
Circadian Clock in Muscle Disease Etiology and Therapeutic Potential for Duchenne Muscular Dystrophy

Abstract
:1. Introduction
2. Circadian Clock Regulation of Skeletal Muscle Structure and Function
3. Circadian Clock in Muscle Stem Cell Biology
4. Circadian Clock Dysregulation in Muscle Disease Etiology
4.1. Involvement of Clock Dysfunction in Muscle Diseases
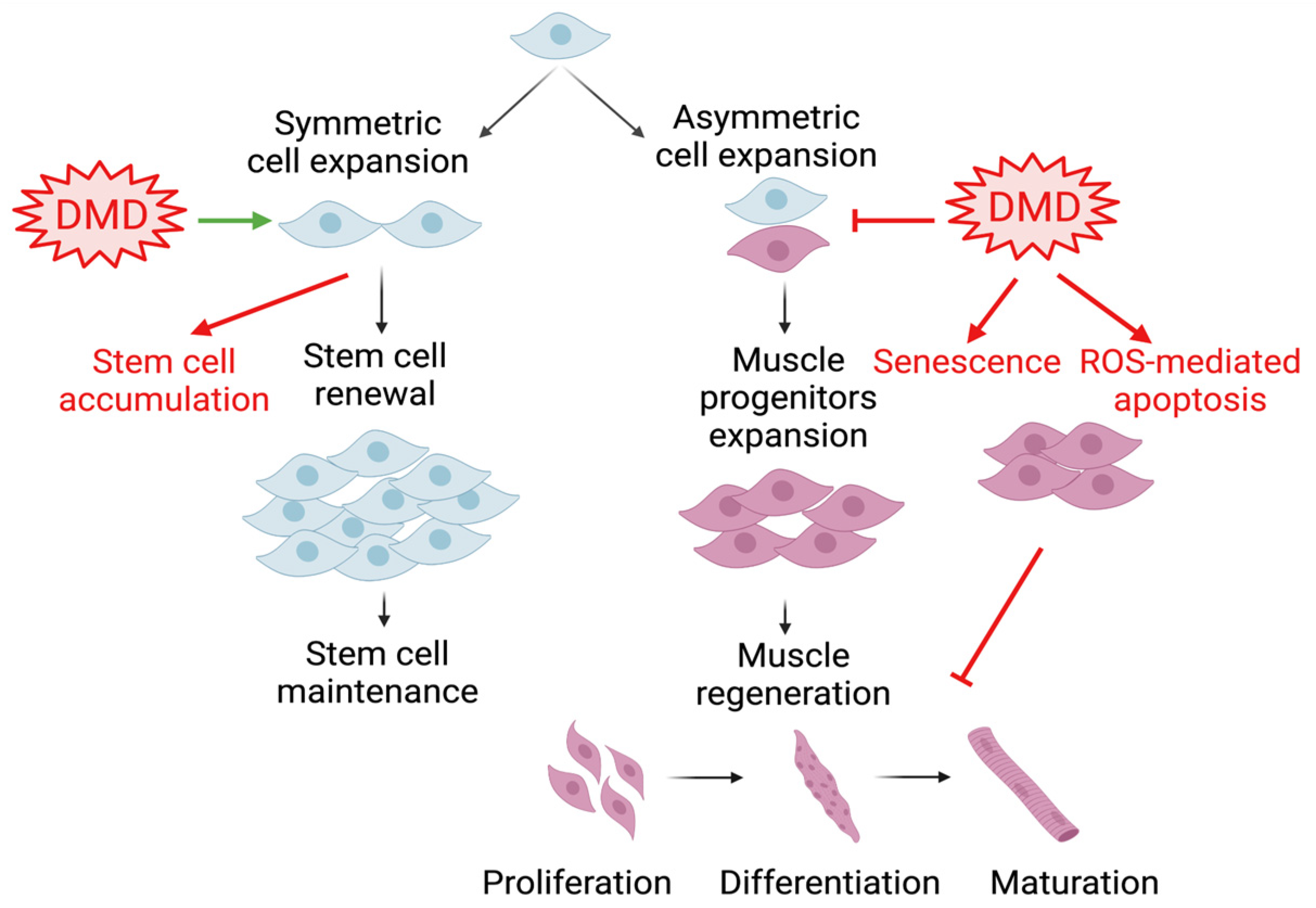
4.2. The Role of the Circadian Clock in Muscular Dystrophy Etiology and Pathogenesis
5. Circadian Clock as a Potential Drug Target for Duchenne Muscular Dystrophy
5.1. Current Research and Therapies for DMD
5.2. Recent Progress in Discovering Circadian Clock Modulators with Potential Applications for DMD
6. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schibler, U.; Sassone-Corsi, P. A web of circadian pacemakers. Cell 2002, 111, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Dibner, C.; Schibler, U.; Albrecht, U. The mammalian circadian timing system: Organization and coordination of central and peripheral clocks. Annu. Rev. Physiol. 2010, 72, 517–549. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, S.; Kannan, N.N. Only time will tell: The interplay between circadian clock and metabolism. Chronobiol. Int. 2021, 38, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Battaglin, F.; Chan, P.; Pan, Y.; Soni, S.; Qu, M.; Spiller, E.R.; Castanon, S.; Roussos Torres, E.T.; Mumenthaler, S.M.; Kay, S.A.; et al. Clocking cancer: The circadian clock as a target in cancer therapy. Oncogene 2021, 40, 3187–3200. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Zhang, G.; Qu, M.; Gimple, R.C.; Wu, Q.; Qiu, Z.; Prager, B.C.; Wang, X.; Kim, L.J.Y.; Morton, A.R.; et al. Targeting Glioblastoma Stem Cells through Disruption of the Circadian Clock. Cancer Discov. 2019, 9, 1556–1573. [Google Scholar] [CrossRef] [PubMed]
- Oshima, T.; Niwa, Y.; Kuwata, K.; Srivastava, A.; Hyoda, T.; Tsuchiya, Y.; Kumagai, M.; Tsuyuguchi, M.; Tamaru, T.; Sugiyama, A.; et al. Cell-based screen identifies a new potent and highly selective CK2 inhibitor for modulation of circadian rhythms and cancer cell growth. Sci. Adv. 2019, 5, eaau9060. [Google Scholar] [CrossRef]
- Sulli, G.; Rommel, A.; Wang, X.; Kolar, M.J.; Puca, F.; Saghatelian, A.; Plikus, M.V.; Verma, I.M.; Panda, S. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature 2018, 553, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Sulli, G.; Lam, M.T.Y.; Panda, S. Interplay between Circadian Clock and Cancer: New Frontiers for Cancer Treatment. Trends Cancer 2019, 5, 475–494. [Google Scholar] [CrossRef]
- Takahashi, J.S.; Hong, H.K.; Ko, C.H.; McDearmon, E.L. The genetics of mammalian circadian order and disorder: Implications for physiology and disease. Nat. Rev. Genet. 2008, 9, 764–775. [Google Scholar] [CrossRef]
- Sulli, G.; Manoogian, E.N.C.; Taub, P.R.; Panda, S. Training the Circadian Clock, Clocking the Drugs, and Drugging the Clock to Prevent, Manage, and Treat Chronic Diseases. Trends Pharmacol. Sci. 2018, 39, 812–827. [Google Scholar] [CrossRef]
- Kiperman, T.; Li, W.; Xiong, X.; Li, H.; Horne, D.; Ma, K. Targeted screening and identification of chlorhexidine as a pro-myogenic circadian clock activator. Stem Cell Res. Ther. 2023, 14, 190. [Google Scholar] [CrossRef] [PubMed]
- Schroder, E.A.; Harfmann, B.D.; Zhang, X.; Srikuea, R.; England, J.H.; Hodge, B.A.; Wen, Y.; Riley, L.A.; Yu, Q.; Christie, A.; et al. Intrinsic muscle clock is necessary for musculoskeletal health. J. Physiol. 2015, 593, 5387–5404. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2017, 18, 164–179. [Google Scholar] [CrossRef] [PubMed]
- Harmer, S.L.; Panda, S.; Kay, S.A. Molecular bases of circadian rhythms. Annu. Rev. Cell Dev. Biol. 2001, 17, 215–253. [Google Scholar] [CrossRef] [PubMed]
- Rey, G.; Cesbron, F.; Rougemont, J.; Reinke, H.; Brunner, M.; Naef, F. Genome-wide and phase-specific DNA-binding rhythms of BMAL1 control circadian output functions in mouse liver. PLoS Biol. 2011, 9, e1000595. [Google Scholar] [CrossRef]
- McCarthy, J.J.; Andrews, J.L.; McDearmon, E.L.; Campbell, K.S.; Barber, B.K.; Miller, B.H.; Walker, J.R.; Hogenesch, J.B.; Takahashi, J.S.; Esser, K.A. Identification of the circadian transcriptome in adult mouse skeletal muscle. Physiol. Genom. 2007, 31, 86–95. [Google Scholar] [CrossRef]
- Miller, B.H.; McDearmon, E.L.; Panda, S.; Hayes, K.R.; Zhang, J.; Andrews, J.L.; Antoch, M.P.; Walker, J.R.; Esser, K.A.; Hogenesch, J.B.; et al. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc. Natl. Acad. Sci. USA 2007, 104, 3342–3347. [Google Scholar] [CrossRef]
- Andrews, J.L.; Zhang, X.; McCarthy, J.J.; McDearmon, E.L.; Hornberger, T.A.; Russell, B.; Campbell, K.S.; Arbogast, S.; Reid, M.B.; Walker, J.R.; et al. CLOCK and BMAL1 regulate MyoD and are necessary for maintenance of skeletal muscle phenotype and function. Proc. Natl. Acad. Sci. USA 2010, 107, 19090–19095. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building muscle: Molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008342. [Google Scholar] [CrossRef]
- Chatterjee, S.; Ma, K. Circadian clock regulation of skeletal muscle growth and repair. F1000Research 2016, 5, 1549. [Google Scholar] [CrossRef]
- Martin, R.A.; Viggars, M.R.; Esser, K.A. Metabolism and exercise: The skeletal muscle clock takes centre stage. Nat. Rev. Endocrinol. 2023, 19, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Harfmann, B.D.; Schroder, E.A.; Esser, K.A. Circadian rhythms, the molecular clock, and skeletal muscle. J. Biol. Rhythm. 2015, 30, 84–94. [Google Scholar] [CrossRef] [PubMed]
