Crystal Structure of Bis(2,4,6-trimethylphenyl)-phosphine Oxide


{kind=link}
{kind=link}
Abstract
:1. Introduction
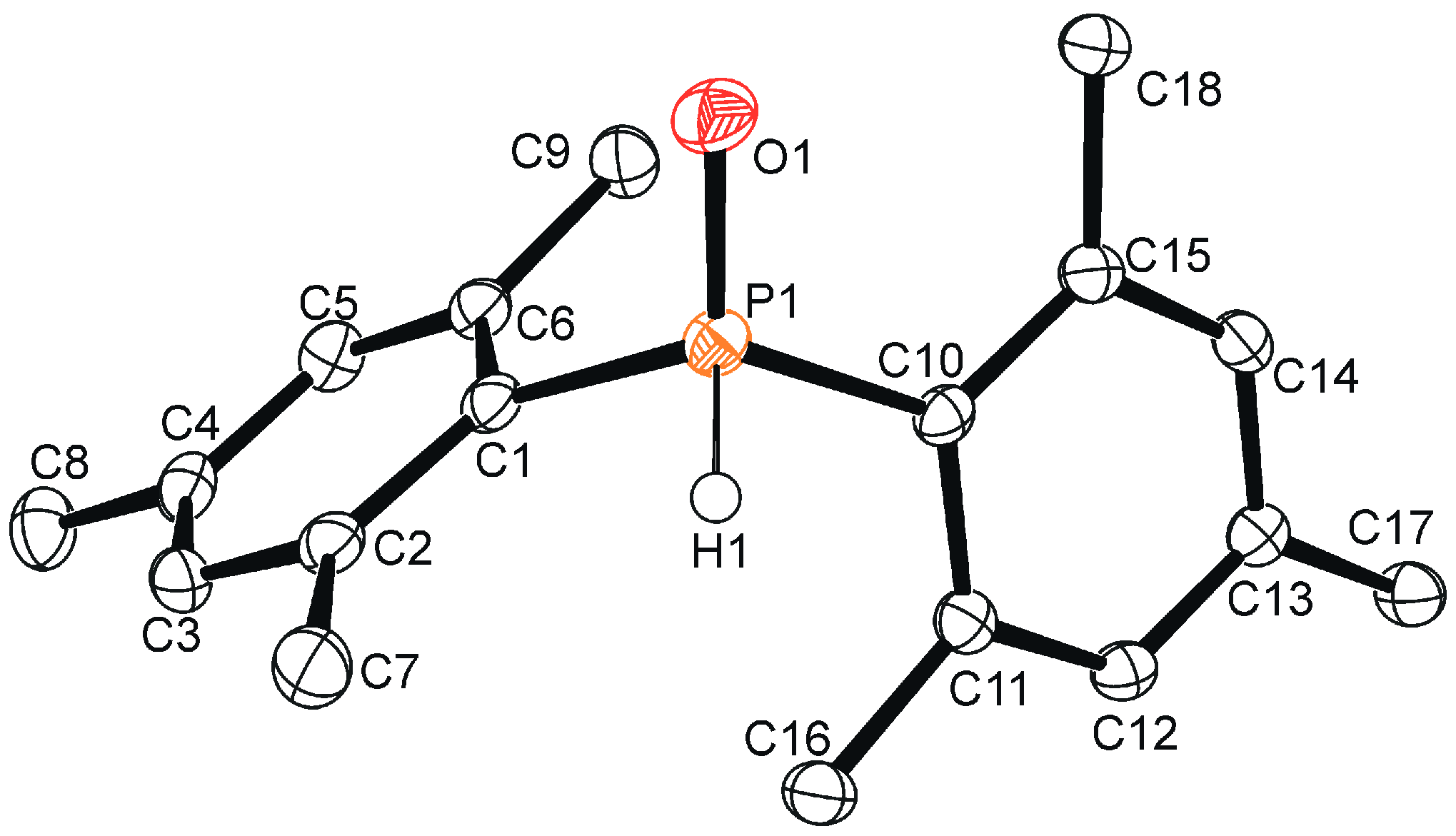
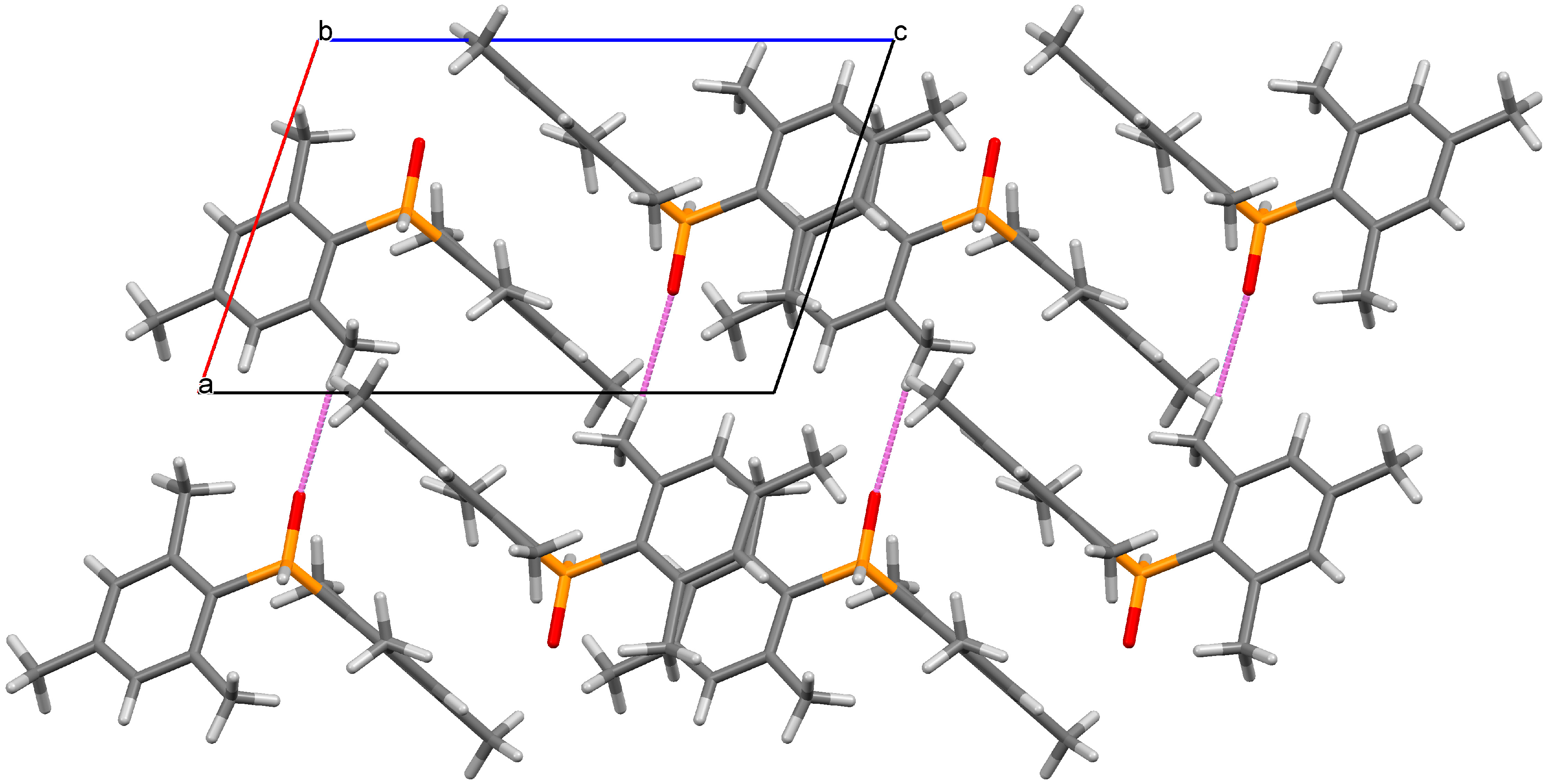
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Instrumentation
3.2.1. General Remarks
3.2.2. X-ray Crystallography
3.3. Synthesis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Boutagy, J.; Thomas, R. Olefin synthesis with organic phosphonate carbanions. Chem. Rev. 1974, 74, 87–99. [Google Scholar] [CrossRef]
- Kitamura, M.; Tokunaga, M.; Noyori, R. Asymmetric Hydrogenation of β-Keto Phosphonates: A Practical Way to Fosfomycin. J. Am. Chem. Soc. 1995, 117, 2931–2932. [Google Scholar] [CrossRef]
- Knowles, W.S. Asymmetric hydrogenations (Nobel lecture 2001). Adv. Synth. Catal. 2003, 345, 3–13. [Google Scholar] [CrossRef]
- Stephan, D.W.; Erker, G. Frustrated Lewis pair chemistry: Development and perspectives. Angew. Chem. Int. Ed. 2015, 54, 6400–6441. [Google Scholar] [CrossRef] [PubMed]
- Sawa, M.; Kiyoi, T.; Kurokawa, K.; Kumihara, H.; Yamamoto, M.; Miyasaka, T.; Ito, Y.; Hirayama, R.; Inoue, T.; Kirii, Y. New type of metalloproteinase inhibitor: Design and synthesis of new phosphonamide-based hydroxamic acids. J. Med. Chem. 2002, 45, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.; Zhang, P.; Murphy, M.E.; Marmorstein, R.; George, D.L. Structural basis for the inhibition of HSP70 and dnak chaperones by small-molecule targeting of a c-terminal allosteric pocket. ACS Chem. Biol. 2014, 9, 2508–2516. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.K.; Budina-Kolomets, A.; Murphy, M.E.; Nefedova, Y. Efficacy of the HSP70 inhibitor PET-16 in multiple myeloma. Cancer Biol. Ther. 2015, 16, 1422–1426. [Google Scholar] [CrossRef] [PubMed]
- Stetz, G.; Verkhivker, G.M. Probing allosteric inhibition mechanisms of the Hsp70 chaperone proteins using molecular dynamics simulations and analysis of the residue interaction networks. J. Chem. Inf. Model. 2016, 56, 1490–1517. [Google Scholar] [CrossRef] [PubMed]
