2. Results and Discussion
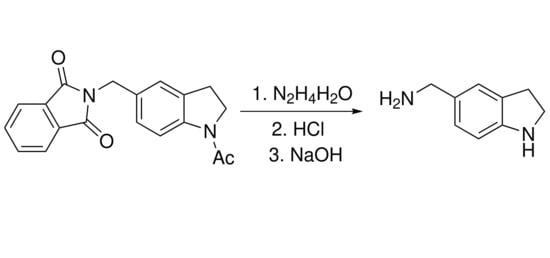
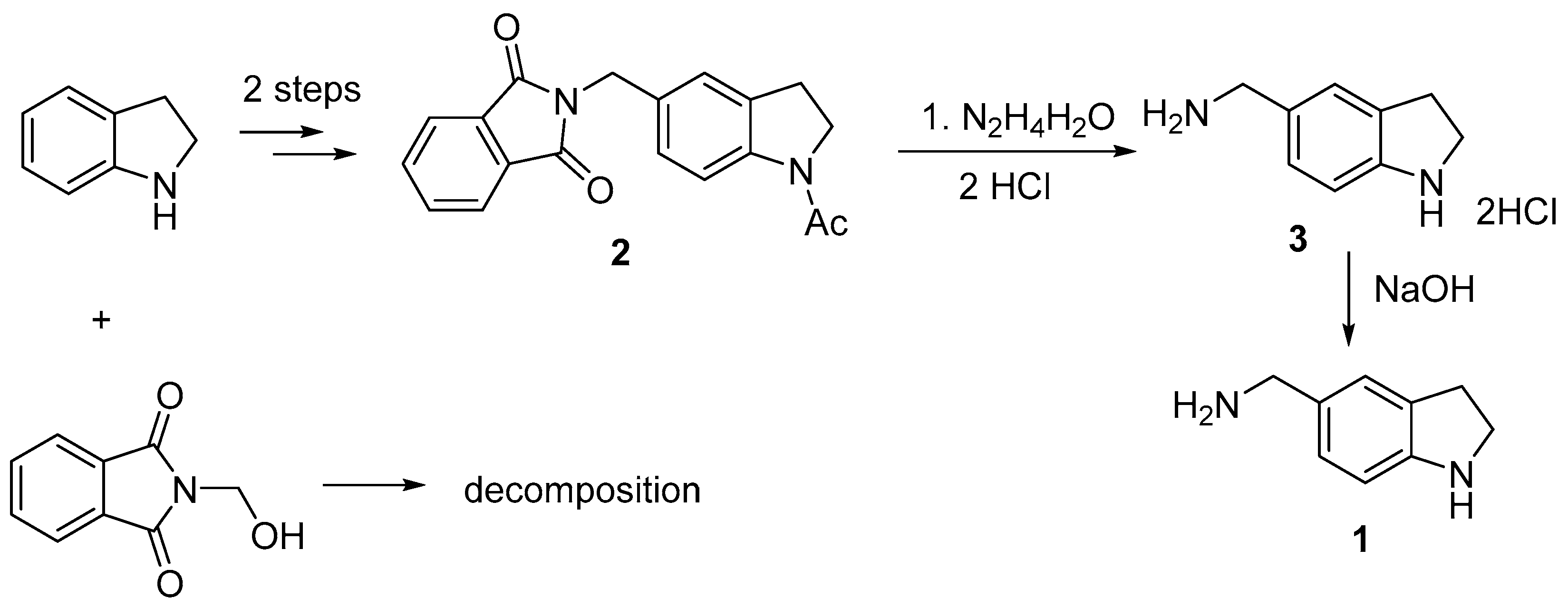
The most direct method for the preparation of (2,3-dihydro-1
H-indol-5-ylmethyl)amine
1 is the Tscherniac-Einhorn reaction of indoline with commercially available 2-(hydroxymethyl)isoindoline-1,3-dione using concentrated sulfuric acid as a catalyst [
4] followed by hydrolysis of phthalimido to amino group. We have shown that this reaction led to a difficult-to-separate mixture of compounds apparently due to the reaction with the unprotected NH indoline group. It has been described that acetyl-protected indoline reacted successfully with 2-(hydroxymethyl)isoindoline-1,3-dione giving compound
2 in good yield [
5]. Refluxing compound
2 with hydrazine hydrate in MeOH, followed by treatment with conc. HCl led to the desired (2,3-dihydro-1
H-indol-5-ylmethyl)amine dihydrochloride
3 in high yield (
Scheme 1). The main feature of this procedure is that two protective (phthalimido and acetyl) groups were removed simultaneously with the formation of unsubstituted heterocycle
3. The target (2,3-dihydro-1
H-indol-5-ylmethyl)amine was obtained by alkalizing the disalt
3.
The structure of (2,3-dihydro-1H-indol-5-ylmethyl)amine 1 and its dihydrochoride salt 3 was confirmed by elemental analysis, high resolution mass-spectrometry, 1H, 13C NMR and IR spectroscopy, and mass-spectrometry. Compared with disubstituted compound 2, the spectral data of compound 1 contain, in addition to signals characteristic of the indoline ring and CH2 group, signals characteristic of the NH2 and NH groups: in 1H NMR spectrum—2.45 (2H) and 5.28 (1H) ppm, and in IR spectrum—3359, 3282, 3012 cm–1.