- McDearmon, E.L.; Patel, K.N.; Ko, C.H.; Walisser, J.A.; Schook, A.C.; Chong, J.L.; Wilsbacher, L.D.; Song, E.J.; Hong, H.; Bradfield, C.A.; et al. Dissecting the Functions of the Mammalian Clock Protein BMAL1 by Tissue-Specific Rescue in Mice. Science 2006, 314, 1304–1308. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Nam, D.; Guo, B.; Kim, J.M.; Winnier, G.E.; Lee, J.; Berdeaux, R.; Yechoor, V.K.; Ma, K. Brain and muscle Arnt-like 1 is a key regulator of myogenesis. J. Cell Sci. 2013, 126, 2213–2224. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Yin, H.; Nam, D.; Li, Y.; Ma, K. Brain and muscle Arnt-like 1 promotes skeletal muscle regeneration through satellite cell expansion. Exp. Cell Res. 2015, 331, 200–210. [Google Scholar] [CrossRef]
- Pastore, S.; Hood, D.A. Endurance training ameliorates the metabolic and performance characteristics of circadian Clock mutant mice. J. Appl. Physiol. 2013, 114, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Xue, T.; Liu, S.B.; Geng, S.; Shi, X.; Qian, P.; He, W.; Zheng, J.; Li, Y.; Lou, J.; et al. Loss of CRY2 promotes regenerative myogenesis by enhancing PAX7 expression and satellite cell proliferation. MedComm 2023, 4, e202. [Google Scholar] [CrossRef] [PubMed]
- Katoku-Kikyo, N.; Paatela, E.; Houtz, D.L.; Lee, B.; Munson, D.; Wang, X.; Hussein, M.; Bhatia, J.; Lim, S.; Yuan, C.; et al. Per1/Per2-Igf2 axis-mediated circadian regulation of myogenic differentiation. J. Cell Biol. 2021, 220, e202101057. [Google Scholar] [CrossRef] [PubMed]
- Welch, R.D.; Billon, C.; Valfort, A.C.; Burris, T.P.; Flaveny, C.A. Pharmacological inhibition of REV-ERB stimulates differentiation, inhibits turnover and reduces fibrosis in dystrophic muscle. Sci. Rep. 2017, 7, 17142. [Google Scholar] [CrossRef]
- Lowe, M.; Lage, J.; Paatela, E.; Munson, D.; Hostager, R.; Yuan, C.; Katoku-Kikyo, N.; Ruiz-Estevez, M.; Asakura, Y.; Staats, J.; et al. Cry2 Is Critical for Circadian Regulation of Myogenic Differentiation by Bclaf1-Mediated mRNA Stabilization of Cyclin D1 and Tmem176b. Cell Rep. 2018, 22, 2118–2132. [Google Scholar] [CrossRef]
- Egan, B.; Zierath, J.R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.; Korf, H.W.; Kuffer, L.; Gross, J.V.; Erren, T.C. Exercise time cues (zeitgebers) for human circadian systems can foster health and improve performance: A systematic review. BMJ Open Sport Exerc. Med. 2018, 4, e000443. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Basse, A.L.; Schonke, M.; Chen, S.; Samad, M.; Altintas, A.; Laker, R.C.; Dalbram, E.; Barres, R.; Baldi, P.; et al. Time of Exercise Specifies the Impact on Muscle Metabolic Pathways and Systemic Energy Homeostasis. Cell Metab. 2019, 30, 92–110 e114. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, A.; Davenne, D.; Gentil, C.; Van Hoecke, J. Circadian rhythm in the torque developed by elbow flexors during isometric contraction. Effect of sampling schedules. Chronobiol. Int. 1997, 14, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, A.; Davenne, D.; Martin, A.; Cometti, G.; Van Hoecke, J. Diurnal rhythm of the muscular performance of elbow flexors during isometric contractions. Chronobiol. Int. 1996, 13, 135–146. [Google Scholar] [CrossRef]
- Gauthier, A.; Davenne, D.; Martin, A.; Van Hoecke, J. Time of day effects on isometric and isokinetic torque developed during elbow flexion in humans. Eur. J. Appl. Physiol. 2001, 84, 249–252. [Google Scholar] [CrossRef]
- Gifford, L.S. Circadian variation in human flexibility and grip strength. Aust. J. Physiother. 1987, 33, 3–9. [Google Scholar] [CrossRef]
- Lucas, S.J.; Anson, J.G.; Palmer, C.D.; Hellemans, I.J.; Cotter, J.D. The impact of 100 hours of exercise and sleep deprivation on cognitive function and physical capacities. J. Sports Sci. 2009, 27, 719–728. [Google Scholar] [CrossRef]
- Reilly, T.; Piercy, M. The effect of partial sleep deprivation on weight-lifting performance. Ergonomics 1994, 37, 107–115. [Google Scholar] [CrossRef]
- Aisbett, B.; Condo, D.; Zacharewicz, E.; Lamon, S. The Impact of Shiftwork on Skeletal Muscle Health. Nutrients 2017, 9, 248. [Google Scholar] [CrossRef]
- Bae, K.; Lee, K.; Seo, Y.; Lee, H.; Kim, D.; Choi, I. Differential effects of two period genes on the physiology and proteomic profiles of mouse anterior tibialis muscles. Mol. Cells 2006, 22, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Woldt, E.; Sebti, Y.; Solt, L.A.; Duhem, C.; Lancel, S.; Eeckhoute, J.; Hesselink, M.K.; Paquet, C.; Delhaye, S.; Shin, Y.; et al. Rev-erb-alpha modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy. Nat. Med. 2013, 19, 1039–1046. [Google Scholar] [CrossRef]
- Charge, S.B.; Rudnicki, M.A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004, 84, 209–238. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493. [Google Scholar] [CrossRef]
- Feige, P.; Tsai, E.C.; Rudnicki, M.A. Analysis of human satellite cell dynamics on cultured adult skeletal muscle myofibers. Skelet. Muscle 2021, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Zammit, P.S.; Heslop, L.; Hudon, V.; Rosenblatt, J.D.; Tajbakhsh, S.; Buckingham, M.E.; Beauchamp, J.R.; Partridge, T.A. Kinetics of myoblast proliferation show that resident satellite cells are competent to fully regenerate skeletal muscle fibers. Exp. Cell Res. 2002, 281, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Gayraud-Morel, B.; Chrétien, F.; Flamant, P.; Gomès, D.; Zammit, P.S.; Tajbakhsh, S. A role for the myogenic determination gene Myf5 in adult regenerative myogenesis. Dev. Biol. 2007, 312, 13–28. [Google Scholar] [CrossRef]
