

Comparative Analysis of the Chloroplast Genomes of Eight Species of the Genus Lirianthe Spach with Its Generic Delimitation Implications


Abstract
:1. Introduction
2. Results
2.1. The Sequence Coverage Analysis of Assembled Chloroplast Genome
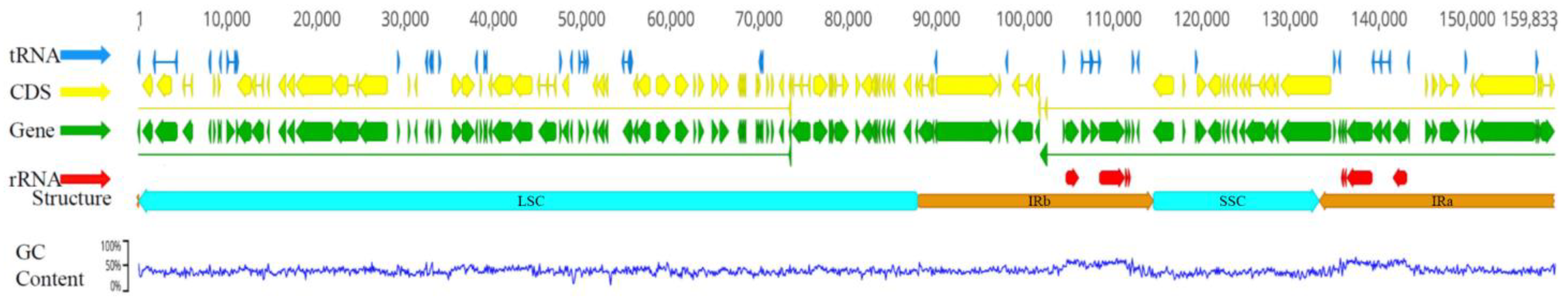
2.2. General Characteristics of Eight Chloroplast Genomes of Lirianthe Species
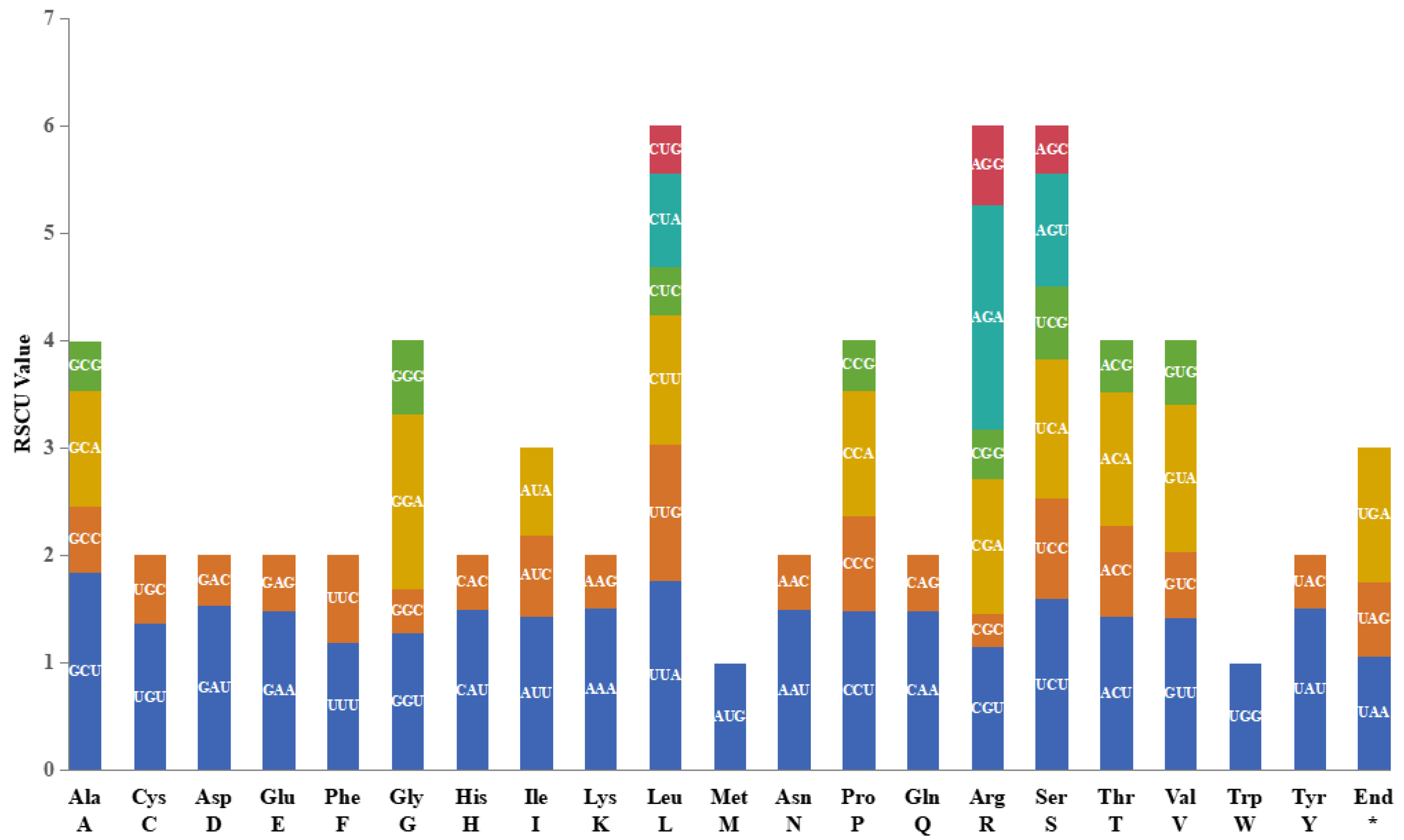
2.3. Codon Usage of Protein-Coding Genes
2.4. Repeat Identification
2.5. DNA Polymorphism
2.6. Contraction and Expansion of IR Region
2.7. Phylogenetic Analysis
2.8. Plant Morphology
3. Discussion
4. Materials and Methods
4.1. Sample DNA Extraction and Sequencing
4.2. Chloroplast Genome Assembly and Annotation
4.3. The Analysis of Structures and Characteristics of CPGs
4.4. Comparitive Analysis of Chloroplast Genomes
4.5. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Azuma, H.; Garcia-Franco, J.G.; Rico-Gray, V.; Thien, L.B. Molecular phylogeny of the Magnoliaceae: The biogeography of tropical and temperate disjunctions. Am. J. Bot. 2001, 88, 2275–2285. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, B.; Nie, Z.; Chen, H.; Chen, F.; Figlar, R.B.; Wen, J. Major clades and a revised classification of Magnolia and Magnoliaceae based on whole plastid genome sequences via genome skimming. J. Syst. Evol. 2020, 58, 673–695. [Google Scholar] [CrossRef]
- Dandy, J.E. The genera of Magnolieae. Bull. Misc. Inf. (R. Bot. Gard. Kew) 1927, 7, 257–264. [Google Scholar] [CrossRef]
- Law, Y.W. A preliminary study on the taxonomy of the family Magnoliaceae. J. Syst. Evol. 1984, 22, 89–109. [Google Scholar]
- Nooteboom, H.P. Notes on Magnoliaceae with a revision of Pachylarnax and Elmerrillia and the Malesian species of Manglietia and Michelia. Blumea Biodivers. Evol. Biogeogr. Plants 1985, 31, 65–121. [Google Scholar]
