
Homologous Recombination Repair Deficiency in Metastatic Prostate Cancer: New Therapeutic Opportunities
 , , ,
, , ,
Abstract
:1. Introduction
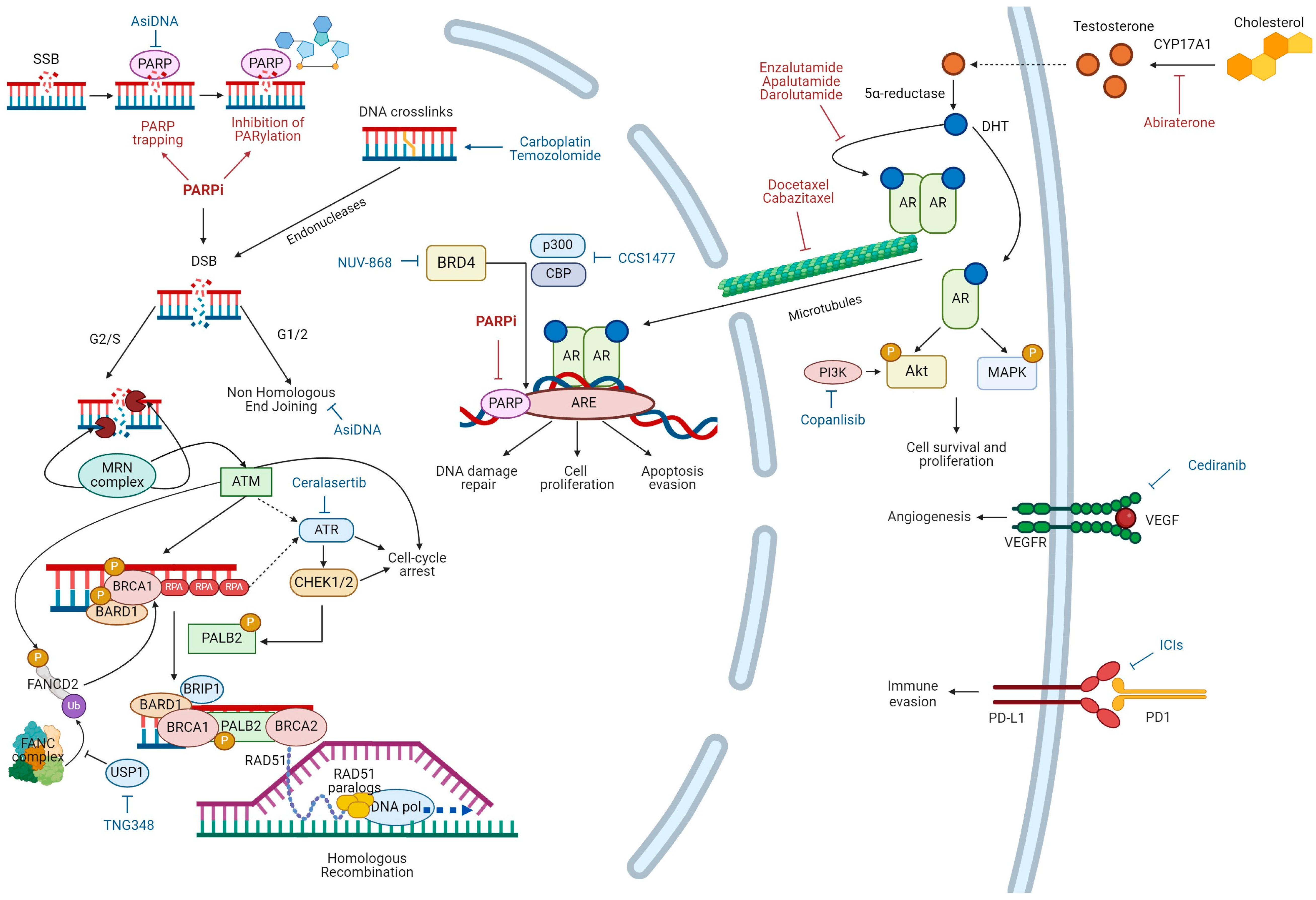
2. Overview on HRR Pathway
3. Identification of HRD in PC
4. PARPis as Single Agents in Metastatic PC
5. Crosstalk between HRR and Androgen Receptor Pathway
6. Therapy Combinations with PARPis in Metastatic PC
7. Mechanisms of Resistance to PARPis in PC
8. Ongoing Trials with PARPis in Metastatic PC
8.1. Phase III Trials
8.2. Phase II Trials
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- National Cancer Institute SEER Program. Cancer Stat Facts: Prostate Cancer. Available online: https://seer.cancer.gov/statfacts/html/prost.html (accessed on 8 March 2024).
