Methyl 2-(1-methyl-3-oxoisoindolin-1-yl)acetate




Abstract
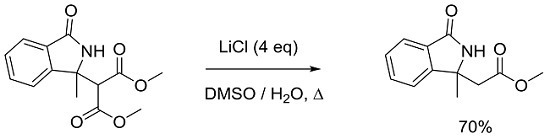
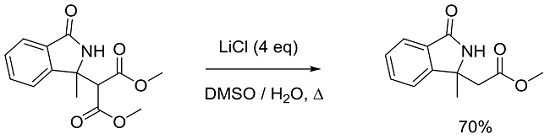
:
1. Introduction
2. Result and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure for Synthesis and Characterization of 2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Speck, K.; Magauer, T. The chemistry of isoindole natural products. Beilstein J. Org. Chem. 2013, 9, 2048–2078. [Google Scholar] [CrossRef] [PubMed]
- Di Mola, A.; Palombi, L.; Massa, A. An Overview on Asymmetric Synthesis of 3-Substituted Isoindolinones. In Targets in Heterocyclic Systems; Attanasi, O.A., Noto, R., Spinelli, D., Eds.; Italian Society of Chemistry: Rome, Italy, 2014; Volume 18, pp. 113–140. [Google Scholar]
- Bella, M.; Gasperi, T. Organocatalytic Formation of Quaternary Stereocenters. Synthesis 2009, 10, 1583–1614. [Google Scholar] [CrossRef]
- Comins, D.L.; Hiebel, A.C. Asymmetric synthesis of 3,3-disubstituted isoindolinones. Tetrahedron Lett. 2005, 46, 5639–5642. [Google Scholar] [CrossRef]
- Di Mola, A.; Di Martino, M.; Capaccio, V.; Pierri, G.; Palombi, L.; Tedesco, C.; Massa, A. Synthesis of 2-Acetylbenzonitriles and Their Reactivity in Tandem Reactions with Carbon and Hetero Nucleophiles: Easy Access to 3,3-Disubstituted Isoindolinones. Eur. J. Org. Chem. 2018, 1699–1708. [Google Scholar] [CrossRef]
- Di Mola, A.; Macchia, A.; Tedesco, C.; Pierri, G.; Palombi, L.; Filosa, R.; Massa, A. Synthetic Strategies and Cascade Reactions of 2-Cyanobenzophenones for the Access to Diverse 3,3-Disubstituted Isoindolinones and 3-Aryl-3-Hydroxyisoindolinones. ChemistrySelect 2019, 4, 4820–4826. [Google Scholar] [CrossRef]
- More, V.; Di Mola, A.; Perillo, M.; De Caprariis, P.; Filosa, R.; Peduto, A.; Massa, A. The aldol addition of readily enolizable 1,3-dicarbonyl compounds to 2-cyanobenzaldehyde in the synthesis of 3-substituted isoindolinones. Synthesis 2011, 18, 3027–3031. [Google Scholar]
- Krapcho, A.P.; Weimaster, J.F.; Eldridge, J.M.; Jahngen, E.G.E., Jr.; Lovey, A.J.; Stephens, W.P. Synthetic applications and mechanism studies of the decarbalkoxylations of geminal diesters and related systems effected in dimethyl sulfoxide by water and/or by water with added salts. J. Org. Chem. 1978, 43, 138–147. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}

| Entry | T (°C) | t (h) | Conv. (%) a | Yield (%) b |
|---|---|---|---|---|
| 1 c | 100 | 18 | -- | -- |
| 2 c | 130 | 5 | 60 | 40 |
| 3 c | 130 | 18 | >95 | 65 |
| 4 d | 130 | 9 | >95 | 70 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mola, A.D.; Macchia, A.; Palombi, L.; Massa, A. Methyl 2-(1-methyl-3-oxoisoindolin-1-yl)acetate. Molbank 2020, 2020, M1131. https://doi.org/10.3390/M1131
Mola AD, Macchia A, Palombi L, Massa A. Methyl 2-(1-methyl-3-oxoisoindolin-1-yl)acetate. Molbank. 2020; 2020(2):M1131. https://doi.org/10.3390/M1131
Chicago/Turabian StyleMola, Antonia Di, Antonio Macchia, Laura Palombi, and Antonio Massa. 2020. "Methyl 2-(1-methyl-3-oxoisoindolin-1-yl)acetate" Molbank 2020, no. 2: M1131. https://doi.org/10.3390/M1131
APA StyleMola, A. D., Macchia, A., Palombi, L., & Massa, A. (2020). Methyl 2-(1-methyl-3-oxoisoindolin-1-yl)acetate. Molbank, 2020(2), M1131. https://doi.org/10.3390/M1131