- Xia, N.H. A new classification system of family Magnoliaceae. In Proceedings of the Second International Symposium on the Family Magnoliaceae, Guangzhou, China, 5–8 May 2009; Huazhong University of Science & Technology Press: Wuhan, China, 2012; pp. 12–38. [Google Scholar]
- Sima, Y.K.; Lu, S.G. A new system for the family Magnoliaceae. In Proceedings of the Second International Symposium on the Family Magnoliaceae, Guangzhou, China, 5–8 May 2009; Huazhong University of Science & Technology Press: Wuhan, China, 2012; pp. 55–71. [Google Scholar]
- Figlar, R.B.; Nooteboom, H.P. Notes on Magnoliaceae IV. Blumea Biodivers. Evol. Biogeogr. Plants 2004, 49, 87–100. [Google Scholar] [CrossRef]
- Li, J. A cladistic analysis of Magnoliaceae. Acta Bot. Yunnanica 1997, 19, 342–356. [Google Scholar]
- Sima, Y.; Yu, H.; Ma, H.; Hao, J.; Chen, S.; Li, S.; Fu, Y. New combinations in Magnoliaceae. J. West China For. Sci. 2020, 49, 29–40. (In Chinese) [Google Scholar]
- Yang, C.; Deng, J.; Zhou, H.; Li, H.; Zheng, Z. The new records of Magnoliaceae in Guizhou province. Guizhou For. Sci. Technol. 2016, 44, 21–23. (In Chinese) [Google Scholar]
- Yang, C.; Deng, J.; Zhou, H. Study on the resources and ornamental characteristics of the native Magnoliaceaes in Guizhou province. Guizhou For. Sci. Technol. 2017, 45, 19–23. (In Chinese) [Google Scholar]
- Yuan, C.; Yang, Y.; Dai, X.; Li, H.; Yang, C.; Wu, Y.; Wang, J. Magnoliaceae plants and its distribution pattern in Guizhou province. J. West China For. Sci. 2017, 46, 68–75. (In Chinese) [Google Scholar]
- Wang, Y.; Wang, D. The complete chloroplast genome and phylogenetic analysis of Manglietia ventii (Magnoliaceae). Mitochondrial DNA Part B-Resour. 2022, 7, 196–198. [Google Scholar] [CrossRef]
- Figlar, R.B.; Nooteboom, H.P. Magnolia Classification. Available online: https://www.magnoliasociety.org/Classification (accessed on 20 November 2023).