- Ganassi, M.; Muntoni, F.; Zammit, P.S. Defining and identifying satellite cell-opathies within muscular dystrophies and myopathies. Exp. Cell Res. 2022, 411, 112906. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Wang, Y.X.; Rudnicki, M.A. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 2015, 142, 1572–1581. [Google Scholar] [CrossRef]
- Chang, N.C.; Chevalier, F.P.; Rudnicki, M.A. Satellite Cells in Muscular Dystrophy—Lost in Polarity. Trends Mol. Med. 2016, 22, 479–496. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Kodippili, K.; Rudnicki, M.A. Satellite cell contribution to disease pathology in Duchenne muscular dystrophy. Front. Physiol. 2023, 14, 1180980. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Yin, H.; Li, W.; Lee, J.; Yechoor, V.K.; Ma, K. The Nuclear Receptor and Clock Repressor Rev-erbalpha Suppresses Myogenesis. Sci. Rep. 2019, 9, 4585. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Cai, Y.; Gao, Z.; Chen, X.; Liu, B.; Zhang, C.; Yu, W.; Cao, Q.; Shen, Y.; Yao, X.; et al. Duchenne muscular dystrophy: Pathogenesis and promising therapies. J. Neurol. 2023, 270, 3733–3749. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Gao, H.; Lin, Y.; Yechoor, V.; Ma, K. Inhibition of Rev-erbalpha ameliorates muscular dystrophy. Exp. Cell Res. 2021, 406, 112766. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Xiong, X.; Lin, Y.; Chatterjee, S.; Ma, K. The clock regulator Bmal1 protects against muscular dystrophy. Exp. Cell Res. 2020, 397, 112348. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Hamlish, N.X.; Thakkar, A.V.; Steffeck, A.W.T.; Rendleman, E.J.; Khan, N.H.; Waldeck, N.J.; DeVilbiss, A.W.; Martin-Sandoval, M.S.; Mathews, T.P.; et al. BMAL1 drives muscle repair through control of hypoxic NAD+ regeneration in satellite cells. Genes Dev. 2022, 36, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Lou, J.; Li, Y.; Qian, P.; He, W.; Hao, Y.; Xue, T.; Li, Y.; Song, Y.H. Satellite cell-specific deletion of Cipc alleviates myopathy in mdx mice. Cell Rep. 2022, 39, 110939. [Google Scholar] [CrossRef]
- Welch, R.D.; Guo, C.; Sengupta, M.; Carpenter, K.J.; Stephens, N.A.; Arnett, S.A.; Meyers, M.J.; Sparks, L.M.; Smith, S.R.; Zhang, J.; et al. Rev-Erb co-regulates muscle regeneration via tethered interaction with the NF-Y cistrome. Mol. Metab. 2017, 6, 703–714. [Google Scholar] [CrossRef]
- Siddle, K. Signalling by insulin and IGF receptors: Supporting acts and new players. J. Mol. Endocrinol. 2011, 47, R1–R10. [Google Scholar] [CrossRef]
- Segales, J.; Islam, A.B.; Kumar, R.; Liu, Q.C.; Sousa-Victor, P.; Dilworth, F.J.; Ballestar, E.; Perdiguero, E.; Munoz-Canoves, P. Chromatin-wide and transcriptome profiling integration uncovers p38alpha MAPK as a global regulator of skeletal muscle differentiation. Skelet. Muscle 2016, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Segales, J.; Perdiguero, E.; Munoz-Canoves, P. Regulation of Muscle Stem Cell Functions: A Focus on the p38 MAPK Signaling Pathway. Front. Cell Dev. Biol. 2016, 4, 91. [Google Scholar] [CrossRef] [PubMed]
- Yamajuku, D.; Shibata, Y.; Kitazawa, M.; Katakura, T.; Urata, H.; Kojima, T.; Nakata, O.; Hashimoto, S. Identification of functional clock-controlled elements involved in differential timing of Per1 and Per2 transcription. Nucleic Acids Res. 2010, 38, 7964–7973. [Google Scholar] [CrossRef]
- Bez Batti Angulski, A.; Hosny, N.; Cohen, H.; Martin, A.A.; Hahn, D.; Bauer, J.; Metzger, J.M. Duchenne muscular dystrophy: Disease mechanism and therapeutic strategies. Front. Physiol. 2023, 14, 1183101. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Chen, Z.; Yu, Y.; Zhang, N.; Jiang, H.; Zhang, G.; Zhang, Z.; Zhang, B. Current Pharmacological Strategies for Duchenne Muscular Dystrophy. Front. Cell Dev. Biol. 2021, 9, 689533. [Google Scholar] [CrossRef]
- Beytia Mde, L.; Vry, J.; Kirschner, J. Drug treatment of Duchenne muscular dystrophy: Available evidence and perspectives. Acta Myol. 2012, 31, 4–8. [Google Scholar]
- Zhang, H.; Liang, J.; Chen, N. Do not neglect the role of circadian rhythm in muscle atrophy. Ageing Res. Rev. 2020, 63, 101155. [Google Scholar] [CrossRef]
- Reeds, P.J.; Palmer, R.M.; Hay, S.M.; McMillan, D.N. Protein synthesis in skeletal muscle measured at different times during a 24 hour period. Biosci. Rep. 1986, 6, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Camera, D.M. Anabolic Heterogeneity Following Resistance Training: A Role for Circadian Rhythm? Front. Physiol. 2018, 9, 569. [Google Scholar] [CrossRef]
- Chang, S.W.; Yoshihara, T.; Machida, S.; Naito, H. Circadian rhythm of intracellular protein synthesis signaling in rat cardiac and skeletal muscles. Biochem. Biophys. Rep. 2017, 9, 153–158. [Google Scholar] [CrossRef]
- Huang, G.; Zhang, F.; Ye, Q.; Wang, H. The circadian clock regulates autophagy directly through the nuclear hormone receptor Nr1d1/Rev-erbalpha and indirectly via Cebpb/(C/ebpbeta) in zebrafish. Autophagy 2016, 12, 1292–1309. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, S.; Kojima, S.; Sasaki, K.; Ishikawa, R.; Tanaka, M.; Shimoda, T.; Hattori, Y.; Aoki, N.; Takahashi, K.; Hirooka, R.; et al. Day-Night Oscillation of Atrogin1 and Timing-Dependent Preventive Effect of Weight-Bearing on Muscle Atrophy. eBioMedicine 2018, 37, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Mayeuf-Louchart, A.; Thorel, Q.; Delhaye, S.; Beauchamp, J.; Duhem, C.; Danckaert, A.; Lancel, S.; Pourcet, B.; Woldt, E.; Boulinguiez, A.; et al. Rev-erb-alpha regulates atrophy-related genes to control skeletal muscle mass. Sci. Rep. 2017, 7, 14383. [Google Scholar] [CrossRef]
