes containing SMARCA2 (BRM) underlie the oncogenic drive of SMARCA4 (BRG1) mutation. Mol. Cell. Biol. 2014, 34, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Helming, K.C.; Wang, X.; Roberts CWM. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell 2014, 26, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Goldfarb, D.; Song, S.; Cannon, C.; Yan, F.; Sakellariou-Thompson, D.; Emanuele, M.; Major, M.B.; Weissman, B.E.; Kuwahara, Y. SNF5/INI1 deficiency redefines chromatin remodeling complex composition during tumor development. Mol. Cancer Res. 2014, 12, 1574–1585. [Google Scholar] [CrossRef] [PubMed]
- Mashtalir, N.; D’Avino, A.R.; Michel, B.C.; Luo, J.; Pan, J.; Otto, J.E.; Zullow, H.J.; McKenzie, Z.M.; Kubiak, R.L.; St Pierre, R.; et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 2018, 175, 1272–1288.e20. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Archer, T.K. Regulating SWI/SNF subunit levels via protein-protein interactions and proteasomal degradation: BAF155 and BAF170 limit expression of BAF57. Mol. Cell. Biol. 2005, 25, 9016–9027. [Google Scholar] [CrossRef]
- Sohn, D.H.; Lee, K.Y.; Lee, C.; Oh, J.; Chung, H.; Jeon, S.H.; Seong, R.H. SRG3 interacts directly with the major components of the SWI/SNF chromatin remodeling complex and protects them from proteasomal degradation. J. Biol. Chem. 2007, 282, 10614–10624. [Google Scholar] [CrossRef]
- Nalawansha, D.A.; Li, K.; Hines, J.; Crews, C.M. Hijacking Methyl Reader Proteins for Nuclear-Specific Protein Degradation. J. Am. Chem. Soc. 2022, 144, 5594–5605. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, H.; Zhang, H. Lysine Methylation-Dependent Proteolysis by the Malignant Brain Tumor (MBT) Domain Proteins. Int. J. Mol. Sci. 2024, 25, 2248. https://doi.org/10.3390/ijms25042248
Sun H, Zhang H. Lysine Methylation-Dependent Proteolysis by the Malignant Brain Tumor (MBT) Domain Proteins. International Journal of Molecular Sciences. 2024; 25(4):2248. https://doi.org/10.3390/ijms25042248
Chicago/Turabian StyleSun, Hong, and Hui Zhang. 2024. "Lysine Methylation-Dependent Proteolysis by the Malignant Brain Tumor (MBT) Domain Proteins" International Journal of Molecular Sciences 25, no. 4: 2248. https://doi.org/10.3390/ijms25042248
APA StyleSun, H., & Zhang, H. (2024). Lysine Methylation-Dependent Proteolysis by the Malignant Brain Tumor (MBT) Domain Proteins. International Journal of Molecular Sciences, 25(4), 2248. https://doi.org/10.3390/ijms25042248