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate Cancer Progression after Androgen Deprivation Therapy: Mechanisms of Castrate Resistance and Novel Therapeutic Approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef] [PubMed]
- Shoag, J.; Barbieri, C.E. Clinical Variability and Molecular Heterogeneity in Prostate Cancer. Asian J. Androl. 2016, 18, 543. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network the Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [CrossRef] [PubMed]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Page, E.C.; Bancroft, E.K.; Brook, M.N.; Assel, M.; Hassan Al Battat, M.; Thomas, S.; Taylor, N.; Chamberlain, A.; Pope, J.; Raghallaigh, H.N.; et al. Interim Results from the IMPACT Study: Evidence for Prostate-Specific Antigen Screening in BRCA2 Mutation Carriers. Eur. Urol. 2019, 76, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Castro, E.; Goh, C.; Olmos, D.; Saunders, E.; Leongamornlert, D.; Tymrakiewicz, M.; Mahmud, N.; Dadaev, T.; Govindasami, K.; Guy, M.; et al. Germline BRCA Mutations Are Associated with Higher Risk of Nodal Involvement, Distant Metastasis, and Poor Survival Outcomes in Prostate Cancer. J. Clin. Oncol. 2013, 31, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Na, R.; Zheng, S.L.; Han, M.; Yu, H.; Jiang, D.; Shah, S.; Ewing, C.M.; Zhang, L.; Novakovic, K.; Petkewicz, J.; et al. Germline Mutations in ATM and BRCA1/2 Distinguish Risk for Lethal and Indolent Prostate Cancer and Are Associated with Early Age at Death. Eur. Urol. 2017, 71, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Fisher, C.; Foster, C.S.; Jameson, C.; Barbachanno, Y.; Bartlett, J.; Bancroft, E.; Doherty, R.; Kote-Jarai, Z.; Peock, S.; et al. Prostate Cancer in Male BRCA1 and BRCA2 Mutation Carriers Has a More Aggressive Phenotype. Br. J. Cancer 2008, 98, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Narod, S.A.; Neuhausen, S.; Vichodez, G.; Armel, S.; Lynch, H.T.; Ghadirian, P.; Cummings, S.; Olopade, O.; Stoppa-Lyonnet, D.; Couch, F.; et al. Rapid Progression of Prostate Cancer in Men with a BRCA2 Mutation. Br. J. Cancer 2008, 99, 371–374. [Google Scholar] [CrossRef]
- Thorne, H.; Willems, A.J.; Niedermayr, E.; Hoh, I.M.Y.; Li, J.; Clouston, D.; Mitchell, G.; Fox, S.; Hopper, J.L.; Kathleen Cunningham Consortium for Research in Familial Breast Cancer Consortium; et al. Decreased Prostate Cancer-Specific Survival of Men with BRCA2 Mutations from Multiple Breast Cancer Families. Cancer Prev. Res. 2011, 4, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Tryggvadóttir, L.; Vidarsdóttir, L.; Thorgeirsson, T.; Jonasson, J.G.; Olafsdóttir, E.J.; Olafsdóttir, G.H.; Rafnar, T.; Thorlacius, S.; Jonsson, E.; Eyfjord, J.E.; et al. Prostate Cancer Progression and Survival in BRCA2 Mutation Carriers. J. Natl. Cancer Inst. 2007, 99, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wu, J.; Gu, W.; Wang, J.; Lin, G.; Qin, X.; Dai, B.; Gan, H.; Ye, D.; Zhu, Y. Prognostic Value of Germline DNA Repair Gene Mutations in De Novo Metastatic and Castration-Sensitive Prostate Cancer. Oncologist 2020, 25, e1042–e1050. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA Damage Response and Cancer Therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.G.; Boulton, S.J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Piombino, C.; Cortesi, L. Insights into the Possible Molecular Mechanisms of Resistance to PARP Inhibitors. Cancers 2022, 14, 2804. [Google Scholar] [CrossRef] [PubMed]
- Cortesi, L.; Piombino, C.; Toss, A. Germline Mutations in Other Homologous Recombination Repair-Related Genes Than BRCA1/2: Predictive or Prognostic Factors? JPM 2021, 11, 245. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific Killing of BRCA2-Deficient Tumours with Inhibitors of Poly(ADP-Ribose) Polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP Inhibitors: Synthetic Lethality in the Clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food & Drug Administration. FDA Approves Olaparib for HRR Gene-Mutated Metastatic Castration-Resistant Prostate Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-hrr-gene-mutated-metastatic-castration-resistant-prostate-cancer (accessed on 8 March 2024).