- Xia, N.; Liu, Y.; Nooteboom, H.P. Magnoliaceae. In Flora of China; Wu, Z.Y., Raven, P.H., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MI, USA, 2008; Volume 7, pp. 48–91. [Google Scholar]
- Shui, Y.; Sima, Y.; Wen, J.; Chen, W. Vouchered Flora of Southeast Yunnan; Yunnan Technology and Sciences Housing: Kunming, China, 2009; Volume 1. [Google Scholar]
- Li, Y.; Li, D. Conservation value and exploitation foreground of the Magnoliaceae plants in Yunnan. J. Beijing For. Univ. 1999, 21, 29–35. [Google Scholar]
- Sima, Y.; Ma, H.; Xu, T.; Yang, J.; Zhang, D. The chemical composition of volatile oils from leaves of three species of Lirianthe Spach in Magnoliaceae and its systematic signifcance. J. West China For. Sci. 2018, 47, 7–14+29. (In Chinese) [Google Scholar]
- Azuma, H.; Thien, L.; Kawano, S. Molecular phylogeny of Magnolia (Magnoliaceae) inferred from cpDNA sequences and evolutionary divergence of the floral scents. J. Plant Res. 1999, 112, 291–306. [Google Scholar] [CrossRef]
- Kim, S.; Suh, Y. Phylogeny of Magnoliaceae based on ten chloroplast DNA regions. J. Plant Biol. 2013, 56, 290–305. [Google Scholar] [CrossRef]
- Wollaeger, H.M. Genetic Variability in Magnolia acuminata (L.) Populations in the Eastern United States. Bachelor’s Thesis, Wittenberg University, LINK Electronic Theses and Dissertations Center, Springfield, OH, USA, 2011. [Google Scholar]
- Chen, S.; Wu, T.; Ma, H.; Fu, Y.; Zhu, Y.; Hao, J.; Jiang, H.; Sima, Y. The complete chloroplast genome sequence of Lirianthe Hodgsonii, a tree species of Magnoliaceae as least concern. Mitochondrial DNA Part B Resour. 2020, 5, 3064–3066. [Google Scholar] [CrossRef]
- Redwan, R.M.; Saidin, A.; Kumar, S.V. Complete chloroplast genome sequence of Md-2 pineapple and its comparative analysis among nine other plants from the Subclass commelinidae. BMC Plant Biol. 2015, 15, 196. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Y.; Henry, R.J.; Rossetto, M.; Wang, Y.; Chen, S. Plant DNA barcoding: From gene to genome. Biol. Rev. Camb. Philos. Soc. 2015, 90, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Li, H.T.; Luo, Y.; Gan, L.; Ma, P.F.; Gao, L.M.; Yang, J.B.; Cai, J.; Gitzendanner, M.A.; Fritsch, P.W.; Zhang, T.; et al. Plastid phylogenomic insights into relationships of all flowering plant families. BMC Biol. 2021, 19, 232. [Google Scholar] [CrossRef]
- Nguyen, V.B.; Linh Giang, V.N.; Waminal, N.E.; Park, H.S.; Kim, N.H.; Jang, W.; Lee, J.; Yang, T.J. Comprehensive comparative analysis of chloroplast genomes from seven Panax species and development of an authentication system based on species-unique single nucleotide polymorphism markers. J. Ginseng Res. 2020, 44, 135–144. [Google Scholar] [CrossRef]
- Kim, K.; Lee, S.C.; Lee, J.; Lee, H.O.; Joh, H.J.; Kim, N.H.; Park, H.S.; Yang, T.J. Comprehensive survey of genetic diversity in chloroplast genomes and 45s nrDNAs within Panax ginseng species. PLoS ONE 2015, 10, e0117159. [Google Scholar] [CrossRef]
- Lang, C.; Weber, N.; Moeller, M.; Schramm, L.; Schelm, S.; Kohlbacher, O.; Fischer, M. Genetic authentication: Differentiation of hazelnut cultivars using polymorphic sites of the chloroplast genome. Food Control. 2021, 130, 108344. [Google Scholar] [CrossRef]