- Neufeld-Cohen, A.; Robles, M.S.; Aviram, R.; Manella, G.; Adamovich, Y.; Ladeuix, B.; Nir, D.; Rousso-Noori, L.; Kuperman, Y.; Golik, M.; et al. Circadian control of oscillations in mitochondrial rate-limiting enzymes and nutrient utilization by PERIOD proteins. Proc. Natl. Acad. Sci. USA 2016, 113, E1673–E1682. [Google Scholar] [CrossRef]
- Schmitt, K.; Grimm, A.; Dallmann, R.; Oettinghaus, B.; Restelli, L.M.; Witzig, M.; Ishihara, N.; Mihara, K.; Ripperger, J.A.; Albrecht, U.; et al. Circadian Control of DRP1 Activity Regulates Mitochondrial Dynamics and Bioenergetics. Cell Metab. 2018, 27, 657–666.e5. [Google Scholar] [CrossRef]
- Jacobi, D.; Liu, S.; Burkewitz, K.; Kory, N.; Knudsen, N.H.; Alexander, R.K.; Unluturk, U.; Li, X.; Kong, X.; Hyde, A.L.; et al. Hepatic Bmal1 Regulates Rhythmic Mitochondrial Dynamics and Promotes Metabolic Fitness. Cell Metab. 2015, 22, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.L.; Abt, M.C.; Gasteiger, G.; Firth, M.A.; O’Connor, M.H.; Geary, C.D.; O’Sullivan, T.E.; van den Brink, M.R.; Pamer, E.G.; Hanash, A.M.; et al. Nfil3 is crucial for development of innate lymphoid cells and host protection against intestinal pathogens. J. Exp. Med. 2014, 211, 1723–1731. [Google Scholar] [CrossRef]
- Nakao, R.; Yamamoto, S.; Horikawa, K.; Yasumoto, Y.; Nikawa, T.; Mukai, C.; Oishi, K. Atypical expression of circadian clock genes in denervated mouse skeletal muscle. Chronobiol. Int. 2015, 32, 486–496. [Google Scholar] [CrossRef]
- Wu, R.; Dang, F.; Li, P.; Wang, P.; Xu, Q.; Liu, Z.; Li, Y.; Wu, Y.; Chen, Y.; Liu, Y. The Circadian Protein Period2 Suppresses mTORC1 Activity via Recruiting Tsc1 to mTORC1 Complex. Cell Metab. 2019, 29, 653–667 e656. [Google Scholar] [CrossRef]
- Larsson, L.A.-O.X.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef]
- Vitale, J.A.; Bonato, M.; La Torre, A.; Banfi, G. The Role of the Molecular Clock in Promoting Skeletal Muscle Growth and Protecting against Sarcopenia. Int. J. Mol. Sci. 2019, 20, 4318. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Park, Y.J.; Cartmell, K.B. Sarcopenia in cancer survivors is associated with increased cardiovascular disease risk. Support. Care Cancer 2018, 26, 2313–2321. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, A.; Yoshimura, Y.; Wakabayashi, H.; Tsuji, Y. Prevalence of stroke-related sarcopenia and its association with poor oral status in post-acute stroke patients: Implications for oral sarcopenia. Clin. Nutr. 2018, 37, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.I.; Park, D.K.; Chung, J.W.; Kim, K.O.; Kwon, K.A.; Kim, Y.J. Circadian rhythm disruption is associated with an increased risk of sarcopenia: A nationwide population-based study in Korea. Sci. Rep. 2019, 9, 12015. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, J.; Sun, Q. Sleep Duration and Sarcopenia: An Updated Systematic Review and Meta-Analysis. J. Am. Med. Dir. Assoc. 2023, 24, 1193–1206.e5. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, A.; Cescon, M.; D’Agostino, C.; Schilardi, F.; Sabatelli, P.; Merlini, L.; Faldini, C. Collagen VI in the Musculoskeletal System. Int. J. Mol. Sci. 2023, 24, 5095. [Google Scholar] [CrossRef] [PubMed]
- Scotton, C.; Bovolenta, M.; Schwartz, E.; Falzarano, M.S.; Martoni, E.; Passarelli, C.; Armaroli, A.; Osman, H.; Rodolico, C.; Messina, S.; et al. Deep RNA profiling identified CLOCK and molecular clock genes as pathophysiological signatures in collagen VI myopathy. J. Cell Sci. 2016, 129, 1671–1684. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E.H. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef]
- Lovering, R.M.; Porter, N.C.; Bloch, R.J. The muscular dystrophies: From genes to therapies. Phys. Ther. 2005, 85, 1372–1388. [Google Scholar] [CrossRef]
- O’Brien, K.F.; Kunkel, L.M. Dystrophin and muscular dystrophy: Past, present, and future. Mol. Genet. Metab. 2001, 74, 75–88. [Google Scholar] [CrossRef]
- Guiraud, S.; Aartsma-Rus, A.; Vieira, N.M.; Davies, K.E.; van Ommen, G.J.; Kunkel, L.M. The Pathogenesis and Therapy of Muscular Dystrophies. Annu. Rev. Genom. Hum. Genet. 2015, 16, 281–308. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Ibraghimov-Beskrovnaya, O.; Ervasti Jm Fau-Leveille, C.J.; Leveille Cj Fau-Slaughter, C.A.; Slaughter Ca Fau-Sernett, S.W.; Sernett Sw Fau-Campbell, K.P.; Campbell, K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 1992, 355, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Campbell, K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell 1991, 66, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Ozawa, E. Glycoprotein complex anchoring dystrophin to sarcolemma. J. Biochem. 1990, 108, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Grozdanovic, Z.; Baumgarten, H.G. Nitric oxide synthase in skeletal muscle fibers: A signaling component of the dystrophin-glycoprotein complex. Histol. Histopathol. 1999, 14, 243–256. [Google Scholar]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [PubMed]
- Pipes, G.T.; Creemers, E.E.; Olson, E.N. The myocardin family of transcriptional coactivators: Versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 2006, 20, 1545–1556. [Google Scholar] [CrossRef]
- Gerber, A.; Esnault, C.; Aubert, G.; Treisman, R.; Pralong, F.; Schibler, U. Blood-borne circadian signal stimulates daily oscillations in actin dynamics and SRF activity. Cell 2013, 152, 492–503. [Google Scholar] [CrossRef]
