Intraphagosomal Free Ca2+ Changes during Phagocytosis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
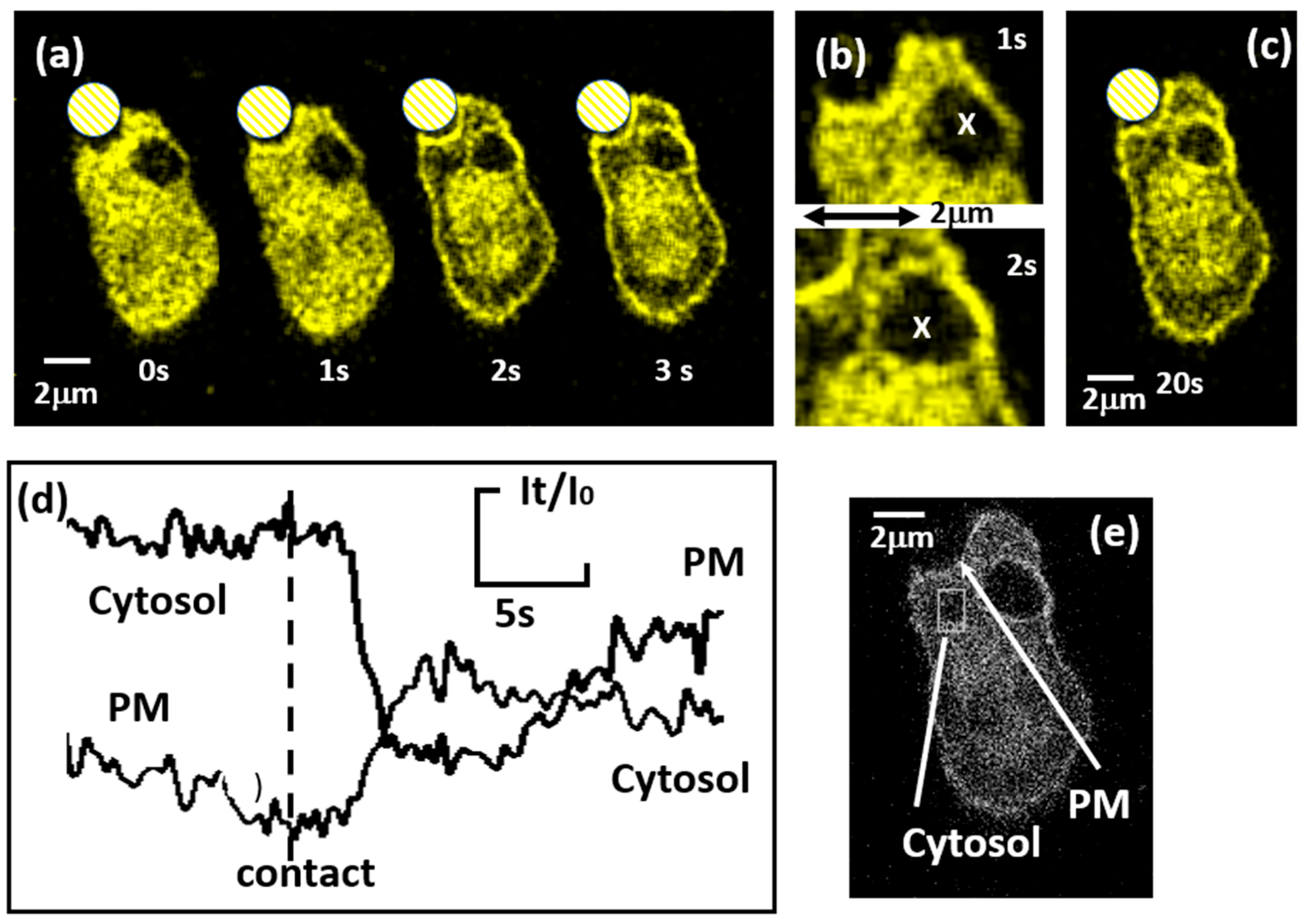{kind=link}
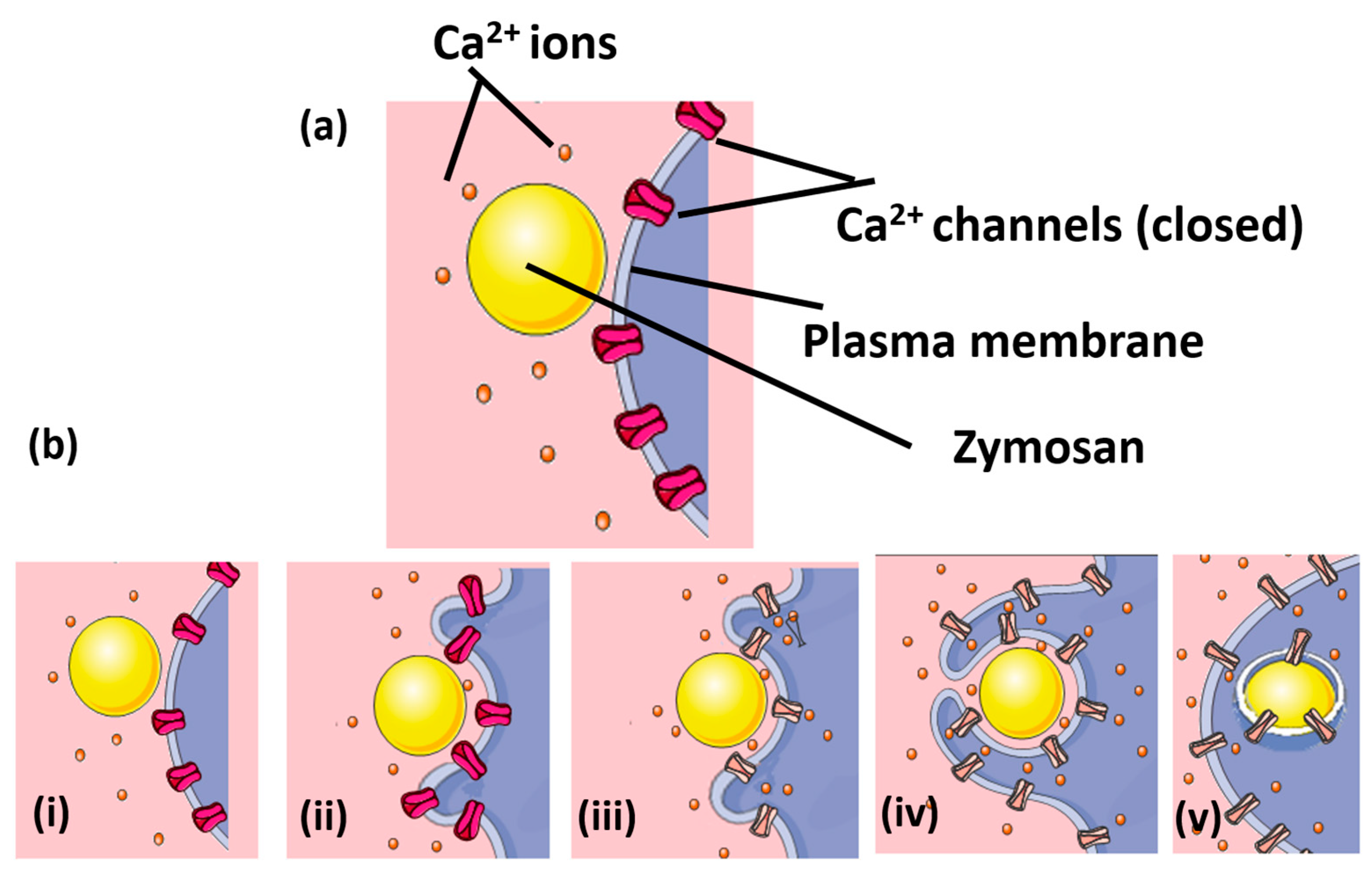{kind=link}
Abstract
:1. Introduction
2. Results
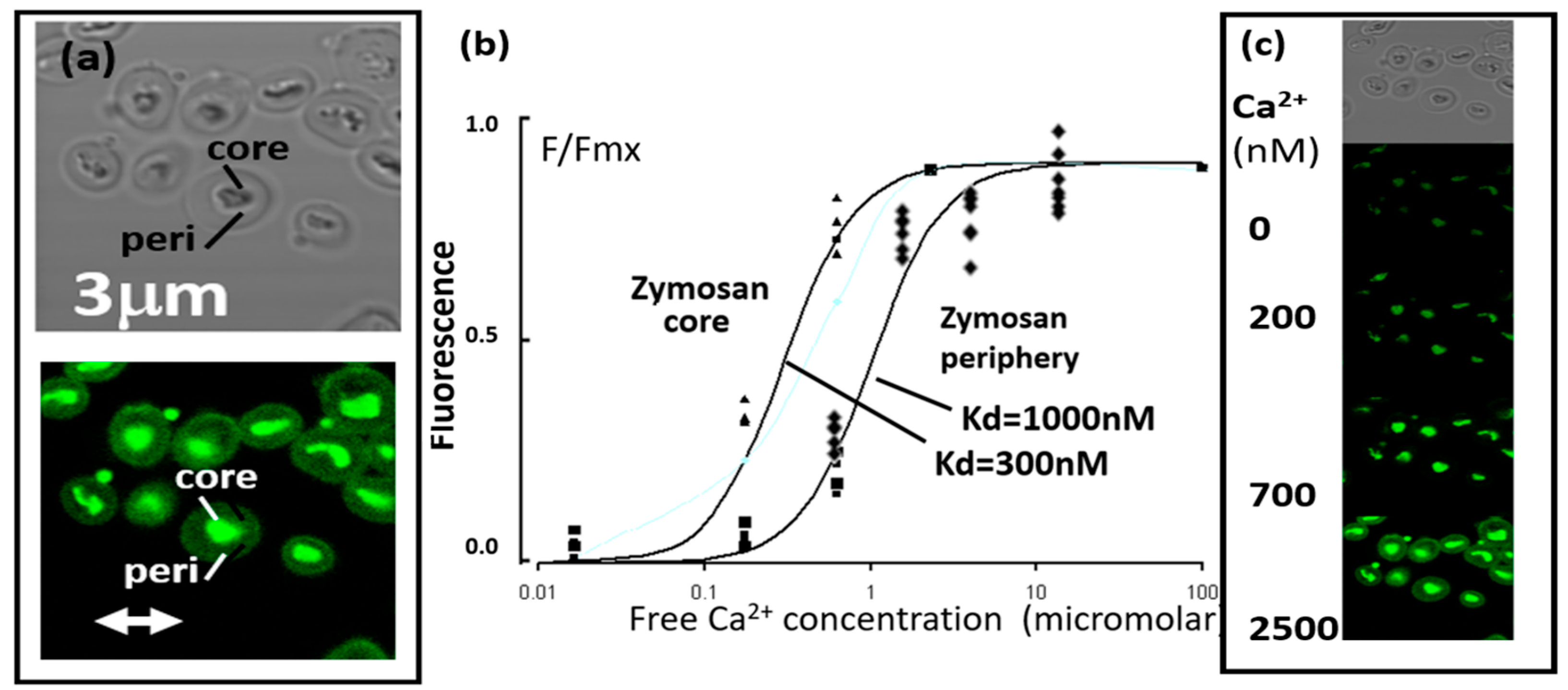
2.1. Fluo4-Zymosan Targets Report Ca2+ Concentration
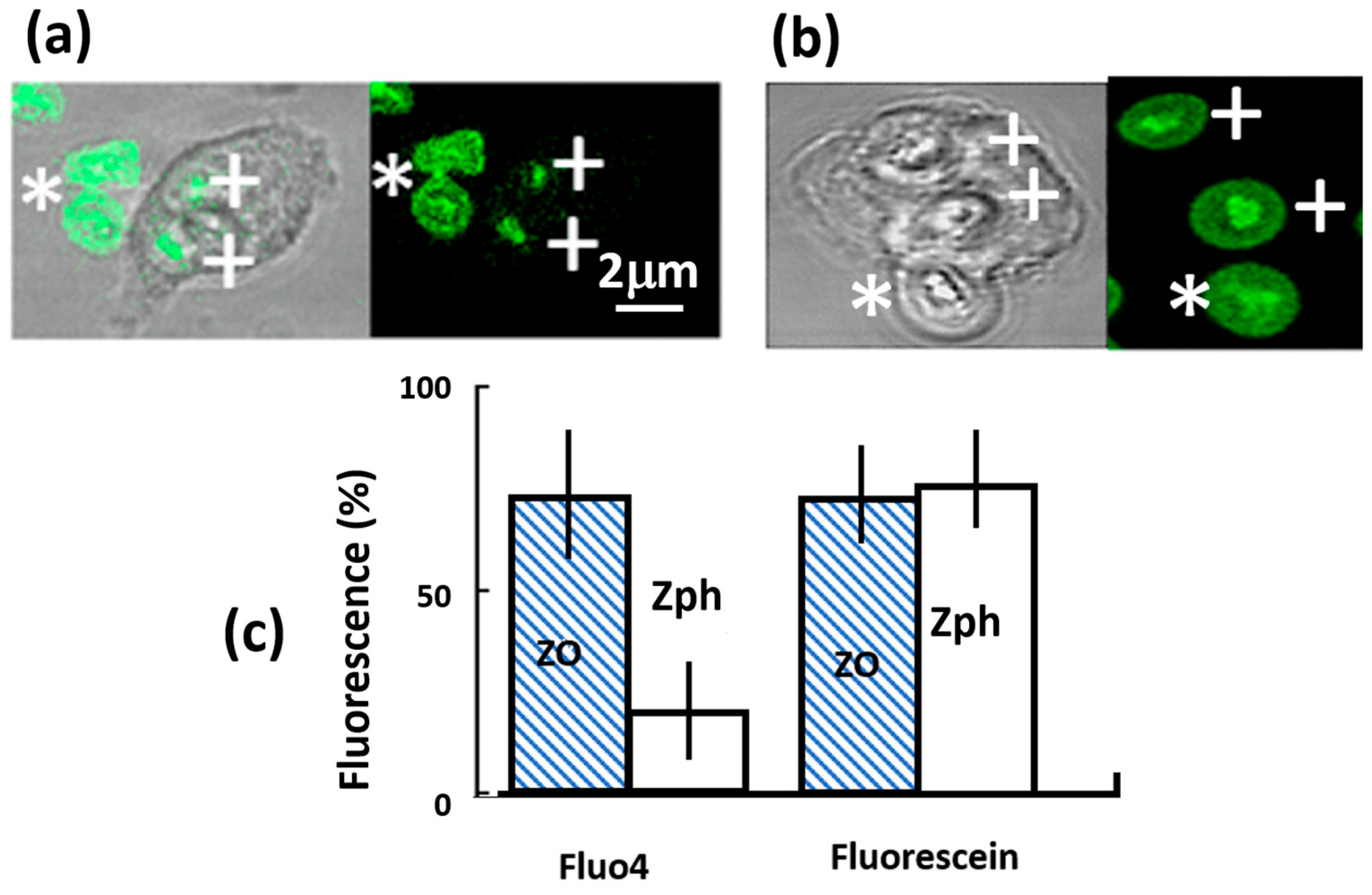
2.2. Phagosomal Fluo4 Signal Is Decreased
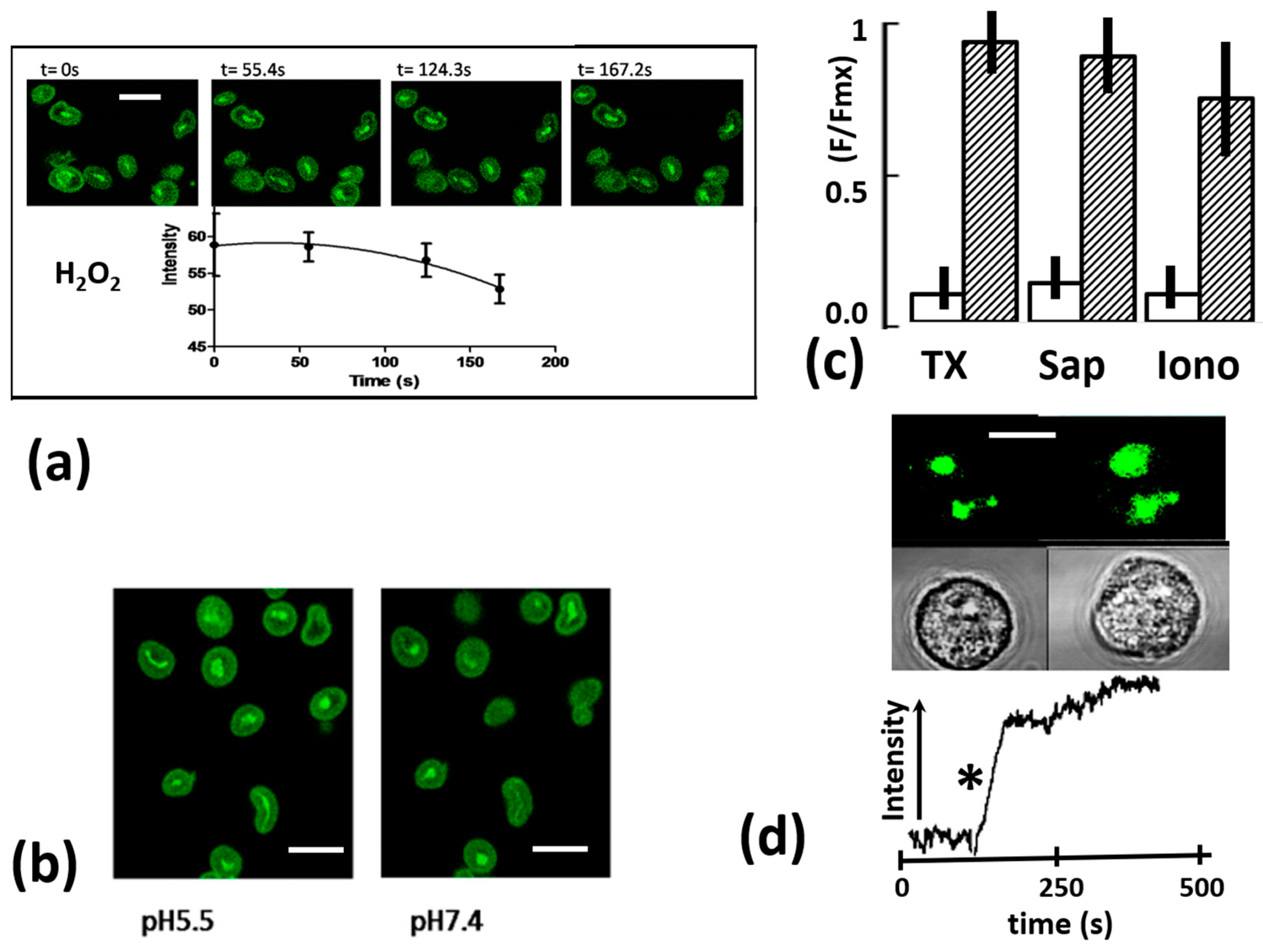
2.3. Phagosomal Fluo4 Signal Decrease Not Due to Oxidants
2.4. Phagosomal Fluo4 Zymosan Remained Responsive to Ca2+ Changes
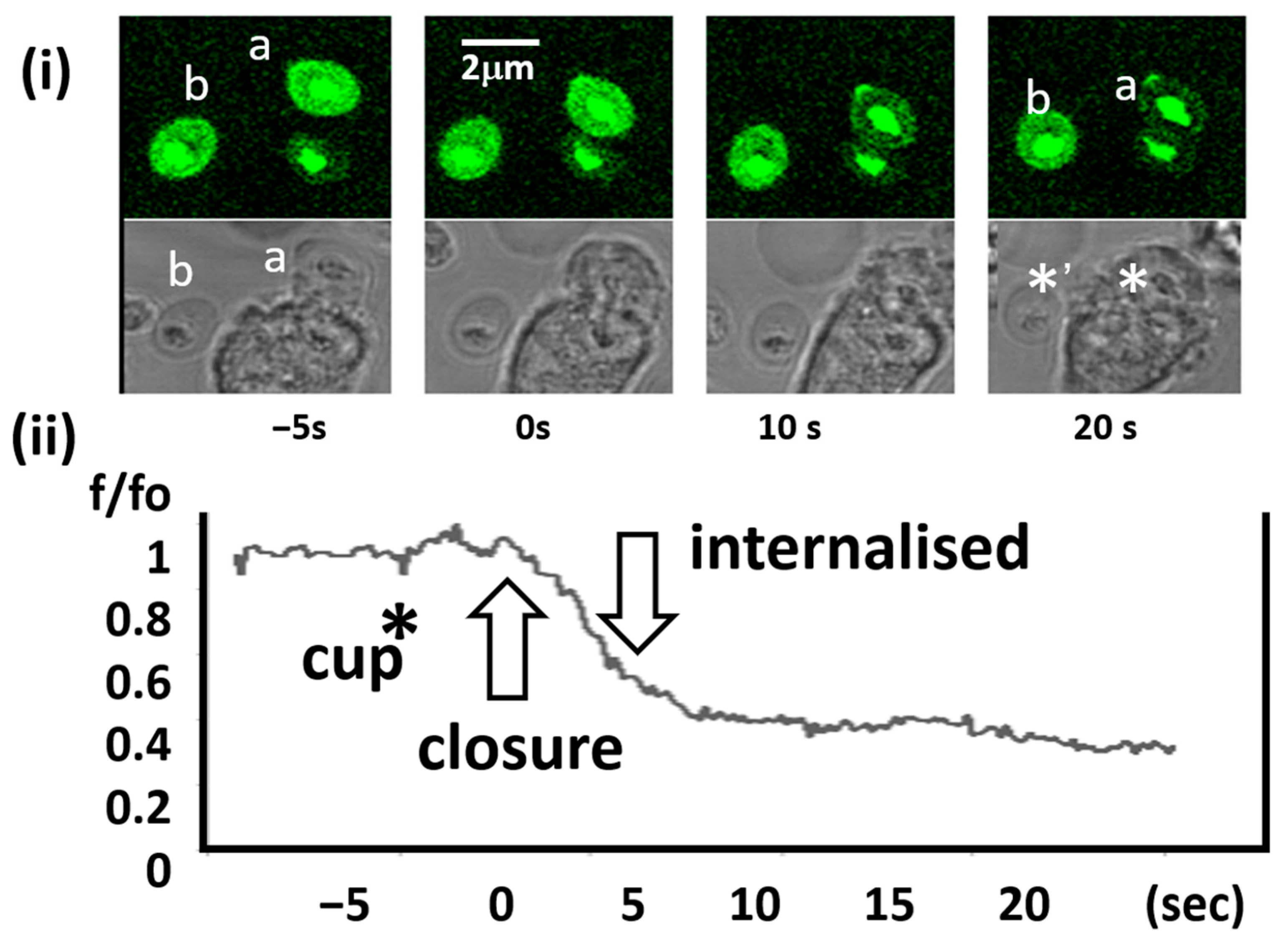
2.5. Kinetics of Intra-Phagosomal Ca2+ Decrease
2.6. Subcellular Location of Ca2+ Leakage Pathways
3. Discussion
4. Materials and Methods
4.1. Cell Preparation
4.2. Properties of the Phagocytic Target
4.3. Imaging and Intra-Phagosomal Ca2+ Monitoring
4.4. Estimation of Intraphagosomal Ca2+ Concentration
4.5. Raw 264.7 Cell Transfection