- Sima, Y.; Wu, T.; Fu, Y.; Hao, J.; Chen, S. Complete chloroplast genome sequence of Lirianthe coco (Loureiro) N. H. Xia & C. Y. Wu (Magnoliaceae), a popular ornamental species. Mitochondrial DNA Part B Resour. 2020, 5, 2410–2412. [Google Scholar] [PubMed]
- Cai, Z.; Penaflor, C.; Kuehl, J.V.; Leebens-Mack, J.; Carlson, J.E.; dePamphilis, C.W.; Boore, J.L.; Jansen, R.K. Complete plastid genome sequences of Drimys, Liriodendron, and Piper: Implications for the phylogenetic relationships of magnoliids. BMC Evol. Biol. 2006, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Li, Z.; Wu, N.; Yang, J.; Yuan, L.; Zhao, T.; Sima, Y.; Xu, T. Comparitive analysis of the chloroplast genomes of three Houpoea plants. Genes 2023, 14, 1262. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Aishan, S.; Zhu, J.; Qin, Y.; Liu, J.; Liu, H.; Tie, J.; Wang, J.; Qin, R. Chloroplast genomes and phylogenetic analysis of three Carthamus (Asteraceae) species. Int. J. Mol. Sci. 2023, 24, 15634. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.Y.; Yang, J.X.; Li, H.K.; Zhao, H.S. Chloroplast genomes of two species of Cypripedium: Expanded genome size and proliferation of at-biased repeat sequences. Front Plant Sci. 2021, 12, 609729. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Lee, S.Y.; Ni, J.; Zhang, X.; Hu, X.; Zou, P.; Wang, W.; Liu, G. Comparative analyses of chloroplast genome provide effective molecular markers for species and cultivar identification in Bougainvillea. Int. J. Mol. Sci. 2023, 24, 15138. [Google Scholar] [CrossRef]
- Zhang, H.; Li, C.; Miao, H.; Xiong, S. Insights from the complete chloroplast genome into the evolution of Sesamum indicum L. PLoS ONE. 2013, 11, e80508. [Google Scholar] [CrossRef]
- China Plant BOL Group; Li, D.Z.; Gao, L.M.; Li, H.T.; Wang, H.; Ge, X.J.; Liu, J.Q.; Chen, Z.D.; Zhou, S.L.; Chen, S.L.; et al. Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc. Natl. Acad. Sci. USA 2011, 108, 19641–19646. [Google Scholar] [PubMed]
- Deng, Y.F. (2679) Proposal to Conserve the Name Talauma Fistulosa (Lirianthe Fistulosa, Magnolia Fistulosa) (Magnoliaceae) with a conserved type. Taxon 2019, 68, 405–406. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.G.; Roper, J.M.; Duffy, A.M. The evolution of chloroplast genome structure in ferns. Genome 2010, 53, 731–738. [Google Scholar] [CrossRef]
- Liang, A.; Luo, W.; Li, Z.; Sima, Y.; Xu, T. The complete chloroplast genome sequence of Magnolia delavayi (Magnoliaceae), a rare ornamental and medical tree endemic to China. Mitochondrial DNA Part B Resour. 2020, 5, 883–884. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. Getorganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. Cpgavas2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. Phylosuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Rozas, R. DnaSP, DNA sequence polymorphism: An interactive program for estimating population genetics parameters from dna sequence data. Comput. Appl. Biosci. 1995, 11, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | L. brevisericea | L. coco | L. confusa | L. delavayi | L. henryi | L. hodgsonii | L. odoratissima | L. phanerophlebia |