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different Roles in a Common Pathway of Genome Protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA Damage Response Pathway: Biomarker and Therapeutic Strategy for Cancer Immunotherapy. Acta. Pharm. Sin. B 2021, 11, 2983–2994. [Google Scholar] [CrossRef] [PubMed]
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA Genes: The Role in Genome Stability, Cancer Stemness and Therapy Resistance. J. Cancer 2019, 10, 2109–2127. [Google Scholar] [CrossRef] [PubMed]
- Ismail, T.; Alzneika, S.; Riguene, E.; Al-maraghi, S.; Alabdulrazzak, A.; Al-Khal, N.; Fetais, S.; Thanassoulas, A.; AlFarsi, H.; Nomikos, M. BRCA1 and Its Vulnerable C-Terminal BRCT Domain: Structure, Function, Genetic Mutations and Links to Diagnosis and Treatment of Breast and Ovarian Cancer. Pharmaceuticals 2024, 17, 333. [Google Scholar] [CrossRef] [PubMed]
- Myler, L.R.; Gallardo, I.F.; Soniat, M.M.; Deshpande, R.A.; Gonzalez, X.B.; Kim, Y.; Paull, T.T.; Finkelstein, I.J. Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair. Mol. Cell 2017, 67, 891–898.e4. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Paull, T.T. Direct Activation of the ATM Protein Kinase by the Mre11/Rad50/Nbs1 Complex. Science 2004, 304, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Cantor, S.B.; Bell, D.W.; Ganesan, S.; Kass, E.M.; Drapkin, R.; Grossman, S.; Wahrer, D.C.; Sgroi, D.C.; Lane, W.S.; Haber, D.A.; et al. BACH1, a Novel Helicase-like Protein, Interacts Directly with BRCA1 and Contributes to Its DNA Repair Function. Cell 2001, 105, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Sy, S.M.H.; Huen, M.S.Y.; Chen, J. PALB2 Is an Integral Component of the BRCA Complex Required for Homologous Recombination Repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef]
- Zhang, F.; Fan, Q.; Ren, K.; Andreassen, P.R. PALB2 Functionally Connects the Breast Cancer Susceptibility Proteins BRCA1 and BRCA2. Mol. Cancer Res. 2009, 7, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Ma, J.; Wu, J.; Ye, L.; Cai, H.; Xia, B.; Yu, X. PALB2 Links BRCA1 and BRCA2 in the DNA-Damage Response. Curr. Biol. 2009, 19, 524–529. [Google Scholar] [CrossRef] [PubMed]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of Eukaryotic Homologous Recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network. 4. 2023 NCCN Clinical Practice Guidelines in Oncology: Prostate Cancer. J. Natl. Compr. Cancer Netw. 2023, 21, 1067–1096. [Google Scholar]
- Catalano, M.; Generali, D.; Gatti, M.; Riboli, B.; Paganini, L.; Nesi, G.; Roviello, G. DNA Repair Deficiency as Circulating Biomarker in Prostate Cancer. Front. Oncol. 2023, 13, 1115241. [Google Scholar] [CrossRef] [PubMed]
- Herzog, T.J.; Vergote, I.; Gomella, L.G.; Milenkova, T.; French, T.; Tonikian, R.; Poehlein, C.; Hussain, M. Testing for Homologous Recombination Repair or Homologous Recombination Deficiency for Poly (ADP-Ribose) Polymerase Inhibitors: A Current Perspective. Eur. J. Cancer 2023, 179, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Foundation Medicine FoundationOne CDx. Available online: https://www.foundationmedicine.com/test/foundationone-cdx (accessed on 17 March 2024).
- Foundation Medicine FoundationOne Liquid CDx. Available online: https://www.foundationmedicine.com/test/foundationone-liquid-cdx (accessed on 17 March 2024).
- Myriad Genetics BRACAnalysis CDx® Germline Companion Diagnostic. Available online: https://myriad.com/genetic-tests/bracanalysiscdx-germline-test/ (accessed on 17 March 2024).