- Xiong, X.; Li, W.; Nam, J.; Qu, M.; Kay, S.A.; Ma, K.A.-O. The actin cytoskeleton-MRTF/SRF cascade transduces cellular physical niche cues to entrain the circadian clock. J. Cell Sci. 2022, 135, jcs260094. [Google Scholar] [CrossRef]
- Rossi, R.; Falzarano, M.S.; Osman, H.; Armaroli, A.; Scotton, C.; Mantuano, P.; Boccanegra, B.; Cappellari, O.; Schwartz, E.; Yuryev, A.; et al. Circadian Genes as Exploratory Biomarkers in DMD: Results From Both the mdx Mouse Model and Patients. Front. Physiol. 2021, 12, 678974. [Google Scholar] [CrossRef] [PubMed]
- Betts, C.A.; Jagannath, A.; van Westering, T.L.; Bowerman, M.; Banerjee, S.; Meng, J.; Falzarano, M.S.; Cravo, L.; McClorey, G.; Meijboom, K.E.; et al. Dystrophin involvement in peripheral circadian SRF signalling. Life Sci. Alliance 2021, 4, e202101014. [Google Scholar] [CrossRef] [PubMed]
- Hardee, J.P.; Caldow, M.K.; Chan, A.S.M.; Plenderleith, S.K.; Trieu, J.; Koopman, R.; Lynch, G.S. Dystrophin deficiency disrupts muscle clock expression and mitochondrial quality control in mdx mice. Cell Physiol. 2021, 321, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Beggs, A.H.; Moyer, M.; Scherpf, S.; Heindrich, K.; Bettecken, T.; Meng, G.; Muller, C.R.; Lindlof, M.; Kaariainen, H.; et al. The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. Am. J. Hum. Genet. 1989, 45, 498–506. [Google Scholar]
- Sato, F.; Kohsaka, A.; Tanimoto, T.; Bhawal, U.K.; Muragaki, Y. Histological analysis of a Becker muscular dystrophy case, diurnal expression of dystrophin in control mice and decreased expression of dystrophin in Bmal1 knockout mice. Histol. Histopathol. 2023, 38, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Dadgar, S.; Wang, Z.; Johnston, H.; Kesari, A.; Nagaraju, K.; Chen, Y.W.; Hill, D.A.; Partridge, T.A.; Giri, M.; Freishtat, R.J.; et al. Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy. J. Cell Biol. 2014, 207, 139–158. [Google Scholar] [CrossRef]
- Balsalobre, A.; Brown, S.A.; Marcacci, L.; Tronche, F.; Kellendonk, C.; Reichardt, H.M.; Schutz, G.; Schibler, U. Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science 2000, 289, 2344–2347. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Muntoni, F.; Urtizberea, A.; Hughes, R.; Griggs, R. Report on the 124th ENMC International Workshop. Treatment of Duchenne muscular dystrophy; defining the gold standards of management in the use of corticosteroids. 2–4 April 2004, Naarden, The Netherlands. Neuromuscul. Disord. 2004, 14, 526–534. [Google Scholar] [CrossRef]
- Budzinska, M.; Zimna, A.; Kurpisz, M. The role of mitochondria in Duchenne muscular dystrophy. J. Physiol. Pharmacol. 2021, 72, 10-26402. [Google Scholar] [CrossRef]
- Manella, G.; Asher, G. The Circadian Nature of Mitochondrial Biology. Front. Endocrinol. 2016, 7, 162. [Google Scholar] [CrossRef]
- Panda, S.; Antoch, M.P.; Miller, B.H.; Su, A.I.; Schook, A.B.; Straume, M.; Schultz, P.G.; Kay, S.A.; Takahashi, J.S.; Hogenesch, J.B. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 2002, 109, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Harfmann, B.D.; Schroder, E.A.; Kachman, M.T.; Hodge, B.A.; Zhang, X.; Esser, K.A. Muscle-specific loss of Bmal1 leads to disrupted tissue glucose metabolism and systemic glucose homeostasis. Skelet. Muscle 2016, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Li, W.; Chatterjee, S.; Xiong, X.; Saha, P.; Yechoor, V.; Ma, K. Metabolic-sensing of the skeletal muscle clock coordinates fuel oxidation. FASEB J. 2020, 34, 6613–6627. [Google Scholar] [CrossRef] [PubMed]
- Peek, C.B.; Levine, D.C.; Cedernaes, J.; Taguchi, A.; Kobayashi, Y.; Tsai, S.J.; Bonar, N.A.; McNulty, M.R.; Ramsey, K.M.; Bass, J. Circadian Clock Interaction with HIF1alpha Mediates Oxygenic Metabolism and Anaerobic Glycolysis in Skeletal Muscle. Cell Metab. 2017, 25, 86–92. [Google Scholar] [CrossRef]
- Nohara, K.; Mallampalli, V.; Nemkov, T.; Wirianto, M.; Yang, J.; Ye, Y.; Sun, Y.; Han, L.; Esser, K.A.; Mileykovskaya, E.; et al. Nobiletin fortifies mitochondrial respiration in skeletal muscle to promote healthy aging against metabolic challenge. Nat. Commun. 2019, 10, 3923. [Google Scholar] [CrossRef] [PubMed]
- Pelin, K.; Wallgren-Pettersson, C. Update on the Genetics of Congenital Myopathies. Semin. Pediatr. Neurol. 2019, 29, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Cardamone, M.; Darras, B.T.; Ryan, M.M. Inherited Myopathies and Muscular Dystrophies. Semin. Neurol. 2008, 28, 250–259. [Google Scholar] [CrossRef]
- Spurney, C.F. Cardiomyopathy of Duchenne muscular dystrophy: Current understanding and future directions. Muscle Nerve 2011, 44, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Cardiopulmonary support in duchenne muscular dystrophy. Lung 2006, 184, 205–215. [Google Scholar] [CrossRef]
- Deng, J.; Zhang, J.; Shi, K.; Liu, Z. Drug development progress in duchenne muscular dystrophy. Front. Pharmacol. 2022, 13, 950651. [Google Scholar] [CrossRef]
- Porter, J.D.; Khanna, S.; Kaminski, H.J.; Rao, J.S.; Merriam, A.P.; Richmonds, C.R.; Leahy, P.; Li, J.; Guo, W.; Andrade, F.H. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2002, 11, 263–272. [Google Scholar] [CrossRef]
- Moore, T.M.; Lin, A.J.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; Ho, T.; Lee, J.L.; Rucker, D.H.; Nguyen, C.Q.; et al. Mitochondrial Dysfunction Is an Early Consequence of Partial or Complete Dystrophin Loss in mdx Mice. Front. Physiol. 2020, 11, 690. [Google Scholar] [CrossRef] [PubMed]
- Partridge, T.A. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 2013, 280, 4177–4186. [Google Scholar] [CrossRef] [PubMed]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, J.S.; Metzger, J.; Reyes, M.; Townsend, D.; Faulkner, J.A. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 2007, 21, 2195–2204. [Google Scholar] [CrossRef]