|---|---|---|---|---|---|---|---|---|
| GetOrganelle | 93.9 | 475.9 | 483.3 | 547.3 | 374.1 | 328.2 | 547.9 | 236.0 |
| Minimap + samtools | 105.79 | 845.20 | 1305.89 | 368.77 | 1577.05 | 300.42 | 1147.52 | 273.37 |
| Species | L. brevisericea | L. coco | L. confusa | L. delavayi | L. henryi | L. hodgsonii | L. odoratissima | L. phanerophlebia |
|---|---|---|---|---|---|---|---|---|
| Name Abbr. | Lbre | Lcoc | Lcon | Ldel | Lhen | Lhod | Lodo | Lpha |
| Accession No. | OR680846 | MT225530 | MT654129 | MT654132 | OR680847 | MT560391 | MT654135 | OR680845 |
| Total Length (bp) | 159,811 | 159,828 | 159,833 | 159,714 | 159,760 | 159,693 | 159,819 | 159,548 |
| LSC (bp) | 87,933 | 87,958 | 87,965 | 87,877 | 87,891 | 87,846 | 87,961 | 87,671 |
| SSC (bp) | 18,758 | 18,760 | 18,752 | 18,761 | 18,757 | 18,745 | 18,772 | 18,761 |
| IR (bp) | 26,560 | 26,555 | 26,558 | 26,538 | 26,556 | 26,551 | 26,543 | 26,558 |
| Total Genes | 130 | |||||||
| CDS | 85 | |||||||
| tRNA | 37 | |||||||
| rRNA | 8 | |||||||
| Total GC% | 39.28 | 39.28 | 39.27 | 39.29 | 39.28 | 39.28 | 39.27 | 39.29 |
| LSC (GC%) | 37.98 | 37.97 | 37.97 | 37.97 | 37.97 | 37.97 | 37.97 | 37.98 |
| SSC (GC%) | 34.36 | 34.37 | 34.39 | 34.46 | 34.38 | 34.37 | 34.32 | 34.41 |
| IR (GC%) | 43.17 | 43.18 | 43.16 | 43.18 | 43.18 | 43.18 | 43.18 | 43.17 |
| A (%) | 47,908 (29.98) | 47,915 (29.98) | 47,913 (29.98) | 47,862 (29.97) | 47,868 (29.96) | 47,851 (29.96) | 47,908 (29.98) | 47,799 (29.96) |
| C (%) | 31,980 (20.01) | 31,983 (20.01) | 31,979 (20.01) | 31,972 (20.02) | 31,980 (20.02) | 31,958 (20.01) | 31,977 (20.01) | 31,940 (20.02) |
| G (%) | 30,790 (19.27) | 30,795 (19.27) | 30,790 (19.26) | 30,779 (19.27) | 30,777 (19.26) | 30,774 (19.27) | 30,782 (19.26) | 30,741 (19.27) |
| T (%) | 49,133 (30.74) | 49,135 (30.74) | 49,151 (30.75) | 49,101 (30.74) | 49,135 (30.76) | 49,110 (30.75) | 49,152 (30.75) | 49,068 (30.75) |
| Species | Lbre | Lcoc | Lcon | Ldel | Lhen | Lhod | Lodo | Lpha | |
|---|---|---|---|---|---|---|---|---|---|
| SSR | A | 17 | 13 | 13 | 17 | 15 | 16 | 16 | 17 |
| C | 2 | 2 | 3 | 3 | 3 | 3 | 2 | 3 | |
| G | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | |
| T | 21 | 23 | 22 | 25 | 24 | 24 | 21 | 24 | |
| TA | 1 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | |
| TC | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | |
| Total No. | 44 | 42 | 43 | 49 | 46 | 48 | 42 | 48 | |
| Tandem Repeats | 16 | 18 | 17 | 17 | 16 | 16 | 17 | 17 | |
| Dispersed Repeats | Complement | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| Forward | 16 | 16 | 15 | 18 | 15 | 16 | 17 | 17 | |
| Palindromic | 27 | 25 | 26 | 25 | 26 | 26 | 25 | 25 | |
| Reverse | 7 | 9 | 9 | 7 | 8 | 8 | 8 | 8 | |
| Total No. | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 | |
| Species | Leaves | Flowers | Fruits |
|---|---|---|---|
| L. brevisericea | Abaxially yellowish-gray sericeous; leaf blade midveins adaxially impressed, lateral veins 12–19 pairs; stipular scars reaching apex of petioles. | Erect; 9 tepals, white; gynoecia densely yellowish-gray sericeous. | Mature carpels dehiscent along dorsal sutures |
