
Design of Dyes Based on the Quinoline or Quinoxaline Skeleton towards Visible Light Photoinitiators


Abstract
:1. Introduction
2. Results and Discussion
2.1. Designing the Structure of the Dyes
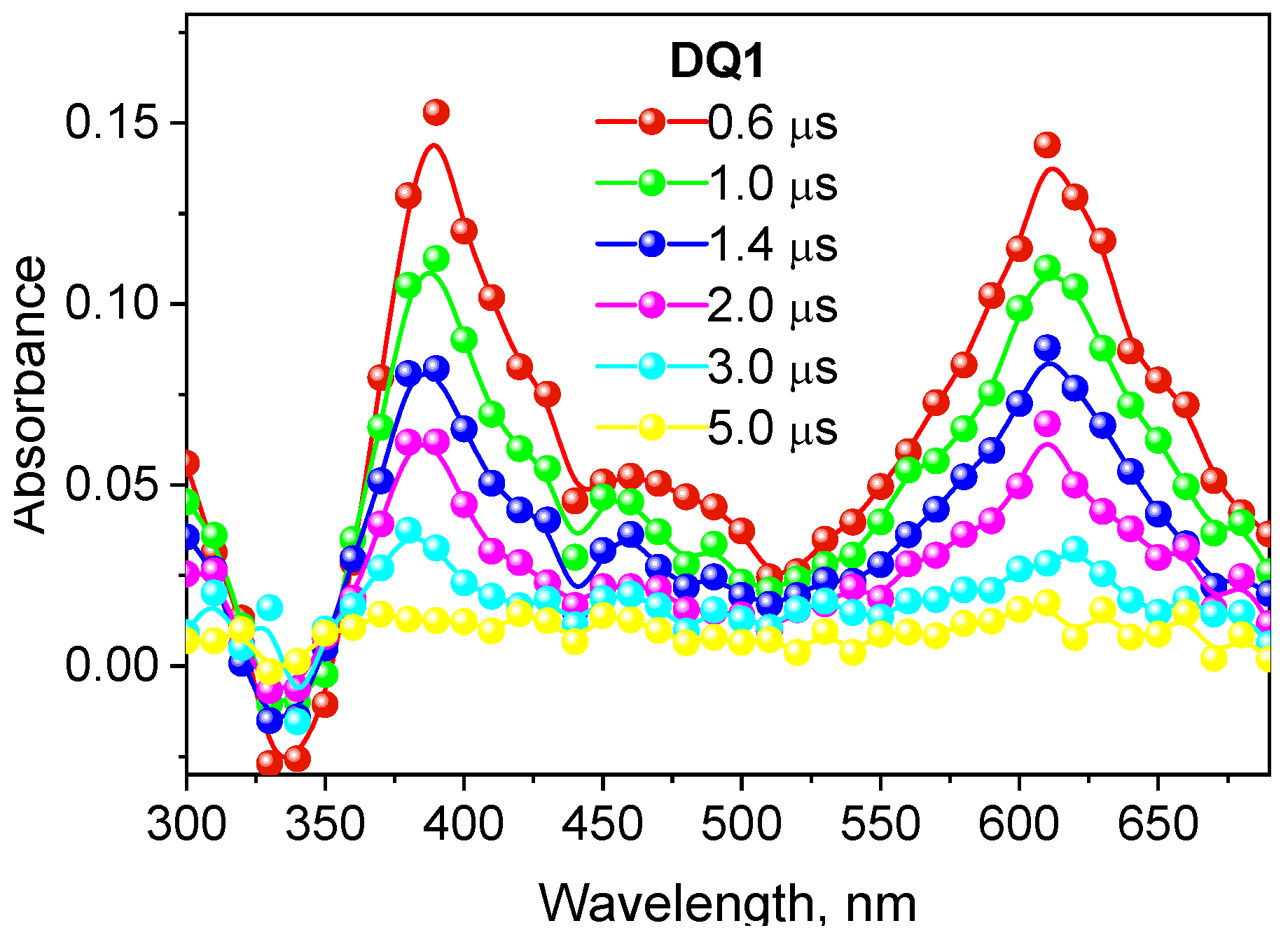
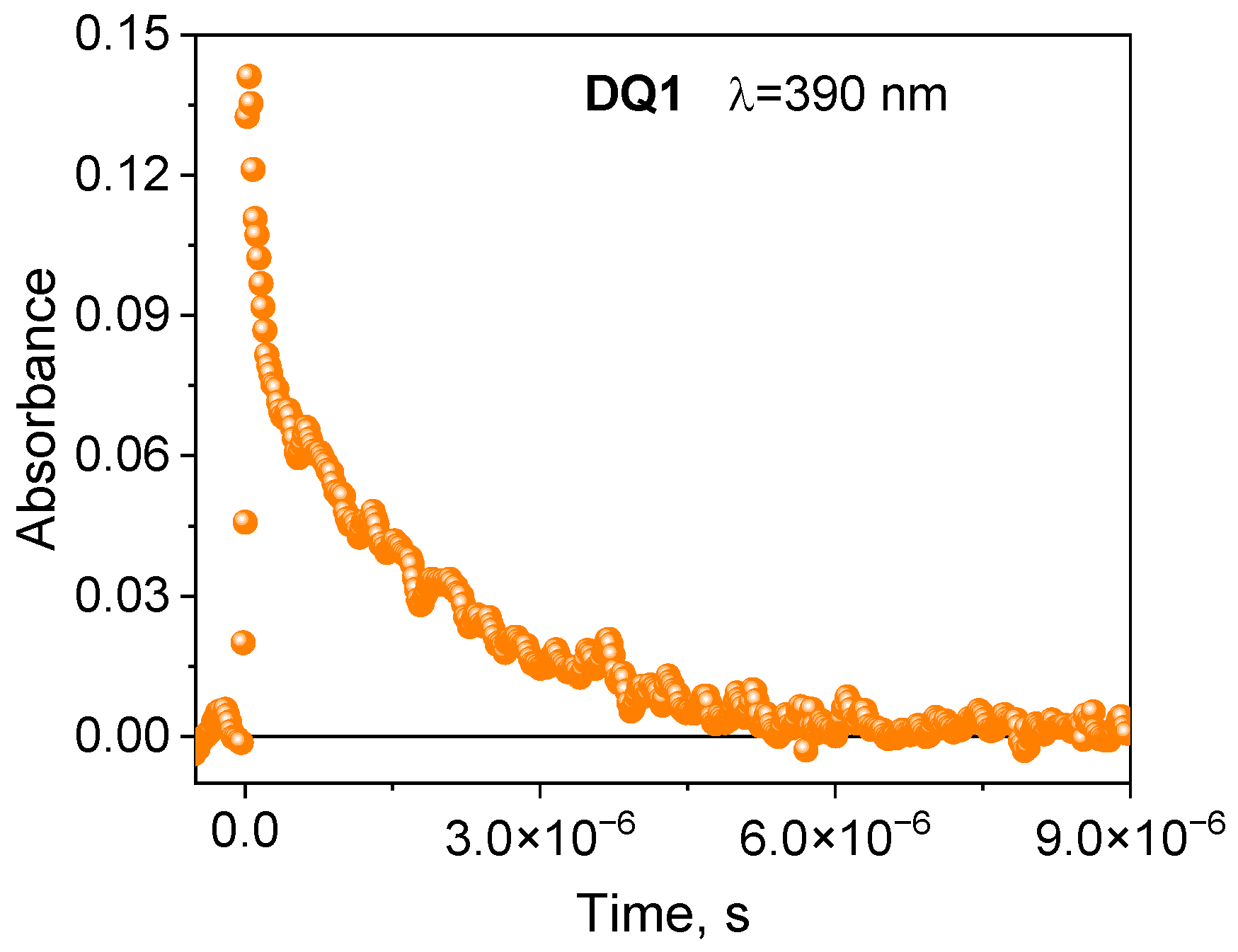
2.2. Spectroscopic Properties