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in Patients with Metastatic Castration-Resistant Prostate Cancer with DNA Repair Gene Aberrations (TOPARP-B): A Multicentre, Open-Label, Randomised, Phase 2 Trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
- de Wit, R.; de Bono, J.; Sternberg, C.N.; Fizazi, K.; Tombal, B.; Wülfing, C.; Kramer, G.; Eymard, J.-C.; Bamias, A.; Carles, J.; et al. Cabazitaxel versus Abiraterone or Enzalutamide in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 2506–2518. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Bianchini, D.; Ileana, E.; Sandhu, S.; Patrikidou, A.; Pezaro, C.; Albiges, L.; Attard, G.; Fizazi, K.; De Bono, J.S.; et al. Antitumour Activity of Abiraterone Acetate against Metastatic Castration-Resistant Prostate Cancer Progressing after Docetaxel and Enzalutamide (MDV3100). Ann. Oncol. 2013, 24, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Oh, W.K.; Cheng, W.Y.; Miao, R.; Vekeman, F.; Gauthier-Loiselle, M.; Duh, M.S.; Drea, E.; Szatrowski, T.P. Real-World Outcomes in Patients with Metastatic Castration-Resistant Prostate Cancer Receiving Second-Line Chemotherapy versus an Alternative Androgen Receptor-Targeted Agent (ARTA) Following Early Progression on a First-Line ARTA in a US Community Oncology Setting. Urol. Oncol. 2018, 36, 500.e1–500.e9. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, D.J.; Annala, M.; Taavitsainen, S.; Finch, D.L.; Oja, C.; Vergidis, J.; Zulfiqar, M.; Sunderland, K.; Azad, A.A.; Kollmannsberger, C.K.; et al. Optimal Sequencing of Enzalutamide and Abiraterone Acetate plus Prednisone in Metastatic Castration-Resistant Prostate Cancer: A Multicentre, Randomised, Open-Label, Phase 2, Crossover Trial. Lancet Oncol. 2019, 20, 1730–1739. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Campbell, D.; Patnaik, A.; Bryce, A.H.; Shapiro, J.; Bambury, R.M.; Zhang, J.; Burke, J.M.; Castellano, D.; Font, A.; et al. Rucaparib for the Treatment of Metastatic Castration-Resistant Prostate Cancer Associated with a DNA Damage Repair Gene Alteration: Final Results from the Phase 2 TRITON2 Study. Eur. Urol. 2023, 84, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Piulats, J.M.; Reaume, M.N.; Ostler, P.; McDermott, R.; Gingerich, J.R.; Pintus, E.; Sridhar, S.S.; Bambury, R.M.; Emmenegger, U.; et al. Rucaparib or Physician’s Choice in Metastatic Prostate Cancer. N. Engl. J. Med. 2023, 388, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 Pathways in DNA Damage Signaling and Cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Brown, L.C.; Antonarakis, E.S.; Armstrong, A.J.; Luo, J. Androgen Receptor Variant-Driven Prostate Cancer II: Advances in Laboratory Investigations. Prostate Cancer Prostatic. Dis. 2020, 23, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Özturan, D.; Morova, T.; Lack, N.A. Androgen Receptor-Mediated Transcription in Prostate Cancer. Cells 2022, 11, 898. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, P.; Di Donato, M.; Giraldi, T.; Migliaccio, A.; Castoria, G.; Auricchio, F. Targeting Rapid Action of Sex-Steroid Receptors in Breast and Prostate Cancers. Front. Biosci. 2012, 4, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Giraldi, T.; Giovannelli, P.; Di Donato, M.; Castoria, G.; Migliaccio, A.; Auricchio, F. Steroid Signaling Activation and Intracellular Localization of Sex Steroid Receptors. J. Cell Commun. Signal. 2010, 4, 161–172. [Google Scholar] [CrossRef]
- Leung, J.K.; Sadar, M.D. Non-Genomic Actions of the Androgen Receptor in Prostate Cancer. Front. Endocrinol. 2017, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Aurilio, G.; Cimadamore, A.; Mazzucchelli, R.; Lopez-Beltran, A.; Verri, E.; Scarpelli, M.; Massari, F.; Cheng, L.; Santoni, M.; Montironi, R. Androgen Receptor Signaling Pathway in Prostate Cancer: From Genetics to Clinical Applications. Cells 2020, 9, 2653. [Google Scholar] [CrossRef] [PubMed]