- Queffélec, C.; Petit, M.; Janvier, P.; Knight, D.A.; Bujoli, B. Surface modification using phosphonic acids and esters. Chem. Rev. 2012, 112, 3777–3807. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, P.; Gao, Y.; Chen, Y.; Tang, G.; Zhao, Y. Copper-catalyzed P-arylation via direct coupling of diaryliodonium salts with phosphorus nucleophiles at room temperature. J. Org. Chem. 2013, 78, 8176–8183. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; Tang, Y.; Yi, H.; Li, Y.; Cheng, Y.; Liu, C.; Lei, A. Copper-catalyzed radical/radical Csp3–H/P–H cross-coupling: α-Phosphorylation of Aryl Ketone O-Acetyloximes. Angew. Chem. Int. Ed. 2015, 54, 6604–6607. [Google Scholar] [CrossRef] [PubMed]
- Noël-Duchesneau, L.; Lagadic, E.; Morlet-Savary, F.; Lohier, J.; Chataigner, I.; Breugst, M.; Lalevée, J.; Gaumont, A.; Lakhdar, S. Metal-Free Synthesis of 6-phosphorylated phenanthridines: Synthetic and mechanistic insights. Org. Lett. 2016, 18, 5900–5903. [Google Scholar] [CrossRef] [PubMed]
- Jessop, C.M.; Parsons, A.F.; Routledge, A.; Irvine, D.J. Radical addition reactions of phosphorus hydrides: tuning the reactivity of phosphorus hydrides, the use of microwaves and horner–wadsworth–emmons-type reactions. Eur. J. Org. Chem. 2006, 2006, 1547–1554. [Google Scholar] [CrossRef]
- Stockland, R.A.; Taylor, R.I.; Thompson, L.E.; Patel, P.B. Microwave-Assisted Regioselective Addition of P (O)−H bonds to alkenes without added solvent or catalyst. Org. Lett. 2005, 7, 851–853. [Google Scholar] [CrossRef] [PubMed]
- Lenker, H.K.; Richard, M.E.; Reese, K.P.; Carter, A.F.; Zawisky, J.D.; Winter, E.F.; Bergeron, T.W.; Guydon, K.S.; Stockland, R.A., Jr. Phospha-michael additions to activated internal alkenes: Steric and electronic effects. J. Org. Chem. 2012, 77, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Dyer, P.W.; Fawcett, J.; Hanton, M.J. Rigid N-phosphino guanidine P, N ligands and their use in nickel-catalyzed ethylene oligomerization. Organometallics 2008, 27, 5082–5087. [Google Scholar] [CrossRef]