2.3. Photopolymerization
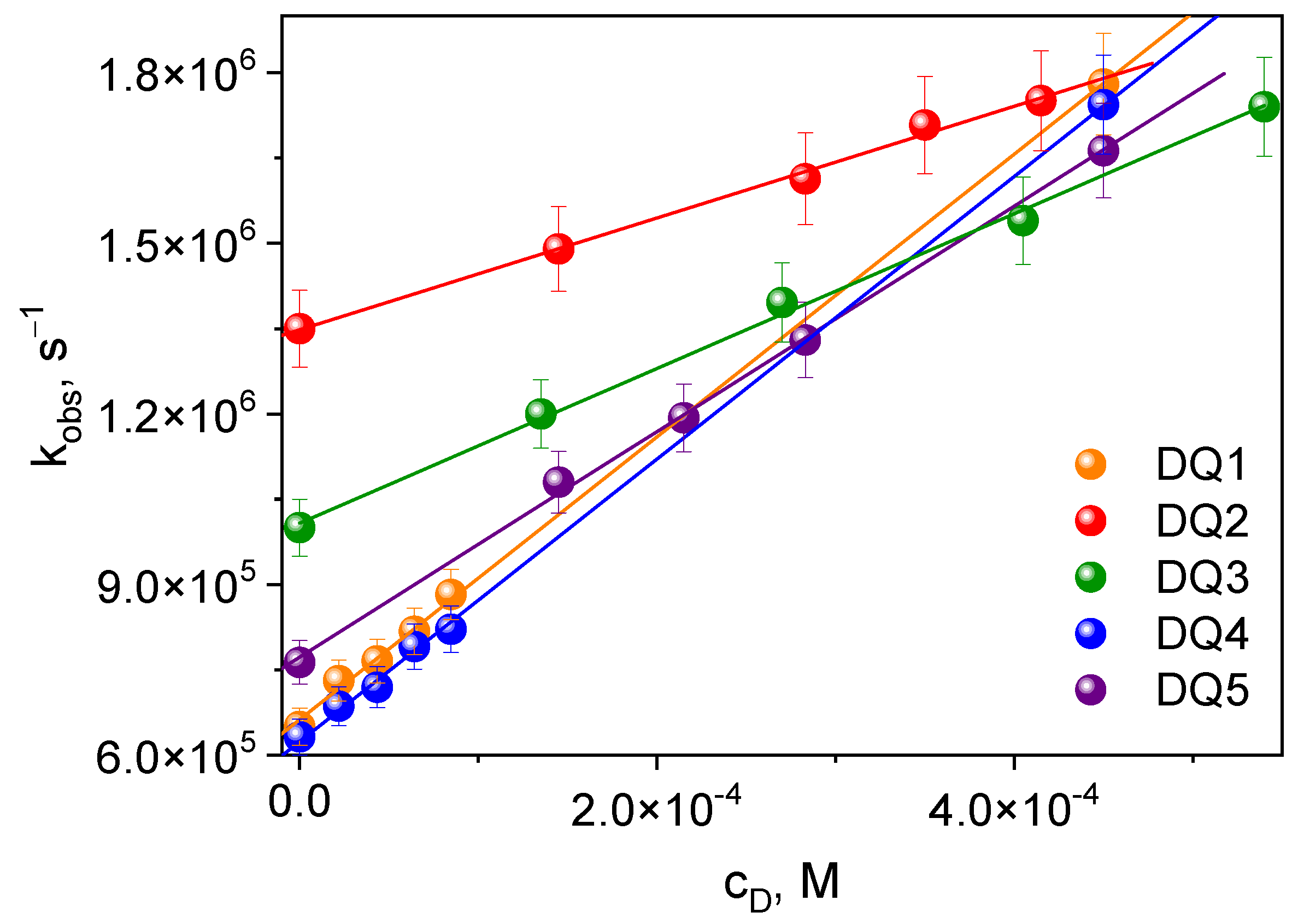
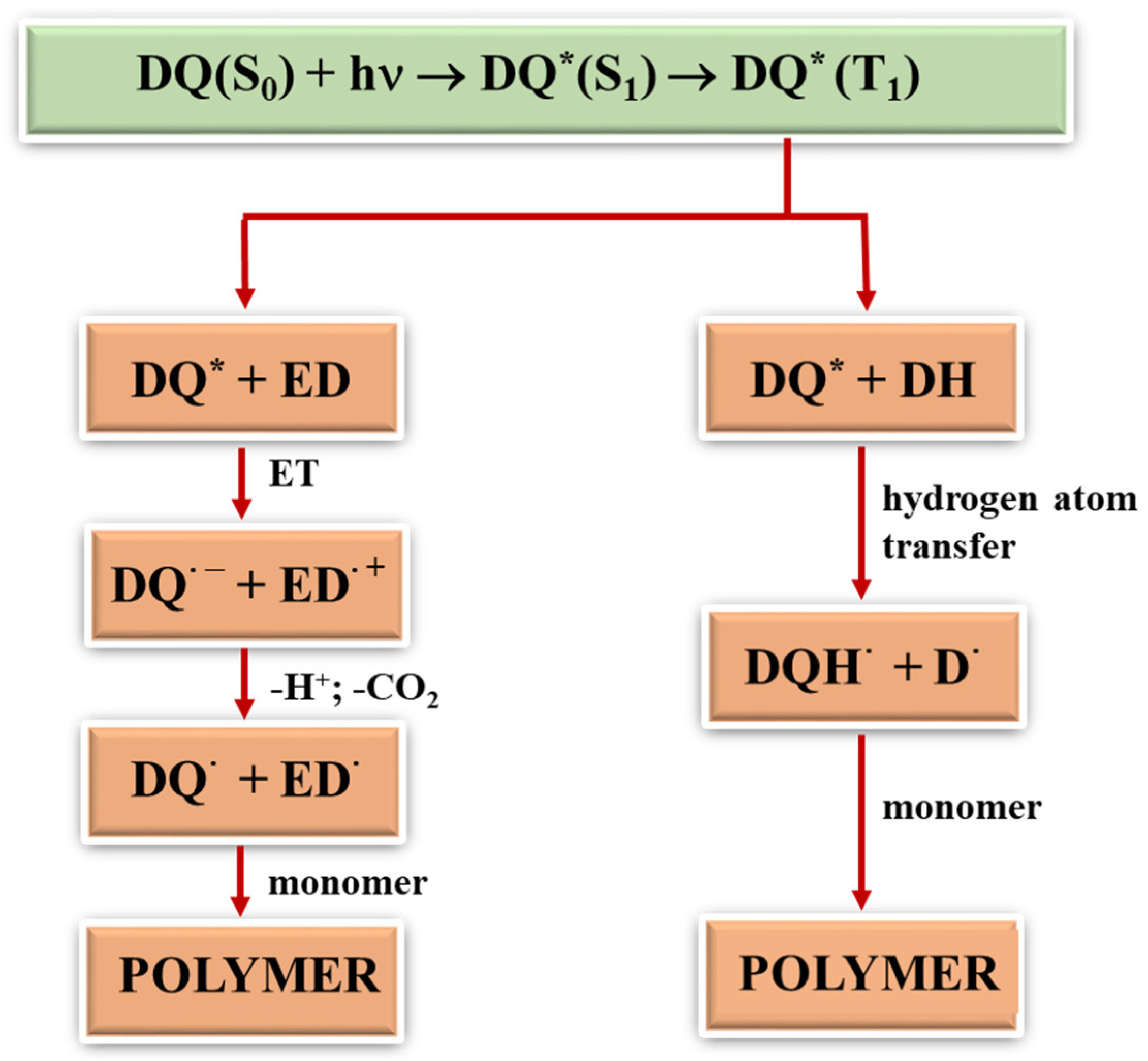
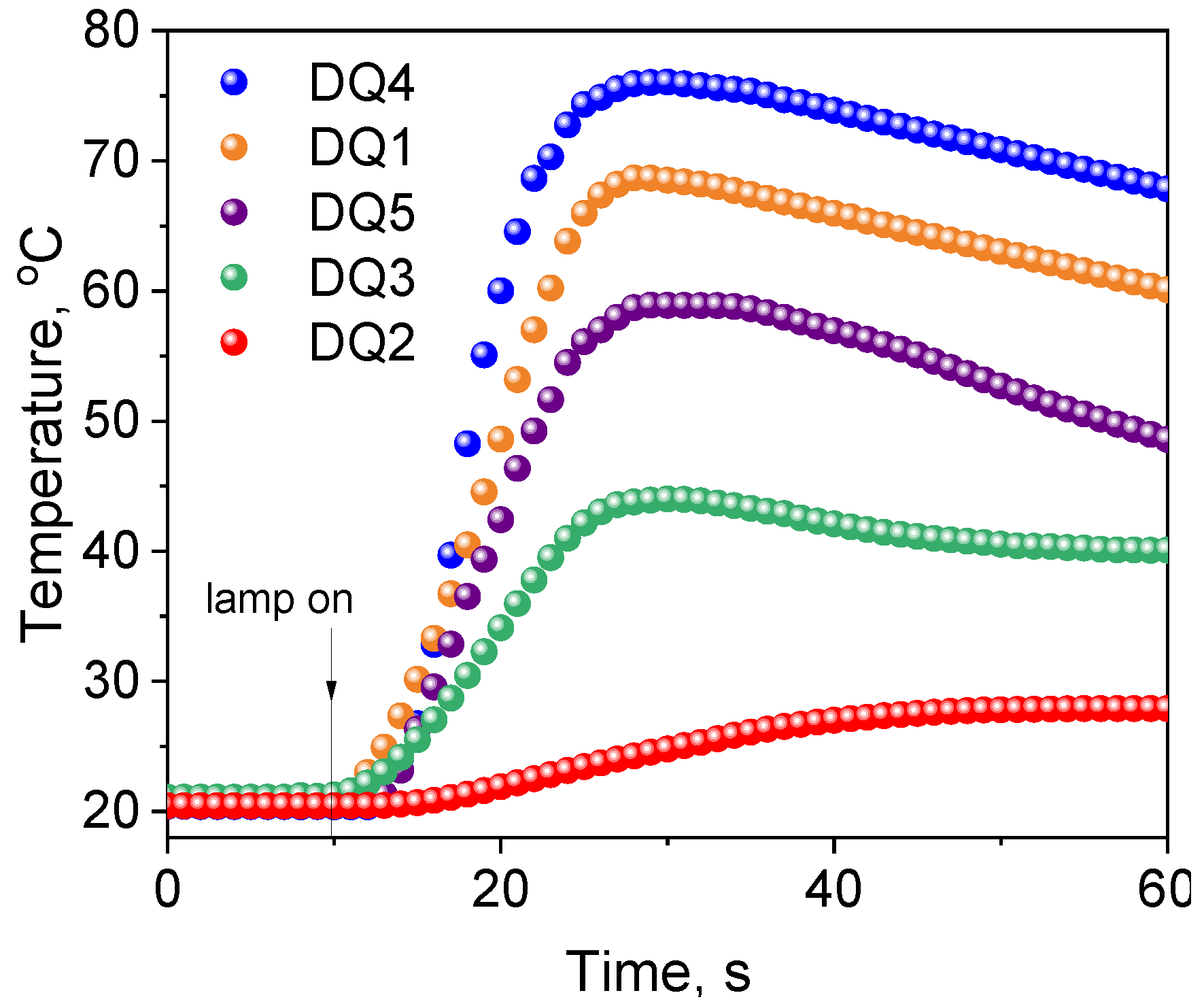
2.3.1. Photoinitiation by the Mechanism of Intermolecular Electron Transfer Influence of the Photoinitiator Structure
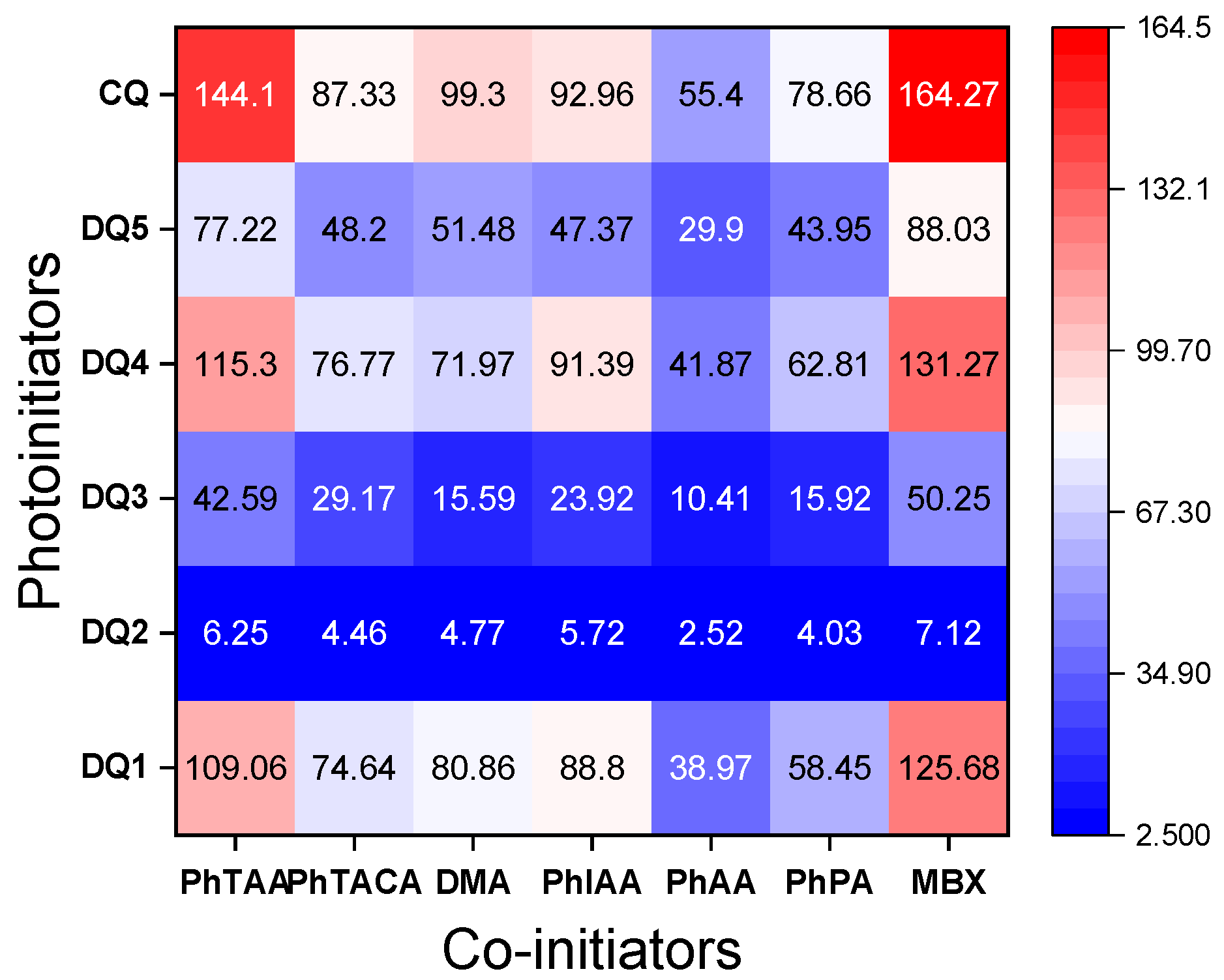