- Maylin, Z.R.; Nicolescu, R.C.; Pandha, H.; Asim, M. Breaking Androgen Receptor Addiction of Prostate Cancer by Targeting Different Functional Domains in the Treatment of Advanced Disease. Transl. Oncol. 2021, 14, 101115. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.N.; Rathkopf, D.; Smith, M.R.; Efstathiou, E.; Attard, G.; Olmos, D.; Lee, J.Y.; Small, E.J.; Pereira De Santana Gomes, A.J.; Roubaud, G.; et al. Niraparib and Abiraterone Acetate for Metastatic Castration-Resistant Prostate Cancer. JCO 2023, 41, 3339–3351. [Google Scholar] [CrossRef] [PubMed]
- Asim, M.; Tarish, F.; Zecchini, H.I.; Sanjiv, K.; Gelali, E.; Massie, C.E.; Baridi, A.; Warren, A.Y.; Zhao, W.; Ogris, C.; et al. Synthetic Lethality between Androgen Receptor Signalling and the PARP Pathway in Prostate Cancer. Nat. Commun. 2017, 8, 374. [Google Scholar] [CrossRef] [PubMed]
- Schiewer, M.J.; Goodwin, J.F.; Han, S.; Brenner, J.C.; Augello, M.A.; Dean, J.L.; Liu, F.; Planck, J.L.; Ravindranathan, P.; Chinnaiyan, A.M.; et al. Dual Roles of PARP-1 Promote Cancer Growth and Progression. Cancer Discov. 2012, 2, 1134–1149. [Google Scholar] [CrossRef] [PubMed]
- Clarke, N.W.; Armstrong, A.J.; Thiery-Vuillemin, A.; Oya, M.; Shore, N.; Loredo, E.; Procopio, G.; De Menezes, J.; Girotto, G.; Arslan, C.; et al. Abiraterone and Olaparib for Metastatic Castration-Resistant Prostate Cancer. NEJM Evid. 2022, 1. [Google Scholar] [CrossRef]
- Saad, F.; Clarke, N.W.; Oya, M.; Shore, N.; Procopio, G.; Guedes, J.D.; Arslan, C.; Mehra, N.; Parnis, F.; Brown, E.; et al. Olaparib plus Abiraterone versus Placebo plus Abiraterone in Metastatic Castration-Resistant Prostate Cancer (PROpel): Final Prespecified Overall Survival Results of a Randomised, Double-Blind, Phase 3 Trial. Lancet Oncol. 2023, 24, 1094–1108. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, N.; Azad, A.A.; Carles, J.; Fay, A.P.; Matsubara, N.; Heinrich, D.; Szczylik, C.; De Giorgi, U.; Young Joung, J.; Fong, P.C.C.; et al. Talazoparib plus Enzalutamide in Men with First-Line Metastatic Castration-Resistant Prostate Cancer (TALAPRO-2): A Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2023, 402, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Bruin, M.A.C.; Sonke, G.S.; Beijnen, J.H.; Huitema, A.D.R. Pharmacokinetics and Pharmacodynamics of PARP Inhibitors in Oncology. Clin. Pharmacokinet 2022, 61, 1649–1675. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Song, X.-L.; Li, X.-A.; Chen, M.; Guo, J.; Yang, D.-H.; Chen, Z.; Zhao, S.-C. Current Therapy and Drug Resistance in Metastatic Castration-Resistant Prostate Cancer. Drug Resist. Updates 2023, 68, 100962. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to Therapy Caused by Intragenic Deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Goodall, J.; Mateo, J.; Yuan, W.; Mossop, H.; Porta, N.; Miranda, S.; Perez-Lopez, R.; Dolling, D.; Robinson, D.R.; Sandhu, S.; et al. Circulating Cell-Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discov. 2017, 7, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.-Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Frankum, J.R.; Punta, M.; Lise, S.; Alexander, J.; Chen, Y.; Yap, T.A.; Haider, S.; Tutt, A.N.J.; Lord, C.J. Clinical BRCA1/2 Reversion Analysis Identifies Hotspot Mutations and Predicted Neoantigens Associated with Therapy Resistance. Cancer Discov. 2020, 10, 1475–1488. [Google Scholar] [CrossRef] [PubMed]