In conclusion, the first representative of indolines containing a methylamine group—(2,3-dihydro-1H-indol-5-ylmethyl)amine 1, was obtained. This compound opens up possibilities for the synthesis of various functional derivatives of disubstituted 1-(indolin-5-yl)methanamines, which may be of interest as compounds with useful pharmacological properties.
3. Materials and Methods
2-((1-Acetylindolin-5-yl)methyl)isoindoline-1,3-dione
2 was prepared according to the published method [
5]. The solvents and reagents were purchased from commercial sources and used as received. Elemental analysis was performed on a 2400 Elemental Analyzer (Perkin ElmerInc., Waltham, MA, USA). Melting point was determined on a Kofler hot-stage apparatus and is uncorrected.
1H and
13C NMR spectra were taken with a Bruker AM-300 machine (Bruker AXS Handheld Inc., Kennewick, WA, USA) (at frequencies of 300 and 75 MHz) with TMS as the standard. J values are given in Hz. MS spectrum (EI, 70 eV) was obtained with a Finnigan MAT INCOS 50 instrument (Hazlet, NJ, USA). IR spectrum was measured with a Bruker “Alpha-T” instrument in KBr pellet. High-resolution MS spectrum was measured on a Bruker micrOTOF II instrument (Bruker Daltonik Gmbh, Bremen, Germany) using electrospray ionization (ESI).
A mixture of 2-((1-acetylindolin-5-yl)methyl)isoindoline-1,3-dione 2 (2 g, 6.3 mmol) and hydrazine hydrate (1 mL, 31.6 mmol) in methanol (20 mL) was refluxed for 3 h. Excess of methanol was removed under reduced pressure. Water (10 mL) and concentrated HCl (10 mL) were added to the residue. The mixture was heated with stirring for 3 h at 70 °C. After filtration of the precipitate the aqueous layer was evaporated, the residue was washed with acetone and dried in air. Yield 1.15 g (83%), white solid, mp 211–213 °C. IR spectrum (KBr), ν, cm–1: 3434, 3003, 2885, 2799 (all NH2 and NH), 2705, 2598, 2466 (all C-H), 1591 and 1576 (N-H), 1508, 1495, 1388, 1294, 1092, 914, 838, 593, 573, 431. 1H-NMR (DMSO-d6 + CF3COOH, ppm, J/Hz): δ 3.17 (2H, t, J = 7.7), 3.69 (2H, t, J = 8.1), 4.01 (2H, d, J = 5.1), 7.41 (1H, d, J = 8.1), 7.49 (1H, m), 7.58 (1H, s), 8.61 (3H, broad s). 13C-NMR (DMSO-d6, ppm): δ 28.9 (CH2), 41.9 (CH2-N), 44.9 (CH2-N), 119.7 (CH-Ar), 126.7 (CH-Ar), 129.1 (CH-Ar), 135.3 (C-Ar), 136.1 (C-Ar) 136.9 (C-Ar). MS (EI, 70 Ev), m/z (I, %): 148 (M+ − 2HCl, 100), 132 (M+ − NH2, 75), 118 (20), 91 (8), 36 (HCl, 33), 30 (16). HRMS (ESI-TOF): calcd for C9H12N2 [M + H]+ 149.1073; found m/z 149.1067. Anal. calcd. for C9H14Cl2N2: C, 48.88; H, 6.38; Cl 32.07; N, 12.67; found: C, 48.25; H, 6.43; Cl 31.96; N, 12.95%.
Salt 3 (1 g, 4.56 mmol) was dissolved in water (8 mL). NaOH solution (40%) was added dropwise at room temperature until pH = 9. The solution was extracted with CH2Cl2 (2 × 10 mL). The combined organic phases were dried over MgSO4, filtered, and concentrated under reduced pressure. Yield 0.52 g (77%), yellow oil. IR spectrum (KBr), ν, cm–1: 3359, 3282 and 3012 (all NH2 and NH), 2927, 2851 (C-H), 1615, 1496 (N-H), 1323, 1251, 1056, 943, 888, 816, 749, 623, 567, 420.1H-NMR (DMSO-d6, ppm, J/Hz): δ 2.45 (2H, broad s), 2.85 (2H, t, J = 8.4), 3.37 (2H, t, J = 8.4), 3.53 (2H, s), 5.28 (1H, broad s), 6.41 (1H, d, J = 8.1), 6.82 (1H, d, J = 7.3), 6.98 (1H s). 13C-NMR (DMSO-d6, ppm): δ 29.3 (CH2), 45.7 (CH2-N), 46.7 (CH2-N), 108.0 (CH-Ar), 123.4 (CH-Ar), 125.8 (CH-Ar), 128.8 (C-Ar), 132.9 (C-Ar) 151.1 (C-Ar). Mass spectrum (EI, 70 Ev), m/z (I, %): 148 (100), 132 (M+ − NH2, 79), 118 (25), 91 (12), 30 (13). HRMS (ESI-TOF): calcd. for C9H12N2 [M + H]+ 149.1073; found m/z 149.1068. Anal. calcd. for C9H12N2: C, 72.94; H, 8.16; N, 18.90; found: C, 72.31; H, 8.23; N, 19.01%.




{kind=link}
{kind=link}