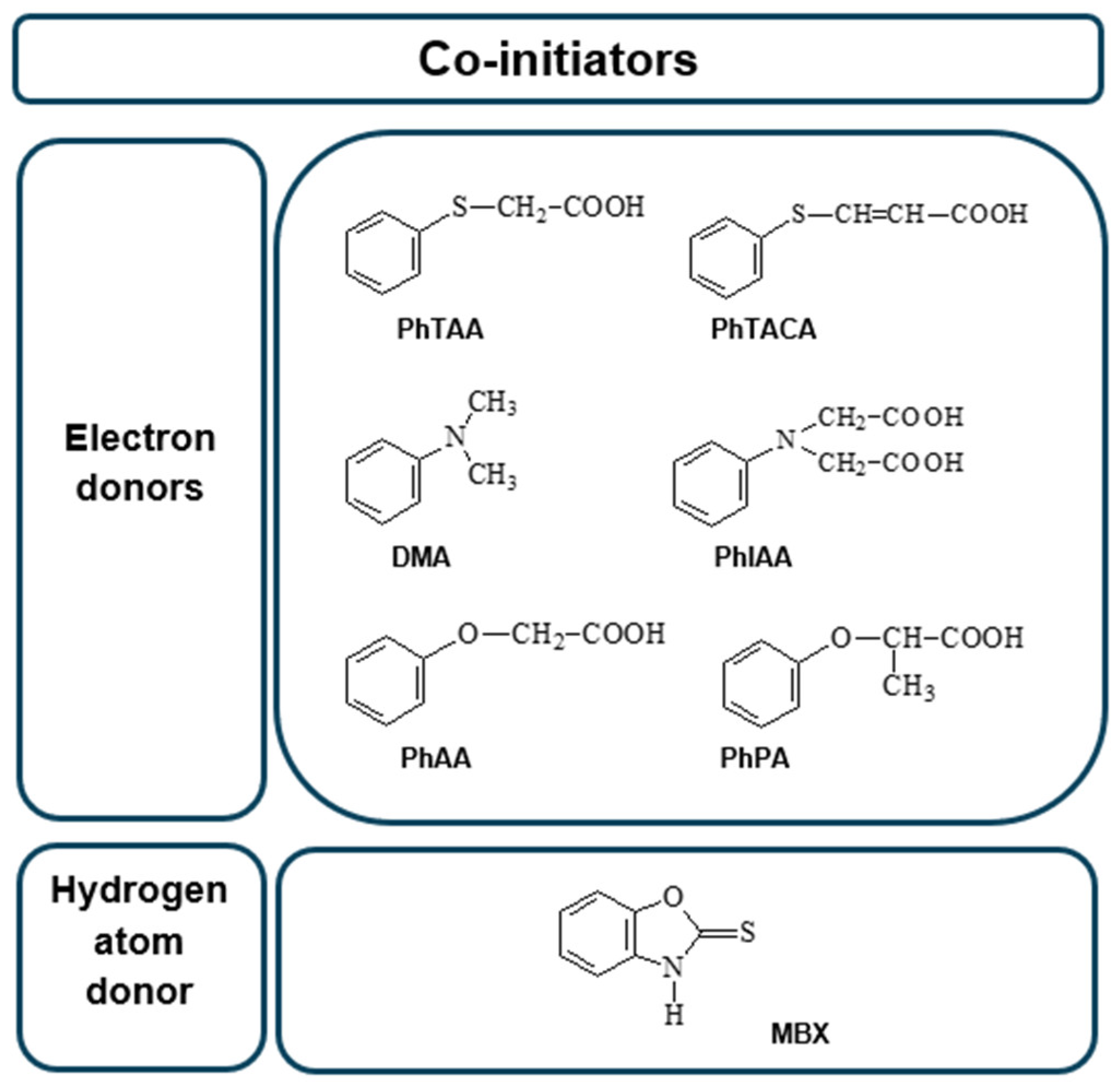
Influence of Co-Initiator Structure
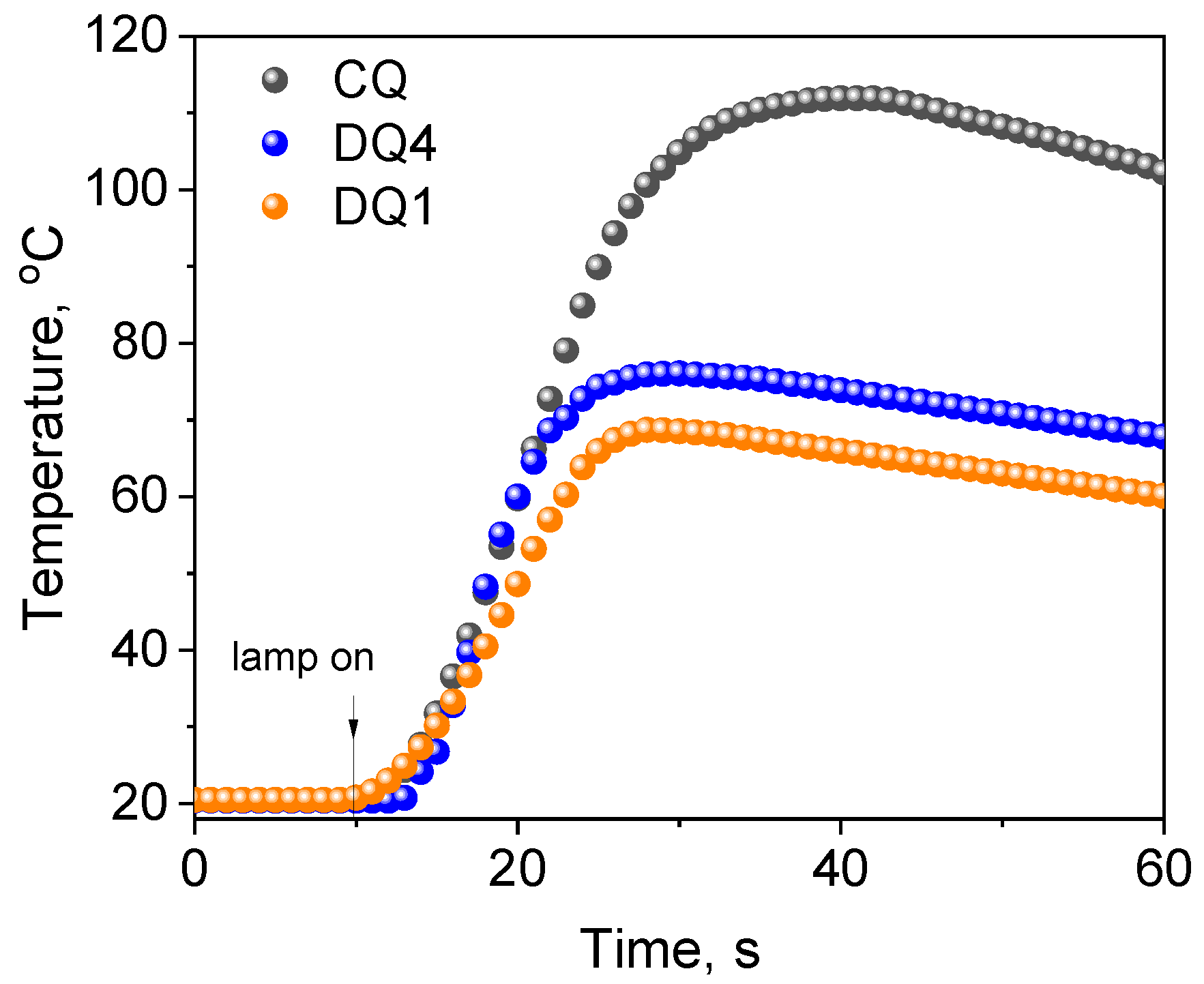
2.3.2. Control Tests Using Commercial Compounds
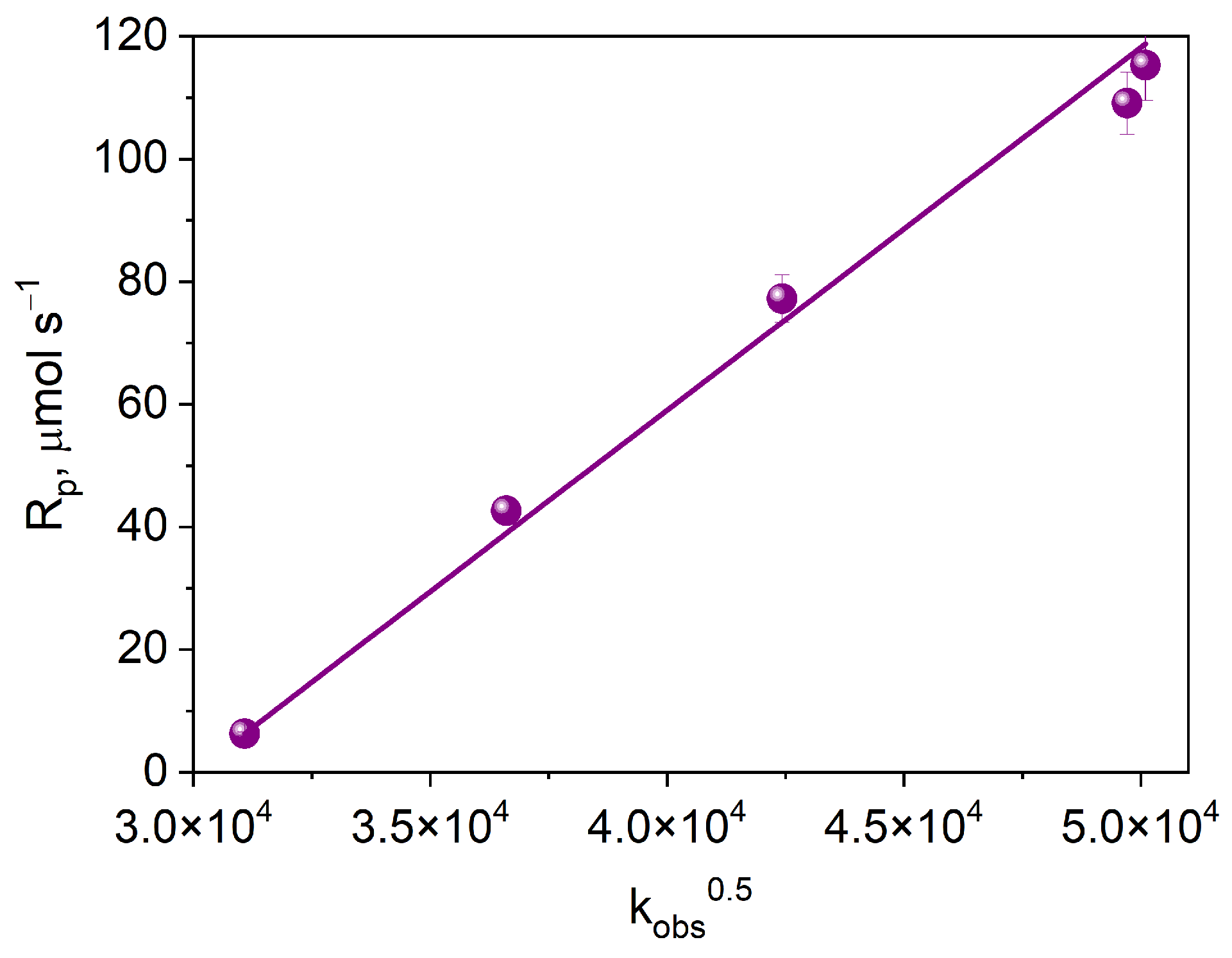
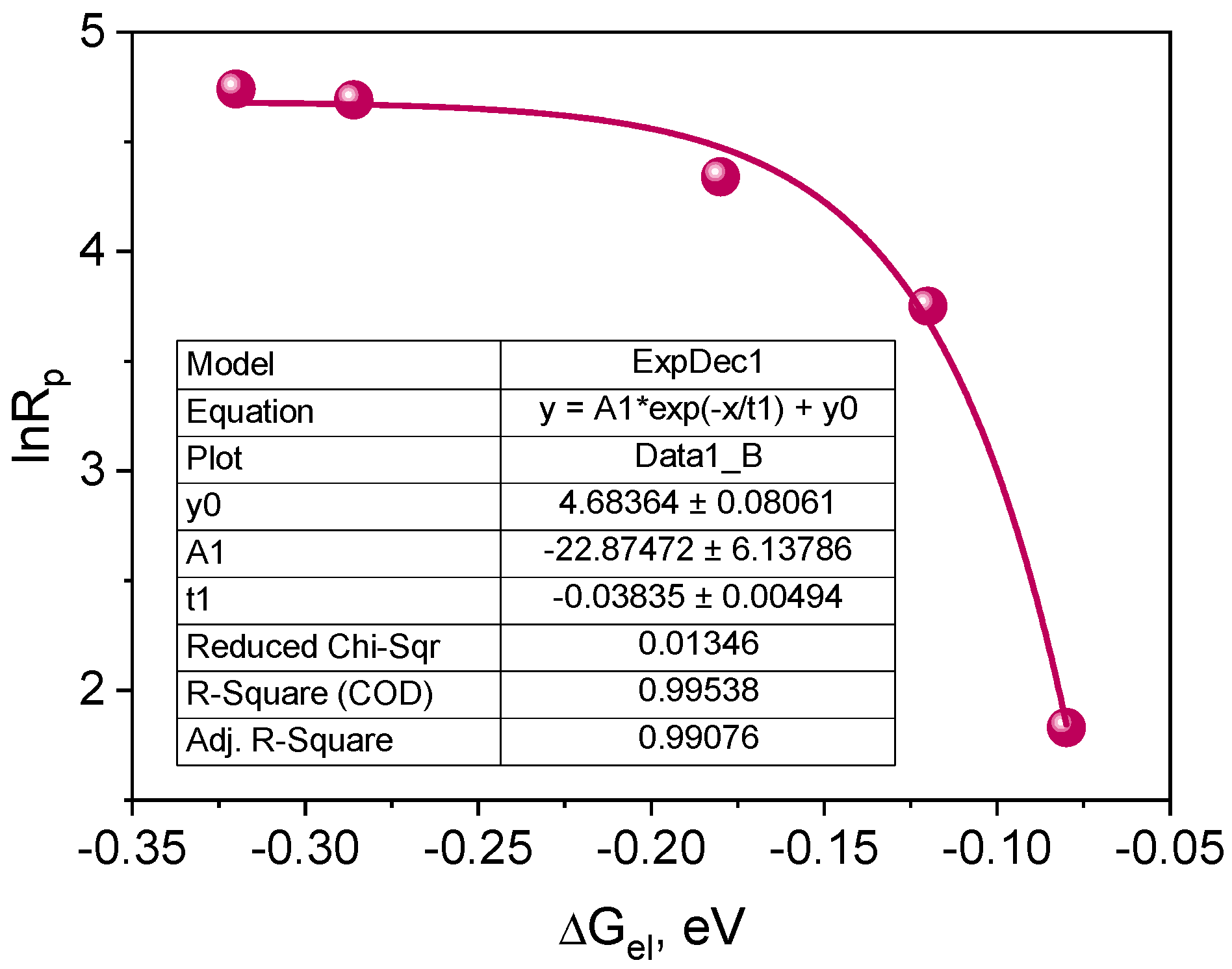
2.3.3. Kinetic and Thermodynamic Conditions of the Polymerization Process