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dewitt, S.; Hallett, M.B. Cytosolic free Ca2+ changes and calpain activation are required for beta integrin-accelerated phagocytosis by human neutrophils. J. Cell Biol. 2002, 159, 181–189. [Google Scholar] [CrossRef]
- Nunes, P.; Demaurex, N. The role of calcium signaling in phagocytosis. J. Leuk. Biol. 2010, 88, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Francis, E.A.; Heinrich, V. Single-cell investigation of the role of calcium bursts in human immune cells. Biophys. J. 2017, 112, 400A. [Google Scholar] [CrossRef]
- Jaumouillé, V.; Grinstein, S. Molecular Mechanisms of Phagosome Formation. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Uribe-Querol, E.; Rosales, C. Phagocytosis: Our current understanding of a universal biological process. Front. Immunol. 2020, 11, 1066. [Google Scholar] [CrossRef] [PubMed]
- Hallett, M.B.; Davies, E.V.; Campbell, A.K. Oxidase activation in individual neutrophils is dependent on the onset and magnitude of the Ca2+ signal. Cell Calcium 1990, 11, 655–663. [Google Scholar] [CrossRef]
- Bei, L.; Hu, T.; Qian, Z.M.; Shen, X. Extracellular Ca2+ regulates the respiratory burst of human neutrophils. Biochim. Biophys. Acta—Mol. Cell Res. 1998, 1404, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Westman, J.; Grinstein, S.; Maxson, M.E. Revisiting the role of calcium in phagosome formation and maturation. J. Leuk. Biol. 2019, 106, 837–851. [Google Scholar] [CrossRef]
- Nunes, P.; Cornut, D.; Demaurex, N. STIM1 juxtaposes ER to phagosomes, generating Ca2+ hotspots that boost phagocytosis. Curr. Biol. 2012, 22, 1990–1997. [Google Scholar] [CrossRef]
- Roberts, R.E.; Vervliet, T.; Bultynck, G.; Parys, J.B.; Hallett, M.B. EPIC3, a novel Ca2+ indicator located at the cell cortex and in microridges, detects high Ca2+ subdomains during Ca2+ influx and phagocytosis. Cell Calcium 2020, 92, 102291. [Google Scholar] [CrossRef]
- Zhang, H.; Clemens, R.A.; Lowell, C.A. STIM1 calcium sensor is required for activation of the phagocyte oxidase during inflammation and host defense. Blood 2014, 123, 2238–2249. [Google Scholar] [CrossRef]