| L. coco | Glabrous, leaf blade midveins adaxially impressed, lateral veins 8–16 pairs;; stipular scars reaching apex of petioles. | Pendulous; 9 tepals, white; gynoecia glabrous. | Mature carpels dehiscent along dorsal sutures. |
| L. confusa | Abaxially yellowish-white curved trichomes; leaf blade midveins adaxially impressed, lateral veins 10–15 pairs; stipular scars reaching apex of petioles. | Pendulous; 9 tepals, white; gynoecia very densely yellowish-white villose. | Mature carpels dehiscent along dorsal sutures |
| L. delavayi | Abaxially densely interwoven tomentose and white powdery but later only with residual trichomes on veins; leaf blade midveins adaxially impressed, lateral veins 11–16 pairs; stipular scars reaching apex of petioles. | Erect; 9 to 12 tepals, white, yellowish-white, pink or red; gynoecia fine yellow villose. | Mature carpels dehiscent along dorsal sutures. |
| L. henryi | Abaxially sparsely appressed pubescent; leaf blade midveins adaxially prominent, lateral veins 14–20 pairs; stipular scars reaching apex of petioles. | Pendulous; 9 tepals, white; gynoecia glabrous. | Mature carpels dehiscent along dorsal sutures |
| L. hodgsonii | Glabrous; leaf blade midveins adaxially prominent, lateral veins 10–20 pairs; stipular scars reaching apex of petioles. | Erect; 9 tepals, white; gynoecia glabrous. | Mature carpels circumscissile |
| L. odoratissima | Abaxially yellowish-white or grayish-brown curved trichomes; leaf blade midvein adaxially impressed, lateral veins 9–14 pairs; stipular scars reaching apex of petioles. | Pendulous; 9 to 10 tepals, white; gynoecia densely grayish-brown pubescent. | Mature carpels dehiscent along dorsal sutures |
| L. phanerophlebia | Glabrous; leaf blade midveins adaxially impressed, lateral veins 11–17 pairs; stipular scars 1/3–1/2 as long as petioles. | Pendulous; 8 to 9 tepals, white; gynoecia glabrous or glaucous. | Mature carpels dehiscent along dorsal sutures |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, T.; Sima, Y.-K.; Chen, S.-Y.; Fu, Y.-P.; Ma, H.-F.; Hao, J.-B.; Zhu, Y.-F. Comparative Analysis of the Chloroplast Genomes of Eight Species of the Genus Lirianthe Spach with Its Generic Delimitation Implications. Int. J. Mol. Sci. 2024, 25, 3506. https://doi.org/10.3390/ijms25063506
Wu T, Sima Y-K, Chen S-Y, Fu Y-P, Ma H-F, Hao J-B, Zhu Y-F. Comparative Analysis of the Chloroplast Genomes of Eight Species of the Genus Lirianthe Spach with Its Generic Delimitation Implications. International Journal of Molecular Sciences. 2024; 25(6):3506. https://doi.org/10.3390/ijms25063506
Chicago/Turabian StyleWu, Tao, Yong-Kang Sima, Shao-Yu Chen, Yu-Pin Fu, Hui-Fen Ma, Jia-Bo Hao, and Yun-Feng Zhu. 2024. "Comparative Analysis of the Chloroplast Genomes of Eight Species of the Genus Lirianthe Spach with Its Generic Delimitation Implications" International Journal of Molecular Sciences 25, no. 6: 3506. https://doi.org/10.3390/ijms25063506
APA StyleWu, T., Sima, Y. -K., Chen, S. -Y., Fu, Y. -P., Ma, H. -F., Hao, J. -B., & Zhu, Y. -F. (2024). Comparative Analysis of the Chloroplast Genomes of Eight Species of the Genus Lirianthe Spach with Its Generic Delimitation Implications. International Journal of Molecular Sciences, 25(6), 3506. https://doi.org/10.3390/ijms25063506