3. Materials and Methods
3.1. Reagents
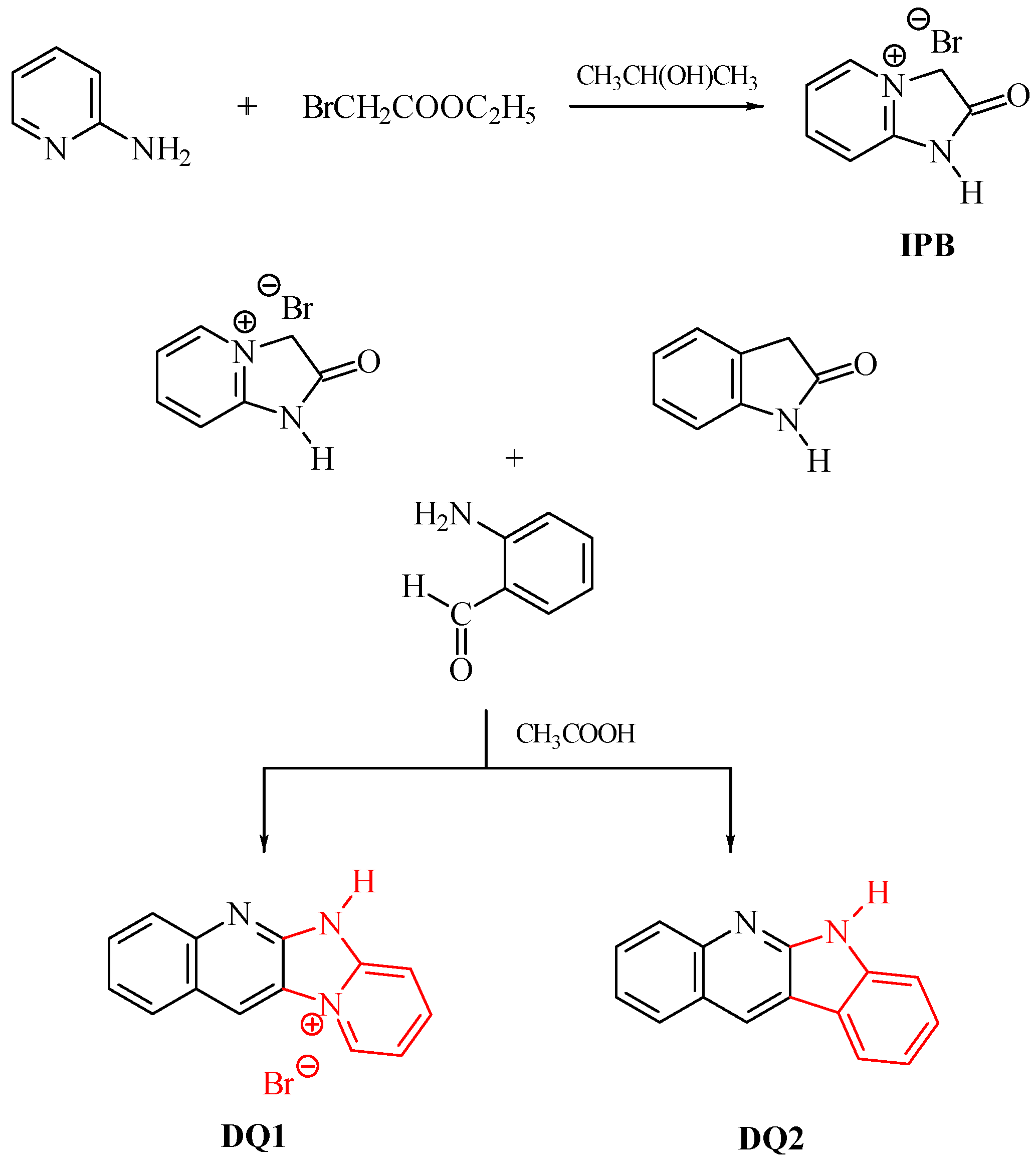
3.2. Synthesis
- DQ1: Quinoline[2,3-b]-1H-imidazo[1,2-a]pyridinium bromide
- Step 1: 2-oxo-2,3-dihydro-1H-imidazo[1,2-a]pyridinium bromide (IPB)
- Step 2
- DQ2; 6H-indolo[2,3-b]quinoline
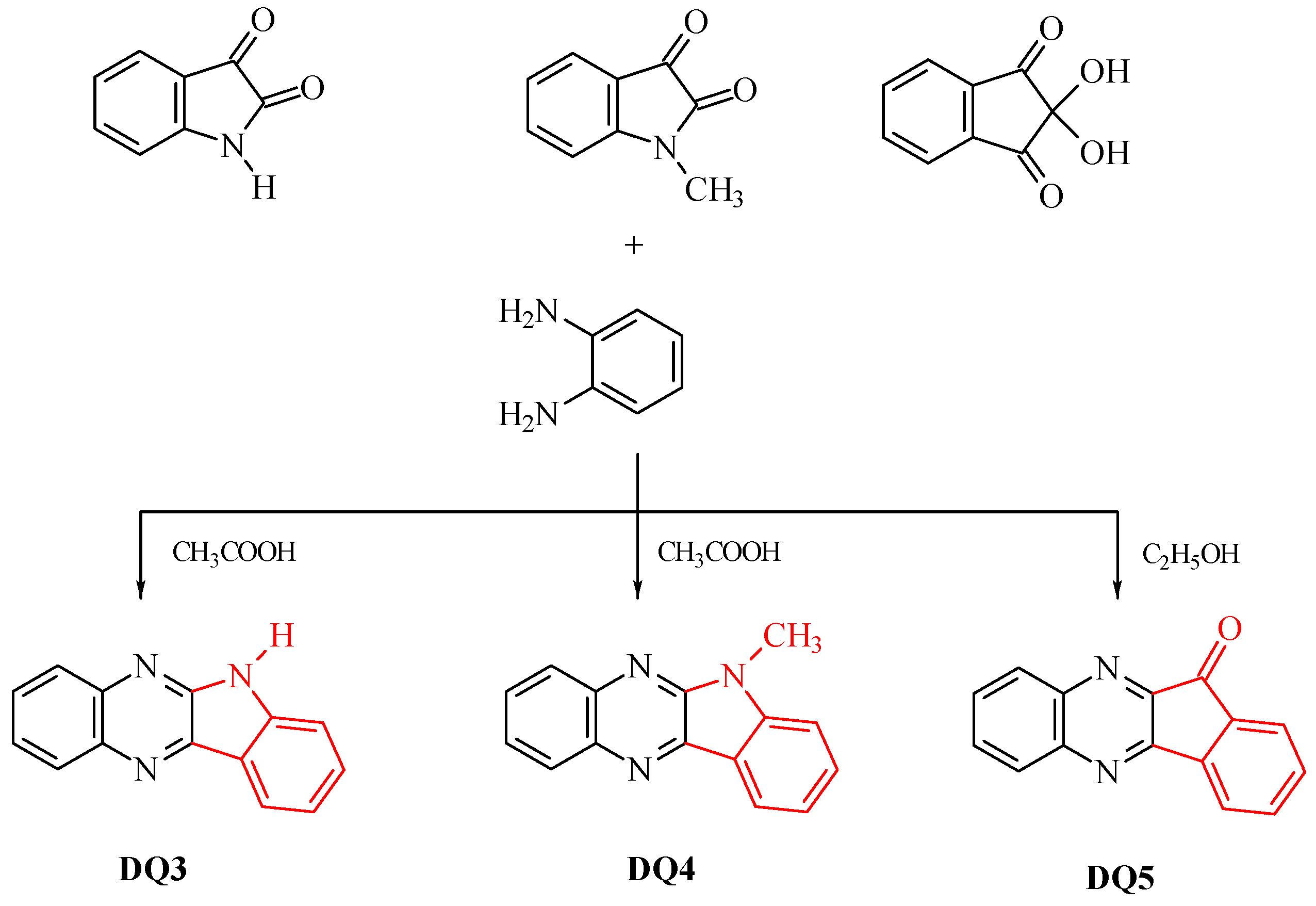
- DQ3; 6H-indolo[2,3-b]quinoxaline
- DQ4; 6-methyl-6H-indolo[2,3-b]quinoxaline
- DQ5; 11H-indeno[1,2-b]qunioxalin-11-on
3.3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sharm El-Sheikh Climate Change Conference, Sharm El-Sheikh, Egypt, 6–20 November 2022. Available online: https://unfccc.int/cop27 (accessed on 5 November 2022).