- Isaac, C.; Wright, A.; Usas, A.; Li, H.; Tang, Y.; Mu, X.; Greco, N.; Dong, Q.; Vo, N.; Kang, J.; et al. Dystrophin and utrophin “double knockout” dystrophic mice exhibit a spectrum of degenerative musculoskeletal abnormalities. J. Orthop. Res. 2013, 31, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Sacco, A.; Mourkioti, F.; Tran, R.; Choi, J.; Llewellyn, M.; Kraft, P.; Shkreli, M.; Delp, S.; Pomerantz, J.H.; Artandi, S.E.; et al. Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell 2010, 143, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- van Ommen, G.J.B.; Bertelson, C.; Ginjaar, H.B.; den Dunnen, J.T.; Bakker, E.; Chelly, J.; Matton, M.; van Essen, A.J.; Bartley, J.; Kunkel, L.M.; et al. Long-range genomic map of the Duchenne muscular dystrophy (DMD) gene: Isolation and use of J66 (DXS268), a distal intragenic marker. Genomics 1987, 1, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, J.R.; Chamberlain, J.S. Progress toward Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2017, 25, 1125–1131. [Google Scholar] [CrossRef]
- Harper, S.Q.; Hauser, M.A.; DelloRusso, C.; Duan, D.; Crawford, R.W.; Phelps, S.F.; Harper, H.A.; Robinson, A.S.; Engelhardt, J.F.; Brooks, S.V.; et al. Modular flexibility of dystrophin: Implications for gene therapy of Duchenne muscular dystrophy. Nat. Med. 2002, 8, 253–261. [Google Scholar] [CrossRef]
- Crawford, G.E.; Faulkner, J.A.; Crosbie, R.H.; Campbell, K.P.; Froehner, S.C.; Chamberlain, J.S. Assembly of the Dystrophin-associated Protein Complex Does Not Require the Dystrophin COOH-terminal Domain. J. Cell Biol. 2000, 150, 1399–1410. [Google Scholar] [CrossRef]
- Rafael, J.A.; Cox, G.A.; Corrado, K.; Jung, D.; Campbell, K.P.; Chamberlain, J.S. Chamberlain. Forced expression of dystrophin deletion constructs reveals structure-function correlations. J. Cell Biol. 1996, 134, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, K.; Miyagoe, Y.; Yamamoto, K.; Nabeshima, Y.; Dickson, G.; Takeda, S. Effective restoration of dystrophin-associated proteins in vivo by adenovirus-mediated transfer of truncated dystrophin cDNAs. FEBS Lett. 1998, 425, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, J.; Xiao, X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc. Natl. Acad. Sci. USA 2000, 97, 13714–13719. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Perez-Pinera, P.; Brown, M.T.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Correction of dystrophin expression in cells from Duchenne muscular dystrophy patients through genomic excision of exon 51 by zinc finger nucleases. Mol. Ther. 2015, 23, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Bowles, D.E.; McPhee, S.W.; Li, C.; Gray, S.J.; Samulski, J.J.; Camp, A.S.; Li, J.; Wang, B.; Monahan, P.E.; Rabinowitz, J.E.; et al. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol. Ther. 2012, 20, 443–455. [Google Scholar] [CrossRef]
- Fan, Y.; Maley, M.; Beilharz, M.; Grounds, M. Rapid death of injected myoblasts in myoblast transfer therapy. Muscle Nerve 1996, 19, 853–860. [Google Scholar] [CrossRef]
- Skuk, D.; Goulet, M.; Roy, B.; Chapdelaine, P.; Bouchard, J.P.; Roy, R.; Dugré, F.J.; Sylvain, M.; Lachance, J.-G.; Deschênes, L.; et al. Dystrophin Expression in Muscles of Duchenne Muscular Dystrophy Patients After High-Density Injections of Normal Myogenic Cells. J. Neuropathol. Exp. Neurol. 2006, 65, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Castellanos Rivera, R.M.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef]
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef]
- El Refaey, M.; Xu, L.; Gao, Y.; Canan, B.D.; Adesanya, T.M.A.; Warner, S.C.; Akagi, K.; Symer, D.E.; Mohler, P.J.; Ma, J.; et al. In Vivo Genome Editing Restores Dystrophin Expression and Cardiac Function in Dystrophic Mice. Circ. Res. 2017, 121, 923–929. [Google Scholar] [CrossRef]
- Zhang, Y.; Nishiyama, T.; Olson, E.N.; Bassel-Duby, R. CRISPR/Cas correction of muscular dystrophies. Exp. Cell Res. 2021, 408, 112844. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.; Deconinck, N.; Fisher, R.; Kahn, D.; Phelps, S.; Gillis, J.M.; Davies, K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat. Med. 1998, 4, 1441–1444. [Google Scholar] [CrossRef]
- Oray, M.; Abu Samra, K.; Ebrahimiadib, N.; Meese, H.; Foster, C.S. Long-term side effects of glucocorticoids. Expert Opin. Drug Saf. 2016, 15, 457–465. [Google Scholar] [CrossRef]
- Schakman, O.; Kalista, S.; Barbe, C.; Loumaye, A.; Thissen, J.P. Glucocorticoid-induced skeletal muscle atrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2163–2172. [Google Scholar] [CrossRef]
- Schakman, O.; Gilson, H.; Thissen, J.P. Mechanisms of glucocorticoid-induced myopathy. J. Endocrinol. 2008, 197, 1–10. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Schuelke, M.; Wagner, K.R.; Stolz, L.E.; Hubner, C.; Riebel, T.; Komen, W.; Braun, T.; Tobin, J.F.; Lee, S.J. Myostatin mutation associated with gross muscle hypertrophy in a child. N. Engl. J. Med. 2004, 350, 2682–2688. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, K. Targeting myostatin for therapies against muscle-wasting disorders. Curr. Opin. Drug Discov. Devel 2008, 11, 487–494. [Google Scholar]
- Bogdanovich, S.; Krag, T.O.; Barton, E.R.; Morris, L.D.; Whittemore, L.A.; Ahima, R.S.; Khurana, T.S. Functional improvement of dystrophic muscle by myostatin blockade. Nature 2002, 420, 418–421. [Google Scholar] [CrossRef]