- Tobalina, L.; Armenia, J.; Irving, E.; O’Connor, M.J.; Forment, J.V. A Meta-Analysis of Reversion Mutations in BRCA Genes Identifies Signatures of DNA End-Joining Repair Mechanisms Driving Therapy Resistance. Ann. Oncol. 2021, 32, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Quigley, D.; Alumkal, J.J.; Wyatt, A.W.; Kothari, V.; Foye, A.; Lloyd, P.; Aggarwal, R.; Kim, W.; Lu, E.; Schwartzman, J.; et al. Analysis of Circulating Cell-Free DNA Identifies Multiclonal Heterogeneity of BRCA2 Reversion Mutations Associated with Resistance to PARP Inhibitors. Cancer Discov. 2017, 7, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; Collier, K.A.; Nagy, R.J.; Pamarthy, S.; Sagar, V.; Fairclough, S.; Odegaard, J.; Lanman, R.B.; Costa, R.; Taxter, T.; et al. Acquired Resistance to Poly (ADP-Ribose) Polymerase Inhibitor Olaparib in BRCA2 -Associated Prostate Cancer Resulting from Biallelic BRCA2 Reversion Mutations Restores Both Germline and Somatic Loss-of-Function Mutations. JCO Precis. Oncol. 2018, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.H.; Salipante, S.J.; Nelson, P.S.; Montgomery, B.; Pritchard, C.C. Polyclonal BRCA2 Reversion Mutations Detected in Circulating Tumor DNA After Platinum Chemotherapy in a Patient with Metastatic Prostate Cancer. JCO Precis. Oncol. 2018, 2, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Simmons, A.D.; Nguyen, M.; Pintus, E. Polyclonal BRCA2 Mutations Following Carboplatin Treatment Confer Resistance to the PARP Inhibitor Rucaparib in a Patient with mCRPC: A Case Report. BMC Cancer 2020, 20, 215. [Google Scholar] [CrossRef] [PubMed]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.-Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of All BRCA1 Copies Predicts Response to the PARP Inhibitor Rucaparib in Ovarian Carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef]
- D’Andrea, A.D. Mechanisms of PARP Inhibitor Sensitivity and Resistance. DNA Repair. 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Mekonnen, N.; Yang, H.; Shin, Y.K. Homologous Recombination Deficiency in Ovarian, Breast, Colorectal, Pancreatic, Non-Small Cell Lung and Prostate Cancers, and the Mechanisms of Resistance to PARP Inhibitors. Front. Oncol. 2022, 12, 880643. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-Wide and High-Density CRISPR-Cas9 Screens Identify Point Mutations in PARP1 Causing PARP Inhibitor Resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.K.; Matulonis, U.A. PARP Inhibitor Resistance Mechanisms and Implications for Post-Progression Combination Therapies. Cancers 2020, 12, 2054. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.N.; Chowdhury, S.; Bjartell, A.; Chung, B.H.; Pereira De Santana Gomes, A.J.; Given, R.; Juárez, A.; Merseburger, A.S.; Özgüroğlu, M.; Uemura, H.; et al. Apalutamide in Patients with Metastatic Castration-Sensitive Prostate Cancer: Final Survival Analysis of the Randomized, Double-Blind, Phase III TITAN Study. JCO 2021, 39, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone Acetate plus Prednisone in Patients with Newly Diagnosed High-Risk Metastatic Castration-Sensitive Prostate Cancer (LATITUDE): Final Overall Survival Analysis of a Randomised, Double-Blind, Phase 3 Trial. Lancet Oncol. 2019, 20, 686–700. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Szmulewitz, R.Z.; Petrylak, D.P.; Holzbeierlein, J.; Villers, A.; Azad, A.; Alcaraz, A.; Alekseev, B.; Iguchi, T.; Shore, N.D.; et al. ARCHES: A Randomized, Phase III Study of Androgen Deprivation Therapy with Enzalutamide or Placebo in Men With Metastatic Hormone-Sensitive Prostate Cancer. JCO 2019, 37, 2974–2986. [Google Scholar] [CrossRef] [PubMed]
- Klein Douwel, D.; Boonen, R.A.C.M.; Long, D.T.; Szypowska, A.A.; Räschle, M.; Walter, J.C.; Knipscheer, P. XPF-ERCC1 Acts in Unhooking DNA Interstrand Crosslinks in Cooperation with FANCD2 and FANCP/SLX4. Mol. Cell 2014, 54, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Jdey, W.; Kozlak, M.; Alekseev, S.; Thierry, S.; Lascaux, P.; Girard, P.-M.; Bono, F.; Dutreix, M. AsiDNA Treatment Induces Cumulative Antitumor Efficacy with a Low Probability of Acquired Resistance. Neoplasia 2019, 21, 863–871. [Google Scholar] [CrossRef]
- Murai, J.; Yang, K.; Dejsuphong, D.; Hirota, K.; Takeda, S.; D’Andrea, A.D. The USP1/UAF1 Complex Promotes Double-Strand Break Repair through Homologous Recombination. Mol. Cell Biol. 2011, 31, 2462–2469. [Google Scholar] [CrossRef] [PubMed]