- Liang, M.; Borjigin, T.; Zhang, Y.; Liu, H.; Liu, B.; Guo, H. Z-Scheme Au@Void@g-C3N4/SnS Yolk–Shell Heterostructures for Superior Photocatalytic CO2 Reduction under Visible Light. ACS Appl. Mater. Interfaces 2018, 10, 34123–34131. [Google Scholar] [CrossRef]
- Billerbeck, K.; Hägele, C.; Träger, J. Relation of the Working Curve and Exposure Intensity in VPP 3D-Printing. Prog. Addit. Manuf. 2023, 2, 1–9. [Google Scholar] [CrossRef]
- de Oliveira, E.R.M.; Vieira, R.P. Synthesis and Characterization of Poly(Limonene) by Photoinduced Controlled Radical Polymerization. J. Polym. Environ. 2020, 28, 2931–2938. [Google Scholar] [CrossRef]
- Tawagi, E.; Ganesh, T.; Cheng, H.-L.M.; Santerre, J.P. Synthesis of Degradable-Polar-Hydrophobic-Ionic Co-Polymeric Microspheres by Membrane Emulsion Photopolymerization: In Vitro and in Vivo Studies. Acta Biomater. 2019, 89, 279–288. [Google Scholar] [CrossRef]
- Messaddeq, S.H.; Bonnet, A.-S.; Santagnelli, S.H.; Salek, G.; Colmenares, Y.N.; Messaddeq, Y. Photopolymerized Hybrids Containing TiO2 Nanoparticles for Gradient-Index Lens. Mater. Chem. Phys. 2019, 236, 121793. [Google Scholar] [CrossRef]
- Wei, L.; Shang, S.; Zheng, Y.; Liu, J.; Zhu, P. Iridescent Structural Colors Printing on Cellulose Fabrics with Robust Structural Coloration. Dye. Pigment. 2024, 221, 111824. [Google Scholar] [CrossRef]
- Dumur, F. Recent Advances on Phenothiazine-Based Oxime Esters as Visible Light Photoinitiators of Polymerization. Eur. Polym. J. 2024, 202, 112597. [Google Scholar] [CrossRef]
- Dumur, F. The Future of Visible Light Photoinitiators of Polymerization for Photocrosslinking Applications. Eur. Polym. J. 2023, 187, 111883. [Google Scholar] [CrossRef]
- Dumur, F. Recent Advances on Carbazole-Based Photoinitiators of Polymerization. Eur. Polym. J. 2020, 125, 109503. [Google Scholar] [CrossRef]
- Ma, Q.; Zhang, Y.; Launay, V.; Le Dot, M.; Liu, S.; Lalevée, J. How to Overcome the Light Penetration Issue in Photopolymerization? An Example for the Preparation of High Content Iron-Containing Opaque Composites and Application in 3D Printing. Eur. Polym. J. 2022, 165, 111011. [Google Scholar] [CrossRef]
- Lalevee, J.; Fouassier, J.P. Dyes and Chromophores in Polymer Science; ISTE Ltd.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2015. [Google Scholar]
- Jasinski, F.; Zetterlund, P.B.; Braun, A.M.; Chemtob, A. Photopolymerization in Dispersed Systems. Prog. Polym. Sci. 2018, 84, 47–88. [Google Scholar] [CrossRef]
- Dickens, S.H.; Stansbury, J.W.; Choi, K.M.; Floyd, C.J.E. Photopolymerization Kinetics of Methacrylate Dental Resins. Macromolecules 2003, 36, 6043–6053. [Google Scholar] [CrossRef]
- Noè, C.; Hakkarainen, M.; Sangermano, M. Cationic UV-Curing of Epoxidized Biobased Resins. Polymers 2021, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Dikova, T.; Maximov, J.; Todorov, V.; Georgiev, G.; Panov, V. Optimization of Photopolymerization Process of Dental Composites. Processes 2021, 9, 779. [Google Scholar] [CrossRef]
- Andreu, A.; Su, P.-C.; Kim, J.-H.; Ng, C.S.; Kim, S.; Kim, I.; Lee, J.; Noh, J.; Subramanian, A.S.; Yoon, Y.-J. 4D Printing Materials for Vat Photopolymerization. Addit. Manuf. 2021, 44, 102024. [Google Scholar] [CrossRef]
- Tomal, W.; Kiliclar, H.C.; Fiedor, P.; Ortyl, J.; Yagci, Y. Visible Light Induced High Resolution and Swift 3D Printing System by Halogen Atom Transfer. Macromol. Rapid Commun. 2023, 44, 2200661. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Noirbent, G.; Zhang, Y.; Sun, K.; Liu, S.; Brunel, D.; Gigmes, D.; Graff, B.; Morlet-Savary, F.; Xiao, P.; et al. Photopolymerization and 3D/4D Applications Using Newly Developed Dyes: Search around the Natural Chalcone Scaffold in Photoinitiating Systems. Dye. Pigment. 2021, 188, 109213. [Google Scholar] [CrossRef]
- Bonardi, A.H.; Dumur, F.; Grant, T.M.; Noirbent, G.; Gigmes, D.; Lessard, B.H.; Fouassier, J.-P.; Lalevée, J. High Performance Near-Infrared (NIR) Photoinitiating Systems Operating under Low Light Intensity and in the Presence of Oxygen. Macromolecules 2018, 51, 1314–1324. [Google Scholar] [CrossRef]
- Corrigan, N.; Yeow, J.; Judzewitsch, P.; Xu, J.; Boyer, C. Seeing the Light: Advancing Materials Chemistry through Photopolymerization. Angew. Chem. Int. Ed. 2019, 58, 5170–5189. [Google Scholar] [CrossRef]
- Lalevée, J.; Allonas, X.; Jradi, S.; Fouassier, J.-P. Role of the Medium on the Reactivity of Cleavable Photoinitiators in Photopolymerization Reactions. Macromolecules 2006, 39, 1872–1879. [Google Scholar] [CrossRef]
- Yagci, Y.; Jockusch, S.; Turro, N.J. Photoinitiated Polymerization: Advances, Challenges, and Opportunities. Macromolecules 2010, 43, 6245–6260. [Google Scholar] [CrossRef]
- Pączkowski, J.; Neckers, D.C. Photoinduced Electron Transfer Initiating Systems for Free-Radical Polymerization. In Electron Transfer in Chemistry; Wiley: Hoboken, NJ, USA, 2008; Volume 5. [Google Scholar]
- Zhang, J.; Zivic, N.; Dumur, F.; Xiao, P.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. A Benzophenone-Naphthalimide Derivative as Versatile Photoinitiator of Polymerization under near UV and Visible Lights. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 445–451. [Google Scholar] [CrossRef]
- Lalevée, J.; Telitel, S.; Tehfe, M.A.; Fouassier, J.P.; Curran, D.P.; Lacôte, E. N-Heterocyclic Carbene Boranes Accelerate Type I Radical Photopolymerizations and Overcome Oxygen Inhibition. Angew. Chem. Int. Ed. 2012, 51, 5958–5961. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewska, E. Photopolymerization kinetics of multifunctional monomers. Prog. Polym. Sci. 2001, 26, 605–665. [Google Scholar] [CrossRef]
- Wrzyszczynski, A.; Bartoszewicz, J.; Hug, G.L.; Marciniak, B.; Paczkowski, J. Photochemical Studies of a Photodissociative Initiator Based on a Benzophenone Derivative Possessing a Thioether Moiety. J. Photochem. Photobiol. A Chem. 2003, 155, 253–259. [Google Scholar] [CrossRef]
- Pyszka, I.; Jędrzejewska, B. Photoinitiation Abilities of Indeno- and Indoloquinoxaline Derivatives and Mechanical Properties of Dental Fillings Based on Multifunctional Acrylic Monomers and Glass Ionomer. Polymer 2023, 266, 125625. [Google Scholar] [CrossRef]
- Pyszka, I.; Jędrzejewska, B. Acenaphthoquinoxaline Derivatives as Dental Photoinitiators of Acrylates Polymerization. Materials 2021, 14, 4881. [Google Scholar] [CrossRef] [PubMed]
- Kucybała, Z.; Wrzyszczyński, A. Photolysis of N-[(4-Benzoyl)Benzenesulfonyl]Benzenesulfonamide. J. Photochem. Photobiol. A Chem. 2002, 153, 109–112. [Google Scholar] [CrossRef]
- Fouassier, J.P.; Allonas, X.; Burget, D. Photopolymerization Reactions under Visible Lights: Principle, Mechanisms and Examples of Applications. Prog. Org. Coat. 2003, 47, 16–36. [Google Scholar] [CrossRef]
- Kabatc, J.; Kucybała, Z.; Pietrzak, M.; Ścigalski, F.; Pączkowski, J. Free Radical Polymerization Initiated via Photoinduced Intermolecular Electron Transfer Process: Kinetic Study 3. Polymer 1999, 40, 735–745. [Google Scholar] [CrossRef]
- Jędrzejewska, B.; Wejnerowska, G. Highly Effective Sensitizers Based on Merocyanine Dyes for Visible Light Initiated Radical Polymerization. Polymers 2020, 12, 1242. [Google Scholar] [CrossRef] [PubMed]