- Guido, D.; Demaurex, N.; Nunes, P. Junctate boosts phagocytosis by recruiting endoplasmic reticulum Ca2+ stores near phagosomes. J. Cell Sci. 2015, 128, 4074–4082. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist-Gustafsson, H.; Gustafsson, M.; Dahlgren, C. Dynamic Ca2+ changes in neutrophil phagosomes. A source for intracellular Ca2+ during phagolysosome formation? Cell Calcium 2000, 27, 353–362. [Google Scholar] [CrossRef]
- Becker, P.L.; Fay, F.S. Photobleaching of fura-2 and its effect on determination of calcium concentrations. Am. J. Physiol. 1987, 253, C613–C618. [Google Scholar] [CrossRef]
- Soto, E.R.; Ostroff, G.R. Characterization of multilayered nanoparticles encapsulated in yeast cell wall particles for DNA delivery. Bioconjug. Chem. 2008, 19, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Gee, K.R.; Brown, K.A.; Chen, W.N.; Bishop-Stewart, J.; Gray, D.; Johnson, I. Chemical and physiological characterization of fluo-4 Ca2+-indicator dyes. Cell Calcium 2000, 27, 97–106. [Google Scholar] [CrossRef]
- Dewitt, S.; Darley, R.L.; Hallett, M.B. Translocation or just location? Pseudopodia affect fluorescent signals. J. Cell Biol. 2009, 184, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.A. Light-scattering from biological cells-dependence of backscatter radiation on membrane thickness and refractive index. Appl. Opt. 1979, 18, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Mie, G. Articles on the optical characteristics of turbid tubes, especially colloidal metal solutions. Ann. Physik 1908, 25, 377–445. [Google Scholar] [CrossRef]
- Mantegazza, A.R.; Savina, A.; Vermeulen, M.; Pérez, L.; Geffner, J.; Hermine, O.; Rosenzweig, S.D.; Faure, F.; Amigorena, S. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood 2008, 112, 4712–4722. [Google Scholar] [CrossRef]
- Segal, A.W. How neutrophils kill microbes. Ann. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Dewitt, S.; Laffafian, I.; Hallett, M.B. Phagosomal oxidative activity during β2 integrin (CR3)-mediated phagocytosis by neutrophils is triggered by a non-restricted Ca2+ signal: Ca2+ controls time not space. J. Cell Sci. 2003, 116, 2857–2865. [Google Scholar] [CrossRef] [PubMed]