- Gargalionis, A.N.; Papavassiliou, K.A.; Papavassiliou, A.G. The Potential of BRD4 Inhibition in Tumour Mechanosignaling. J. Cell Mol. Med. 2023, 27, 4215–4218. [Google Scholar] [CrossRef]
- Pegg, N.; Brooks, N.; Worthington, J.; Young, B.; Prosser, A.; Lane, J.; Taddei, D.; Brown, R.; Harbottle, G.; Shannon, J.; et al. Characterisation of CCS1477: A Novel Small Molecule Inhibitor of P300/CBP for the Treatment of Castration Resistant Prostate Cancer. JCO 2017, 35, 11590. [Google Scholar] [CrossRef]
- Peyraud, F.; Italiano, A. Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers 2020, 12, 1502. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Study | Treatment Arms | Median Follow-Up | Median rPFS in ITT Population | HR (95%CI) | Median rPFS in HRD Group | HR (95%CI) | Tests to Determine HRR Status |
|---|---|---|---|---|---|---|---|
| PROpel | Abiraterone + olaparib Abiraterone + placebo | 19.3 mo 19.4 mo | 24.8 mo 16.6 mo | 0.66 (0.54–0.81) | NR 13.9 mo | 0.5 (0.34–0.73) | Tissue and ctDNA (FoundationOne CDx) |
| TALAPRO-2 | Enzalutamide + talazoparib Enzalutamide + placebo | 24.9 mo 24.6 mo | NR 21.9 mo | 0.63 (0.51–0.78) | 27.9 mo 16.4 mo | 0.46 (0.30–0.70) | Tissue and ctDNA (FoundationOne CDx) |
| MAGNITUDE * | Enzalutamide + niraparib Enzalutamide + placebo | 18.6 mo 18.6 mo | - - | - - | 16.5 mo 13.7 mo | 0.73 (0.56–0.96) | Tissue and ctDNA (FoundationOne CDx, Resolution HRD, AmoyDx) |
| Official Title NCT Number | Setting | Experimental Arm | Control Arm | Primary Endpoints | Status | Enrolment | Primary Completion |
|---|---|---|---|---|---|---|---|
| CASPAR NCT04455750 | I-line mCRPC, regardless of HRR status | Enzalutamide + rucaparib | Enzalutamide + placebo | rPFS, OS | Active, not recruiting | 61 (actual) | May 2024 |
| TALAPRO-3 NCT04821622 | I-line mHSPC with HRD | Enzalutamide + talazoparib | Enzalutamide + placebo | rPFS | Active, not recruiting | 599 (actual) | September 2025 |
| AMPLITUDE NCT04497844 | I-line mHSPC with HRD | Abiraterone + niraparib | Abiraterone + placebo | rPFS | Active, not recruiting | 696 (actual) | November 2024 |
| NCT Number | Setting | Treatment | Primary Endpoints | Status | Enrolment | Primary Completion |
|---|---|---|---|---|---|---|
| NCT05501548 | ≥II-line mCRPC HRR-proficient | Olaparib + ascorbate | PSA50 | Recruiting | 15 (estimated) | March 2028 |
| TRAP trial NCT03787680 | ≥II-line mCRPC, regardless of HRR status | Olaparib + ceralasertib | ORR in HRR-proficient patients | Active, not recruiting | 49 (actual) | January 2023 |
| FAALCON NCT04748042 | Oligometastatic HSPC | Olaparib + abiraterone + radiotherapy | Percentage of patients without treatment failure at 24 months | Recruiting | 29 (estimated) | May 2025 |
| NCT03263650 | Aggressive variants of metastatic PC | Olaparib maintenance after six cycles of cabazitaxel and carboplatin | PFS | Active, not recruiting | 119 (actual) | June 2024 |
| NCT02893917 | >II-line mCRPC | Olaparib ± cediranib | rPFS | Active, not recruiting | 90 (estimated) | December 2023 |
| NCT05167175 | I-line mHSPC with HRD | Olaparib + abiraterone | rPFS | Recruiting | 30 (estimated) | December 2024 |
| NCT05005728 Cohort C | ≥II-line mCRPC with HRD/CDK12 biallelic loss tumors | Olaparib + vudalimab | Incidence of treatment-related AEs | Recruiting | 85 * (estimated) | June 2023 |
| NCT05700669 mCRPC cohort | mCRPC progressed to previous PARPi | Olaparib + AsiDNA | ORR | Recruiting | 115 * (estimated) | December 2026 |
| NCT05252390 mCRPC cohort | mCRPC progressed to a previous ARSI | NUV-868 ± olaparib or enzalutamide | ORR, PSA50, rPFS | Recruiting | 657 * (estimated) | June 2026 |
| NCT03682289 Cohort D | mCRPC progressed to a previous ARSI | Ceralasertib ± olaparib or durvalumab | ORR, PSA50 | Recruiting | 89 * (estimated) | July 2025 |