- Härling, S.; Greiser, J.; Al-Shboul, T.M.; Görls, H.; Krieck, S.; Westerhausen, M. Calcium-mediated hydrophosphorylation of organic isocyanates with diphenylphosphane oxide. Aust. J. Chem. 2013, 66, 1264–1273. [Google Scholar] [CrossRef]
- Christiansen, A.; Li, C.; Garland, M.; Selent, D.; Ludwig, R.; Spannenberg, A.; Baumann, W.; Franke, R.; Börner, A. On the tautomerism of secondary phosphane oxides. Eur. J. Org. Chem. 2010, 2010, 2733–2741. [Google Scholar] [CrossRef]
- Fleming, C.G.; Slawin, A.M.; Arachchige, K.S.A.; Randall, R.; Bühl, M.; Kilian, P. Synthetic and computational study of geminally bis (supermesityl) substituted phosphorus compounds. Dalton Trans. 2013, 42, 1437–1450. [Google Scholar] [CrossRef] [PubMed]
- APEX 3, v. 2016.1-0; Bruker AXS Inc.: Madison, WI, USA, 2016.
- SAINT, v. 8. 37A; Bruker AXS Inc.: Madison, WI, USA, 2015.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Cryst. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Streek, J.V.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veinot, A.J.; Ramgoolam, K.; Giffin, N.A.; Masuda, J.D. Crystal Structure of Bis(2,4,6-trimethylphenyl)-phosphine Oxide. Molbank 2017, 2017, M957. https://doi.org/10.3390/M957
Veinot AJ, Ramgoolam K, Giffin NA, Masuda JD. Crystal Structure of Bis(2,4,6-trimethylphenyl)-phosphine Oxide. Molbank. 2017; 2017(3):M957. https://doi.org/10.3390/M957
Chicago/Turabian StyleVeinot, Alex J., Ketnavi Ramgoolam, Nick A. Giffin, and Jason D. Masuda. 2017. "Crystal Structure of Bis(2,4,6-trimethylphenyl)-phosphine Oxide" Molbank 2017, no. 3: M957. https://doi.org/10.3390/M957
APA StyleVeinot, A. J., Ramgoolam, K., Giffin, N. A., & Masuda, J. D. (2017). Crystal Structure of Bis(2,4,6-trimethylphenyl)-phosphine Oxide. Molbank, 2017(3), M957. https://doi.org/10.3390/M957