- Jacob, M.C.; Favre, M.; Bensa, J.C. Membrane cell permeabilization with Saponin and multiparametric flow cytometry. Cytometry 1991, 12, 550–558. [Google Scholar] [CrossRef]
- Hallett, M.B.; Al-Jumaa, M.; Dewitt, S. Optical methods for the measurement and manipulation of cytosolic calcium signals in neutrophils. Methods Mol. Biol. 2014, 1124, 107–120. [Google Scholar] [PubMed]
- Francis, E.A.; Heinrich, V. Mechanistic understanding of single-cell behavior is essential for transformative advances in biomedicine. Yale J. Biol. Med. 2018, 91, 279–289. [Google Scholar]
- Anke, D.; Kiya, T.; Gong, H.; Gao, X.; Malik, A.B. Role of the phagosomal redox-sensitive TRP channel TRPM2 in regulating bactericidal activity of macrophages. J. Cell Sci. 2017, 130, 735–744. [Google Scholar] [CrossRef]
- Starkus, J.G.; Fleig, A.; Reinhold, P. The calcium-permeable non-selective cation channel TRPM2 is modulated by cellular. J. Physiol. 2010, 588, 1227–1240. [Google Scholar] [CrossRef]
- Oancea, E.; Meyer, T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell 1998, 95, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Teruel, M.N.; Meyer, T. Parallel single-cell monitoring of receptor-triggered membrane translocation of a calcium-sensing protein module. Science 2002, 295, 1910–1912. [Google Scholar] [CrossRef]
- Kilpatrick, B.S.; Eden, E.R.; Schapira, A.H.; Futter, C.F.; Patel, S. Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J. Cell Sci. 2013, 126, 60–66. [Google Scholar] [CrossRef]
- Roberts, R.E.; Martin, M.; Marion, S.; Elumalai, G.L.; Lewis, K.; Hallett, M.B. Ca2+-activated cleavage of ezrin visualised dynamically in living myeloid cells during cell surface area expansion. J. Cell Sci. 2020, 133, jcs236968. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, D.W.; Sullivan, J.A.; Mandell, G.L. Intracellular free calcium localization in neutrophils during phagocytosis. Science 1985, 230, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.R.; Dewitt, S.; Laffafian, I.; Hallett, M.B. Phagocytosis by inflammatory phagocytes: Experimental