- Pyszka, I.; Kucybała, Z.; Jędrzejewska, B. Effective Singlet Oxygen Sensitizers Based on the Phenazine Skeleton as Efficient Light Absorbers in Dye Photoinitiating Systems for Radical Polymerization of Acrylates. Materials 2021, 14, 3085. [Google Scholar] [CrossRef] [PubMed]
- Jędrzejewska, B.; Ośmiałowski, B. Difluoroboranyl Derivatives as Efficient Panchromatic Photoinitiators in Radical Polymerization Reactions. Polym. Bull. 2018, 75, 3267–3281. [Google Scholar] [CrossRef]
- Strzelczyk, R.; Podsiadły, R. Naphthoylenebenzimidazolone Dyes as One-Component Photoinitiators. Color. Technol. 2017, 133, 178–183. [Google Scholar] [CrossRef]
- Kabatc, J. The Influence of a Radical Structure on the Kinetics of Photopolymerization. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 1575–1589. [Google Scholar] [CrossRef]
- Balcerak, A.; Kabatc, J. The Photooxidative Sensitization of Bis(p-Substituted Diphenyl)Iodonium Salts in the Radical Polymerization of Acrylates. RSC Adv. 2019, 9, 28490–28499. [Google Scholar] [CrossRef] [PubMed]
- Mousawi, A.A.; Dietlin, C.; Graff, B.; Morlet-Savary, F.; Toufaily, J.; Hamieh, T.; Fouassier, J.P.; Chachaj-Brekiesz, A.; Ortyl, J.; Lalevée, J. Meta-Terphenyl Derivative/Iodonium Salt/9H-Carbazole-9-Ethanol Photoinitiating Systems for Free Radical Promoted Cationic Polymerization upon Visible Lights. Macromol. Chem. Phys. 2016, 217, 1955–1965. [Google Scholar] [CrossRef]
- Pyszka, I.; Kucybała, Z.; Pączkowski, J. Reinvestigation of the Mechanism of the Free Radical Polymerization Photoinitiation Process by Camphorquinone–Coinitiator Systems: New Results. Macromol. Chem. Phys. 2004, 205, 2371–2375. [Google Scholar] [CrossRef]
- Allen, N.S. Photochemistry and Photophysics of Polymer Materials; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Borjigin, T.; Schmitt, M.; Giacoletto, N.; Rico, A.; Bidotti, H.; Nechab, M.; Zhang, Y.; Graff, B.; Morlet-Savary, F.; Xiao, P.; et al. The Blue-LED-Sensitive Naphthoquinone-Imidazolyl Derivatives as Type II Photoinitiators of Free Radical Photopolymerization. Adv. Mater. Interfaces 2023, 10, 2202352. [Google Scholar] [CrossRef]
- Kucková, L.; Jomová, K.; Švorcová, A.; Valko, M.; Segľa, P.; Moncoľ, J.; Kožíšek, J. Synthesis, Crystal Structure, Spectroscopic Properties and Potential Biological Activities of Salicylate–Neocuproine Ternary Copper(II) Complexes. Molecules 2015, 20, 2115–2137. [Google Scholar] [CrossRef]
- Abu Ali, H.; Abu Shamma, A.; Kamel, S. New Mixed Ligand Cobalt(II/III) Complexes Based on the Drug Sodium Valproate and Bioactive Nitrogen-Donor Ligands. Synthesis, Structure and Biological Properties. J. Mol. Struct. 2017, 1142, 40–47. [Google Scholar] [CrossRef]
- Dhumwad, S.D. Synthesis, Characterization and Biological Studies of Co(II), Ni(II), Cu(II) and Zn(II) Complexes of Schiff Bases Derived from 3-Formyl-2-Mercaptoquinolines. J. Chem. Pharm. Res. 2011, 3, 504–517. [Google Scholar]
- Jaman, Z.; Karim, M.R.; Siddiquee, T.A.; Mirza, A.H.; Ali, M.A. Synthesis of 5-Substituted 2, 9-Dimethyl-1,10-Phenanthroline Dialdehydes and Their Schiff Bases with Sulfur-Containing Amines. Int. J. Org. Chem. 2013, 3, 214–219. [Google Scholar] [CrossRef]
- Abu Ali, H.; Kamel, S.; Abu Shamma, A. Novel Structures of Zn(II) Biometal Cation with the Biologically Active Substituted Acetic Acid and Nitrogen Donor Ligands: Synthesis, Spectral, Phosphate Diester Catalytic Hydrolysis and Anti-Microbial Studies. Appl. Organomet. Chem. 2017, 31, e3829. [Google Scholar] [CrossRef]
- Jabali, B.; Abu Ali, H. New Zinc(II) Complexes of the Non-Steroidal Anti-Inflammatory Drug (Indomethacin) and Various Nitrogen Donor Ligands. Synthesis, Characterization and Biological Activity. Polyhedron 2016, 117, 249–258. [Google Scholar] [CrossRef]
- Pereira, J.A.; Pessoa, A.M.; Cordeiro, M.N.D.S.; Fernandes, R.; Prudêncio, C.; Noronha, J.P.; Vieira, M. Quinoxaline, Its Derivatives and Applications: A State of the Art Review. Eur. J. Med. Chem. 2015, 97, 664–672. [Google Scholar] [CrossRef]
- Segoviano-Garfias, J.J.N.; Zanor, G.A.; Ávila-Ramos, F.; Bivián-Castro, E.Y. Equilibrium Studies of Iron (III) Complexes with Either Pyrazine, Quinoxaline, or Phenazine and Their Catecholase Activity in Methanol. Molecules 2022, 27, 3257. [Google Scholar] [CrossRef]
- Murov, S.L.; Carmichael, I.; Hug, G.L. Handbook of Photochemistry; Marcel Dekker: New York, NY, USA, 1993. [Google Scholar]
- Lament, B.; Karpiuk, J.; Waluk, J. Determination of Triplet Formation Efficiency from Kinetic Profiles of the Ground State Recovery. Photochem. Photobiol. Sci. 2003, 2, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Stern, O.; Volmer, M. Über Die Abklingungszeit Der Fluoreszenz. Physik. Zeitschr. 1919, 20, 183–188. [Google Scholar]
- Ghorbani, F.; Harry, S.A.; Capilato, J.N.; Pitts, C.R.; Joram, J.; Peters, G.N.; Tovar, J.D.; Smajlagic, I.; Siegler, M.A.; Dudding, T.; et al. Carbonyl-Directed Aliphatic Fluorination: A Special Type of Hydrogen Atom Transfer Beats Out Norrish II. J. Am. Chem. Soc. 2020, 142, 14710–14724. [Google Scholar] [CrossRef]
- Zhu, J.L.; Schull, C.R.; Tam, A.T.; Rentería-Gómez, Á.; Gogoi, A.R.; Gutierrez, O.; Scheidt, K.A. Photoinduced Acylations Via Azolium-Promoted Intermolecular Hydrogen Atom Transfer. J. Am. Chem. Soc. 2023, 145, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Gruber, H.F. Photoinitiators for Free Radical Polymerization. Prog. Polym. Sci. 1992, 17, 953–1044. [Google Scholar] [CrossRef]
- Jakubiak, J.; Allonas, X.; Fouassier, J.P.; Sionkowska, A.; Andrzejewska, E.; Linden, L.Å.; Rabek, J.F. Camphorquinone–Amines Photoinitating Systems for the Initiation of Free Radical Polymerization. Polymer 2003, 44, 5219–5226. [Google Scholar] [CrossRef]
- Encinas, M.V.; Majmud, C.; Lissi, E.A. Polymerization Photoinitiated by Carbonyl Compounds. IX. Methyl Methacrylate Polymerization Photoinitiated by Anthraquinone in Presence of Triethylamine. J. Polym. Sci. Part A Polym. Chem. 1990, 28, 2465–2474. [Google Scholar] [CrossRef]
- Kucybała, Z.; Pietrzak, M.; Osmańska, K.; Pączkowski, J. Mechanistic and Kinetic Study of Free Radical Polymerization Photoinitiated by Xanthene Dye-n-Phenylglycine Derivatives Photoredox Pairs. Pol. J. Chem. 2005, 79, 851–866. [Google Scholar]
- Linden, S.M.; Neckers, D.C. Fundamental Properties of Rose Bengal. 25. Bleaching Studies of Rose Bengal Onium Salts. J. Am. Chem. Soc. 1988, 110, 1257–1260. [Google Scholar] [CrossRef]
- Kabatc, J.; Pietrzak, M.; Pączkowski, J. Cyanine Borates Revisited. Application of the Marcus Equation for the Description of the Kinetics of Photoinitiated Free Radical Polymerization. IV. Macromolecules 1998, 31, 4651–4654. [Google Scholar] [CrossRef]
- Bartholomew, R.F.; Davidson, R.S. The Photosensitised Oxidation of Amines. Part I. The Use of Benzophenone as a Sensitiser. J. Chem. Soc. C 1971, 1, 2342–2346. [Google Scholar] [CrossRef]
- Brimage, D.R.G.; Davidson, R.S.; Steiner, P.R. Use of Heterocyclic Compounds as Photosensitisers for the Decarboxylation of Carboxylic Acids. J. Chem. Soc. Perkin Trans. C 1973, 1, 526–529. [Google Scholar] [CrossRef]
- Davidson, R.S.; Harrison, K.; Steiner, P.R. The Photosensitised Decarboxylation of Carboxylic Acids by Aromatic Ketones. J. Chem. Soc. C 1971, 1, 3480–3482. [Google Scholar] [CrossRef]
- Davidson, R.S.; Steiner, P.R. The Photosensitized Decarboxylation of Carboxylic Acids by Benzophenone and Quinones. J. Chem. Soc. C 1971, 1, 1682–1689. [Google Scholar] [CrossRef]