- Bogdanovich, S.; Perkins, K.J.; Krag, T.O.; Whittemore, L.A.; Khurana, T.S. Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. FASEB J. 2005, 19, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Rybalka, E.; Timpani, C.A.; Debruin, D.A.; Bagaric, R.M.; Campelj, D.G.; Hayes, A. The Failed Clinical Story of Myostatin Inhibitors against Duchenne Muscular Dystrophy: Exploring the Biology behind the Battle. Cells 2020, 9, 2657. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Song, S.; Tien, C.L.; Qi, L.; Graves, A.; Nasiotis, E.; Burris, T.P.; Zhao, Y.; Sun, Z.; Zhang, L. SR9009 improves heart function after pressure overload independent of cardiac REV-ERB. Front. Cardiovasc. Med. 2022, 9, 952114. [Google Scholar] [CrossRef] [PubMed]
- Keller, H.L.; Schneider, B.S.P.; Eppihimer, L.A.; Cannon, J.G. Association of IGF-I and IGF-II with myofiber regeneration in vivo. Muscle Nerve 1999, 22, 347–354. [Google Scholar] [CrossRef]
- Huang, S.; Jiao, X.; Lu, D.; Pei, X.; Qi, D.; Li, Z. Recent advances in modulators of circadian rhythms: An update and perspective. J. Enzyme Inhib. Med. Chem. 2020, 35, 1267–1286. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Lee, J.W.; St John, P.C.; Sawa, M.; Iwaisako, K.; Noguchi, T.; Pongsawakul, P.Y.; Sonntag, T.; Welsh, D.K.; Brenner, D.A.; et al. Identification of small molecule activators of cryptochrome. Science 2012, 337, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Solt, L.A.; Wang, Y.; Banerjee, S.; Hughes, T.; Kojetin, D.J.; Lundasen, T.; Shin, Y.; Liu, J.; Cameron, M.D.; Noel, R.; et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature 2012, 485, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Kojetin, D.; Wang, Y.; Kamenecka, T.M.; Burris, T.P. Identification of SR8278, a synthetic antagonist of the nuclear heme receptor REV-ERB. ACS Chem. Biol. 2011, 6, 131–134. [Google Scholar] [CrossRef]
- Becker-André, M.; Wiesenberg, I.; Schaeren-Wiemers, N.; Andre, E.; Missbach, M.; Saurat, J.H.; Carlberg, C. Pineal gland hormone melatonin binds and activates an orphan of the nuclear receptor superfamily. J. Biol. Chem. 1994, 269, 28531–28534. [Google Scholar] [CrossRef]
- Fernández-Martínez, J.; Ramirez-Casas, Y.; Yang, Y.; Aranda-Martinez, P.; Martinez-Ruiz, L.; Escames, G.; Acuna-Castroviejo, D. From Chronodisruption to Sarcopenia: The Therapeutic Potential of Melatonin. Biomolecules 2023, 13, 1779. [Google Scholar] [CrossRef]
- Stein, R.M.; Kang, H.J.; McCorvy, J.D.; Glatfelter, G.C.; Jones, A.J.; Che, T.; Slocum, S.; Huang, X.P.; Savych, O.; Moroz, Y.S.; et al. Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature 2020, 579, 609–614. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Nohara, K.; Park, N.; Park, Y.S.; Guillory, B.; Zhao, Z.; Garcia, J.M.; Koike, N.; Lee, C.C.; Takahashi, J.S.; et al. The Small Molecule Nobiletin Targets the Molecular Oscillator to Enhance Circadian Rhythms and Protect against Metabolic Syndrome. Cell Metab. 2016, 23, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Bugge, A.; Feng, D.; Everett, L.J.; Briggs, E.R.; Mullican, S.E.; Wang, F.; Jager, J.; Lazar, M.A. Rev-erbalpha and Rev-erbbeta coordinately protect the circadian clock and normal metabolic function. Genes Dev. 2012, 26, 657–667. [Google Scholar] [CrossRef]
- Amador, A.; Huitron-Resendiz, S.; Roberts, A.J.; Kamenecka, T.M.; Solt, L.A.; Burris, T.P. Pharmacological Targeting the REV-ERBs in Sleep/Wake Regulation. PLoS ONE 2016, 11, e0162452. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Atkins, A.R.; Yu, R.T.; Downes, M.; Evans, R.M. Road to exercise mimetics: Targeting nuclear receptors in skeletal muscle. J. Mol. Endocrinol. 2013, 51, T87–T100. [Google Scholar] [CrossRef] [PubMed]
- Geldof, L.; Deventer, K.; Roels, K.; Tudela, E.; Van Eeno, P. In Vitro Metabolic Studies of REV-ERB Agonists SR9009 and SR9011. Int. J. Mol. Sci. 2016, 17, 1676. [Google Scholar] [CrossRef] [PubMed]
- Jetten, A.M. Retinoid-related orphan receptors (RORs): Critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl. Recept. Signal 2009, 7, e003. [Google Scholar] [CrossRef] [PubMed]
- Sayed, R.K.A.; Fernandez-Ortiz, M.; Diaz-Casado, M.E.; Rusanova, I.; Rahim, I.; Escames, G.; Lopez, L.C.; Mokhtar, D.M.; Acuna-Castroviejo, D. The Protective Effect of Melatonin Against Age-Associated, Sarcopenia-Dependent Tubular Aggregate Formation, Lactate Depletion, and Mitochondrial Changes. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Solt, L.A.; Kumar, N.; Nuhant, P.; Wang, Y.; Lauer, J.L.; Liu, J.; Istrate, M.A.; Kamenecka, T.M.; Roush, W.R.; Vidovic, D.; et al. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature 2011, 472, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Kojetin, D.J.; Solt, L.A.; Kumar, K.G.; Nuhant, P.; Duckett, D.R.; Cameron, M.D.; Butler, A.A.; Roush, W.R.; Griffin, P.R.; et al. Identification of SR3335 (ML-176): A synthetic RORalpha selective inverse agonist. ACS Chem. Biol. 2011, 6, 218–222. [Google Scholar] [CrossRef]
- Raichur, S.; Fitzsimmons, R.L.; Myers, S.A.; Pearen, M.A.; Lau, P.; Eriksson, N.; Wang, S.M.; Muscat, G.E. Identification and validation of the pathways and functions regulated by the orphan nuclear receptor, ROR alpha1, in skeletal muscle. Nucleic Acids Res. 2010, 38, 4296–4312. [Google Scholar] [CrossRef] [PubMed]
- Nangle, S.; Xing, W.; Zheng, N. Crystal structure of mammalian cryptochrome in complex with a small molecule competitor of its ubiquitin ligase. Cell Res. 2013, 23, 1417–1419. [Google Scholar] [CrossRef]