strategies for stimulation and quantification. Methods Mol. Biol. 2003, 225, 35–46. [Google Scholar] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dewitt, S.; Green, J.; Laffafian, I.; Lewis, K.J.; Hallett, M.B. Intraphagosomal Free Ca2+ Changes during Phagocytosis. Int. J. Mol. Sci. 2024, 25, 4254. https://doi.org/10.3390/ijms25084254
Dewitt S, Green J, Laffafian I, Lewis KJ, Hallett MB. Intraphagosomal Free Ca2+ Changes during Phagocytosis. International Journal of Molecular Sciences. 2024; 25(8):4254. https://doi.org/10.3390/ijms25084254
Chicago/Turabian StyleDewitt, Sharon, Joanna Green, Iraj Laffafian, Kimberly J. Lewis, and Maurice B. Hallett. 2024. "Intraphagosomal Free Ca2+ Changes during Phagocytosis" International Journal of Molecular Sciences 25, no. 8: 4254. https://doi.org/10.3390/ijms25084254
APA StyleDewitt, S., Green, J., Laffafian, I., Lewis, K. J., & Hallett, M. B. (2024). Intraphagosomal Free Ca2+ Changes during Phagocytosis. International Journal of Molecular Sciences, 25(8), 4254. https://doi.org/10.3390/ijms25084254