- Ikeda, S.; Murata, S.; Ishii, K.; Hamaguchi, H. Mechanistic Studies of the Pyrene-Sensitized Photodecomposition of N-Phenylglycine: Acceleration of the Photodecomposition by the Addition of an Electron Acceptor. Bull. Chem. Soc. Jpn. 2000, 73, 2783–2792. [Google Scholar] [CrossRef]
- Rehm, D.; Weller, A. Kinetics of Fluorescence Quenching by Electron and H-Atom Transfer. Isr. J. Chem. 1970, 8, 259–271. [Google Scholar] [CrossRef]
- Weller, A. Photoinduced Electron Transfer in Solution: Exciplex and Radical Ion Pair Formation Free Enthalpies and Their Solvent Dependence. Z. Phys. Chem. 1982, 133, 93–98. [Google Scholar] [CrossRef]
- Marcus, R.A. On the Theory of Electron-Transfer Reactions. VI. Unified Treatment for Homogeneous and Electrode Reactions. J. Chem. Phys. 2004, 43, 679–701. [Google Scholar] [CrossRef]
- Eberson, L. Electron Transfer in Organic Chemistry; Springer: New York, NY, USA, 1987. [Google Scholar]
- Marcus, R.A. Chemical and Electrochemical Electron-Transfer Theory. Annu. Rev. Phys. Chem. 1964, 15, 155–196. [Google Scholar] [CrossRef]
- Pączkowski, J.; Pietrzak, M.; Kucybała, Z. Generalization of the Kinetic Scheme for Photoinduced Polymerization via an Intermolecular Electron Transfer Process. 2. Application of the Marcus Theory. Macromolecules 1996, 29, 5057–5064. [Google Scholar] [CrossRef]
- Sarkis, G.Y.; Al-Badri, H.T. Synthesis and Spectroscopic Studies of Some New Substituted 6H-Indolo[2,3-b]Quinoxalines. J. Heterocycl. Chem. 1980, 17, 813–815. [Google Scholar] [CrossRef]
- Huynh, T.N.; Tran, D.T.; Nguyen, T.T. Elemental Sulfur Promoted Condensation of Indoles and 1,2-Phenylenediamines. Tetrahedron Lett. 2023, 116, 154360. [Google Scholar] [CrossRef]
- Manna, K.; Agrawal, Y.K. Microwave Assisted Synthesis of New Indophenazine 1,3,5-Trisubstruted Pyrazoline Derivatives of Benzofuran and Their Antimicrobial Activity. Bioorganic Med. Chem. Lett. 2009, 19, 2688–2692. [Google Scholar] [CrossRef]
- Avula, S.; Komsani, J.; Koppireddi, S.; Yadla, R.; Kanugula, A.; Kotamraju, S. Synthesis and Cytotoxicity of Novel 6H-Indolo[2,3-b]Quinoxaline Derivatives. Med. Chem. Res. 2012, 22, 3712–3718. [Google Scholar] [CrossRef]
- Van Dormael, A. Colorants Polyméthiniques Dérivés Du Noyau de La Pyrimidazolone. Bull. Des. Sociétés Chim. Belg. 1949, 58, 167–182. [Google Scholar] [CrossRef]
- Pyszka, I.; Kucybała, Z. Quinolineimidazopyridinium Derivatives as Visible-Light Photoinitiators of Free Radical Polymerization. Polymer 2007, 48, 959–965. [Google Scholar] [CrossRef]
- Majhi, B.; Parwez, A.; Palit, S.; Dutta, S. One-Pot Cascade Annulation-Triggered Synthesis of N-6-Substituted Norcryptotackieine Alkaloids and Evaluation of Their Antileishmanial Activities. J. Org. Chem. 2022, 87, 14695–14705. [Google Scholar] [CrossRef] [PubMed]
- Rong, G.-Q.; Zhao, J.-Q.; Zhang, X.-M.; Xu, X.-Y.; Yuan, W.-C.; Zhou, M.-Q. One-Pot Access to Indolylchromeno[2,3-b]Indoles via Iodine-Mediated Friedel-Crafts Alkylation/Oxidative Coupling Reaction of Indoles and Salicylaldehydes. Tetrahedron 2018, 74, 2383–2390. [Google Scholar] [CrossRef]
- Ivashchenko, A.V.; Drushlyak, A.G.; Titov, V.V. Reaction of O-Phenylenediamine with Isatins. Chem. Heterocycl. Compd. 1984, 20, 537–542. [Google Scholar] [CrossRef]
- Takekuma, S.; Katayama, S.; Takekuma, H. Preparation and Structure of Tetrakis(Indolo[2,3-b]Quinoxalinato)Dinickel(II). Chem. Lett. 2000, 29, 614–615. [Google Scholar] [CrossRef]
- Kumbhar, A.; Kanase, D.; Mohite, S.; Salunkhe, R.; Lohar, T. Brönsted Acid Hydrotrope Combined Catalysis in Water: A Green Approach for the Synthesis of Indoloquinoxalines and Bis-Tetronic Acids. Res. Chem. Intermed. 2021, 47, 2263–2278. [Google Scholar] [CrossRef]
- Kanhed, A.M.; Patel, D.V.; Patel, N.R.; Sinha, A.; Thakor, P.S.; Patel, K.B.; Prajapati, N.K.; Patel, K.V.; Yadav, M.R. Indoloquinoxaline Derivatives as Promising Multi-Functional Anti-Alzheimer Agents. J. Biomol. Struct. Dyn. 2022, 40, 2498–2515. [Google Scholar] [CrossRef]
- Edayadulla, N.; Lee, Y.R. Cerium Oxide Nanoparticle-Catalyzed Three-Component Protocol for the Synthesis of Highly Substituted Novel Quinoxalin-2-Amine Derivatives and 3,4-Dihydroquinoxalin-2-Amines in Water. RSC Adv. 2014, 4, 11459–11468. [Google Scholar] [CrossRef]
- Pyszka, I.; Skowroński, Ł.; Jędrzejewska, B. Study on New Dental Materials Containing Quinoxaline-Based Photoinitiators in Terms of Exothermicity of the Photopolymerization Process. Int. J. Mol. Sci. 2023, 24, 2752. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dyes | a (nm) | ε (M−1 cm−1) | b (nm) | ΦFl | (nm) | (eV) | Ered (V) | c ΔGel (eV) |
|---|---|---|---|---|---|---|---|---|
| DQ1 | 387 | 2610 | 447 | 0.230 | 526 | 2.79 | −1.054 | −0.286 |
| 408 | 3040 | 477 | 542 | |||||
| 432 | 2360 | 511 | 572 | |||||
| DQ2 | 316 | 14,400 | 406 | 0.176 | 510 | 2.51 | −0.98 | −0.08 |
| 330 | 20,150 | |||||||
| 367 | 3980 | |||||||
| DQ3 | 334 | 14,100 | 465 | 0.039 | 543 | 2.74 | −1.17 | −0.12 |
| 350 | 16,300 | |||||||
| 386 | 4200 | 588 | ||||||
| DQ4 | 334 | 14,100 | 476 | 0.040 | 515 | 2.95 | −1.18 | −0.32 |
| 351 | 16,500 | |||||||
| 405 | 3400 | 561 | ||||||
| DQ5 | 287 | 34,600 | 443 | 0.123 | 529 | 2.72 | −1.09 | −0.18 |
| 381 | 3500 | |||||||
| CQ | 472 | 40 | - | - | - | - | - | - |
| Dyes | ΦT | τT, μs |
|---|---|---|
| DQ1 | 0.5490 | 1.52 |
| DQ2 | 0.0060 | 0.74 |
| DQ3 | 0.1061 | 1.00 |
| DQ4 | 0.6320 | 1.58 |
| DQ5 | 0.2704 | 1.31 |
| Co-Initiators | kq × 10−8, M−1 s−1 | ||||
|---|---|---|---|---|---|
| DQ1 | DQ2 | DQ3 | DQ4 | DQ5 | |
| PhTAA | 25.10 | 9.66 | 13.40 | 24.71 | 18.00 |
| PhTACA | 5.20 | 3.01 | 3.12 | 6.25 | 5.30 |
| DMA | 6.94 | 3.16 | 3.98 | 7.09 | 5.92 |
| PhIAA | 16.20 | 6.62 | 8.92 | 17.80 | 12.30 |
| PhAA | 2.79 | 2.33 | 2.63 | 2.65 | 4.61 |
| PhPA | 3.02 | 2.75 | 2.98 | 3.12 | 5.05 |
| MBX | 3.36 | 1.97 | 2.01 | 2.98 | 3.11 |
| Co-Initiators | Rp, μmol·s−1 | |||||
|---|---|---|---|---|---|---|
| DQ1 | DQ2 | DQ3 | DQ4 | DQ5 | CQ | |
| PhTAA | 109.06 | 6.25 | 42.59 | 115.30 | 77.22 | 144.10 |
| PhTACA | 74.64 | 4.46 | 29.17 | 76.77 | 48.20 | 87.33 |
| DMA | 80.86 | 4.77 | 15.59 | 71.97 | 51.48 | 99.30 |
| PhIAA | 88.80 | 5.72 | 23.92 | 91.39 | 47.37 | 92.96 |
| PhAA | 38.97 | 2.52 | 10.41 | 41.87 | 29.90 | 55.40 |
| PhPA | 58.45 | 4.03 | 15.92 | 62.81 | 43.95 | 78.66 |
| MBX | 125.68 | 7.12 | 50.25 | 131.27 | 88.03 | 164.27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pyszka, I.; Jędrzejewska, B. Design of Dyes Based on the Quinoline or Quinoxaline Skeleton towards Visible Light Photoinitiators. Int. J. Mol. Sci. 2024, 25, 4289. https://doi.org/10.3390/ijms25084289
Pyszka I, Jędrzejewska B. Design of Dyes Based on the Quinoline or Quinoxaline Skeleton towards Visible Light Photoinitiators. International Journal of Molecular Sciences. 2024; 25(8):4289. https://doi.org/10.3390/ijms25084289
Chicago/Turabian StylePyszka, Ilona, and Beata Jędrzejewska. 2024. "Design of Dyes Based on the Quinoline or Quinoxaline Skeleton towards Visible Light Photoinitiators" International Journal of Molecular Sciences 25, no. 8: 4289. https://doi.org/10.3390/ijms25084289
APA StylePyszka, I., & Jędrzejewska, B. (2024). Design of Dyes Based on the Quinoline or Quinoxaline Skeleton towards Visible Light Photoinitiators. International Journal of Molecular Sciences, 25(8), 4289. https://doi.org/10.3390/ijms25084289