- Rosenberg, A.S.; Puig, M.; Nagaraju, K.; Hoffman, E.P.; Villalta, S.A.; Rao, V.A.; Wakefield, L.M.; Woodcock, J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med. 2015, 7, 299rv294. [Google Scholar] [CrossRef] [PubMed]
- Cermakian, N.; Lange, T.; Golombek, D.; Sarkar, D.; Nakao, A.; Shibata, S.; Mazzoccoli, G. Crosstalk between the circadian clock circuitry and the immune system. Chronobiol. Int. 2013, 30, 870–888. [Google Scholar] [CrossRef] [PubMed]
- Timmons, G.A.; O’Siorain, J.R.; Kennedy, O.D.; Curtis, A.M.; Early, J.O. Innate Rhythms: Clocks at the Center of Monocyte and Macrophage Function. Front. Immunol. 2020, 11, 1743. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, L. The Many Roles of Macrophages in Skeletal Muscle Injury and Repair. Front. Cell Dev. Biol. 2022, 10, 952249. [Google Scholar] [CrossRef]
- Yang, X.O.; Pappu, B.P.; Nurieva, R.; Akimzhanov, A.; Kang, H.S.; Chung, Y.; Ma, L.; Shah, B.; Panopoulos, A.D.; Schluns, K.S.; et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 2008, 28, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Loo, C.S.; Zhao, X.; Solt, L.A.; Liang, Y.; Bapat, S.P.; Cho, H.; Kamenecka, T.M.; Leblanc, M.; Atkins, A.R.; et al. The nuclear receptor REV-ERBalpha modulates Th17 cell-mediated autoimmune disease. Proc. Natl. Acad. Sci. USA 2019, 116, 18528–18536. [Google Scholar] [CrossRef] [PubMed]
- Kojetin, D.J.; Burris, T.P. REV-ERB and ROR nuclear receptors as drug targets. Nat. Rev. Drug Discov. 2014, 13, 197–216. [Google Scholar] [CrossRef]
- Wang, R.; Campbell, S.; Amir, M.; Mosure, S.A.; Bassette, M.A.; Eliason, A.; Sundrud, M.S.; Kamenecka, T.M.; Solt, L.A. Genetic and pharmacological inhibition of the nuclear receptor RORalpha regulates T(H)17 driven inflammatory disorders. Nat. Commun. 2021, 12, 76. [Google Scholar] [CrossRef]
- Sunildutt, N.; Parihar, P.; Chethikkattuveli Salih, A.R.; Lee, S.H.; Choi, K.H. Revolutionizing drug development: Harnessing the potential of organ-on-chip technology for disease modeling and drug discovery. Front. Pharmacol. 2023, 14, 1139229. [Google Scholar] [CrossRef] [PubMed]




 : denotes activation and
: denotes activation and  denotes inhibition.
: denotes activation and denotes inhibition.
denotes inhibition.
: denotes activation and denotes inhibition.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Clock Target | Clock-Modulatory and Associated Effects | Muscle Disease Relevance | Human Studies | References |
|---|---|---|---|---|---|
| SR8278 | Rev-erbα/β | Antagonist Activation of clock gene expression | Stimulation of muscle regeneration Promotion of myoblast proliferation and differentiation | N/A | Bugge et al. 2012 [163], Chatterjee et al. 2019 [53], Kiperman et al. 2023 [11], Kojetin et al. 2011 [158], Welch et al. 2017 [59] |
| SR9011/SR9009 | Rev-erbα/β | Agonist Suppression of clock genes Induction of wakefulness Increase in energy expenditure and weight loss | Increase in mitochondrial content Promotion of muscle oxidation | N/A | Amador et al. 2016 [164], Fan et al. 2013 [165], Geldof et al. 2016 [166], Solt et al. 2012 [157], Woldt et al. 2013 [42] |
| Chlorhexidine | CLOCK | Activator Promotion of Bmal1/CLOCK interaction Induction of clock gene expression | Promotion of satellite cell proliferation and myogenic differentiation | Antiseptic/antimicrobial, mouth wash | Chatterjee et al. 2013 and 2015 [24,25], Kiperman et al. 2023 [11] |
| N-acetyl-5-methoxytryptamine (Melatonin) | RORα/γ | Inhibition of ubiquitin–proteasome protein degradation to modulate circadian clock loop Stabilization of circadian rhythm | Mitigation of age-related sarcopenia | Insomnia, sleep-related pathologies, cancer, neuroprotection | Becker-André et al. 1994 [159], Fernández-Martínez et al. 2023 [160], Jetten 2009 [167], Sayed et al. 2018 [168] |
| SR1078 | RORα/γ | Agonist Promotion of core clock target gene expression | Not determined | N/A | Kojetin et al. 2010 [158] |
| SR1001 | RORα/γ | Inverse agonist Inhibition of clock oscillation | Not determined | N/A | Solt et al. 2011 [169] |
| SR3335 | RORα | Selective inverse agonist Suppression of RORα target gene expression Inhibition of gluconeogenesis | Not determined | N/A | Kumar et al. 2012 [170] |
| Nobiletin | RORα/γ | Agonist Enhancement of clock oscillatory amplitude Activation of circadian clock Promotion of energy homeostasis | Increase in muscle mitochondrial oxidation Promotion of lipid metabolism | Weight loss, Alzheimer’s disease | He et al. 2016 [162], Raichur et al. 2010 [171] |
| KL001 | Cry1/2 | Cry1/2 stabilizer Period lengthening Inhibition of glucagon-induced gluconeogenesis | Not determined | Hemophilia B | Hirota et al. 2012 [156], Nangle et al. 2013 [172] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiperman, T.; Ma, K. Circadian Clock in Muscle Disease Etiology and Therapeutic Potential for Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2024, 25, 4767. https://doi.org/10.3390/ijms25094767
Kiperman T, Ma K. Circadian Clock in Muscle Disease Etiology and Therapeutic Potential for Duchenne Muscular Dystrophy. International Journal of Molecular Sciences. 2024; 25(9):4767. https://doi.org/10.3390/ijms25094767
Chicago/Turabian StyleKiperman, Tali, and Ke Ma. 2024. "Circadian Clock in Muscle Disease Etiology and Therapeutic Potential for Duchenne Muscular Dystrophy" International Journal of Molecular Sciences 25, no. 9: 4767. https://doi.org/10.3390/ijms25094767
APA StyleKiperman, T., & Ma, K. (2024). Circadian Clock in Muscle Disease Etiology and Therapeutic Potential for Duchenne Muscular Dystrophy. International Journal of Molecular Sciences, 25(9), 4767. https://doi.org/10.3390/ijms25094767