| NCT06065059 PC cohort | Metastatic PC BRCA1/2 mutant or with HRD | TNG348 ± Olaparib | ORR | Recruiting | 140 * (estimated) | December 2025 |
| NCT03568656 mCRPC cohort | mCRPC progressed to a previous ARSI and docetaxel | CCS1477 ± olaparib or abiraterone or enzalutamide or darolutamide | Incidence of treatment-related AEs, laboratory assessments | Recruiting | 350 * (estimated) | March 2024 |
| NCT02484404 Cohort 4 | mCRPC progressed to a previous ARSI and/or docetaxel | Olaparib + durvalumab | ORR, AEs, PSA response | Recruiting | 384 * (estimated) | December 2024 |
| NCT04332744 | I-line high-volume mHSPC | Enzalutamide ± talazoparib | PSA-CR | Active, not recruiting | 54 (actual) | April 2025 |
| NCT04734730 | I-line mHSPC | Talazoparib + abiraterone | PSA-CR | Recruiting | 70 (estimated) | August 2027 |
| NCT04019327 | mCRPC without DDR mutations progressed to a previous ARSI | Talazoparib + temozolomide | ORR | Recruiting | 44 (estimated) | July 2027 |
| NCT04550494 PC cohort | Metastatic PC with DDR mutations | Talazoparib | Rate of patients with Rad51 activation | Recruiting | 30 * (estimated) | December 2024 |
| KNIGHTS NCT06212583 | Recurrent oligometastatic HSPC with high-risk DDR mutations | Radiotherapy ± niraparib and abiraterone | PSA at the 18-month progression | Not yet recruiting | 88 (estimated) | December 2028 |
| NCT04592237 | Aggressive variants of metastatic PC | Niraparib ± cetrelimab maintenance after six cycles of cabazitaxel + carboplatin + cetrelimab | PFS | Recruiting | 120 (estimated) | December 2025 |
| NCT05689021 | mCRPC with SPOP mutations progressed to a previous ARSI | Niraparib + abiraterone | PSA50 | Recruiting | 30 (estimated) | September 2024 |
| PLATPARP NCT04288687 | Platinum-sensitive mCRPC with DDR mutations | Niraparib maintenance | 6-month rPFS | Active, not recruiting | 12 (actual) | June 2024 |
| TRIUMPH NCT03413995 | mHSPC with HRD in patients refusing ADT | Rucaparib | PSA50 | Active, not recruiting | 30 (estimated) | November 2023 |
| NCT04253262 | mCRPC progressed to a previous ARSI with HRD | Rucaparib + Copanlisib | ORR | Active, not recruiting | 13 (actual) | January 2024 |
| PLATI-PARP NCT03442556 | mCRPC with HRD | Rucaparib maintenance after 4 cycles of docetaxel and carboplatin | rPFS | Active, not recruiting | 18 (actual) | May 2025 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piombino, C.; Pipitone, S.; Tonni, E.; Mastrodomenico, L.; Oltrecolli, M.; Tchawa, C.; Matranga, R.; Roccabruna, S.; D’Agostino, E.; Pirola, M.; et al. Homologous Recombination Repair Deficiency in Metastatic Prostate Cancer: New Therapeutic Opportunities. Int. J. Mol. Sci. 2024, 25, 4624. https://doi.org/10.3390/ijms25094624
Piombino C, Pipitone S, Tonni E, Mastrodomenico L, Oltrecolli M, Tchawa C, Matranga R, Roccabruna S, D’Agostino E, Pirola M, et al. Homologous Recombination Repair Deficiency in Metastatic Prostate Cancer: New Therapeutic Opportunities. International Journal of Molecular Sciences. 2024; 25(9):4624. https://doi.org/10.3390/ijms25094624
Chicago/Turabian StylePiombino, Claudia, Stefania Pipitone, Elena Tonni, Luciana Mastrodomenico, Marco Oltrecolli, Cyrielle Tchawa, Rossana Matranga, Sara Roccabruna, Elisa D’Agostino, Marta Pirola, and et al. 2024. "Homologous Recombination Repair Deficiency in Metastatic Prostate Cancer: New Therapeutic Opportunities" International Journal of Molecular Sciences 25, no. 9: 4624. https://doi.org/10.3390/ijms25094624
APA StylePiombino, C., Pipitone, S., Tonni, E., Mastrodomenico, L., Oltrecolli, M., Tchawa, C., Matranga, R., Roccabruna, S., D’Agostino, E., Pirola, M., Bacchelli, F., Baldessari, C., Baschieri, M. C., Dominici, M., Sabbatini, R., & Vitale, M. G. (2024). Homologous Recombination Repair Deficiency in Metastatic Prostate Cancer: New Therapeutic Opportunities. International Journal of Molecular Sciences, 25(9), 4624. https://doi.org/10.3390/ijms25094624