Genotyping by Sequencing Reasserts the Close Relationship between Tef and Its Putative Wild Eragrostis Progenitors


Abstract
:1. Introduction
2. Materials and Methods
2.1. Germplasm Panel
2.2. DNA Extraction, Library Preparation, and Genotyping by Sequencing
2.3. GBS Raw Data Processing
2.4. Mapping Reads to the Tef Reference Genome and SNP Calling
2.5. Mapping Reads to the Tef Pseudo-Chromosomes
2.6. Population Structure Analysis
2.7. Molecular Phylogenetic Analysis
3. Results
3.1. Genotyping by Sequencing of the ApeKI Eragrostis Species Library
3.2. Number of SNPs Correlates with Chromosome Length
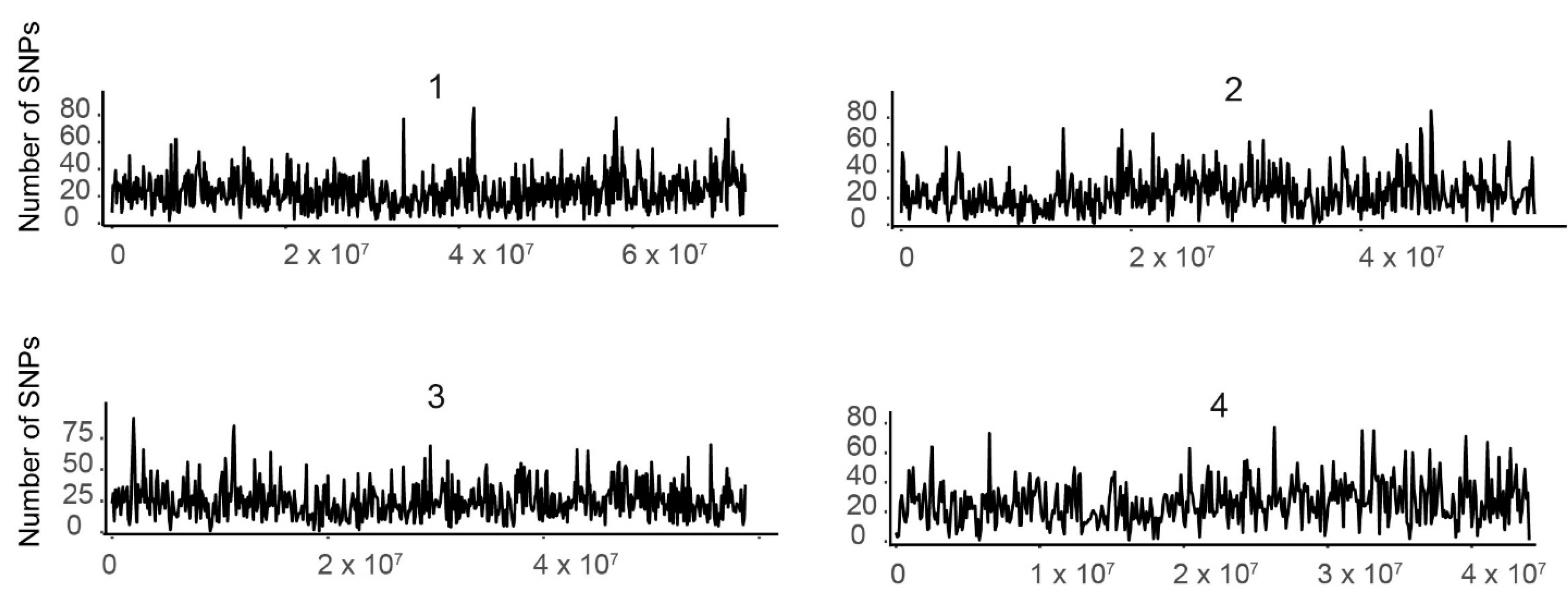
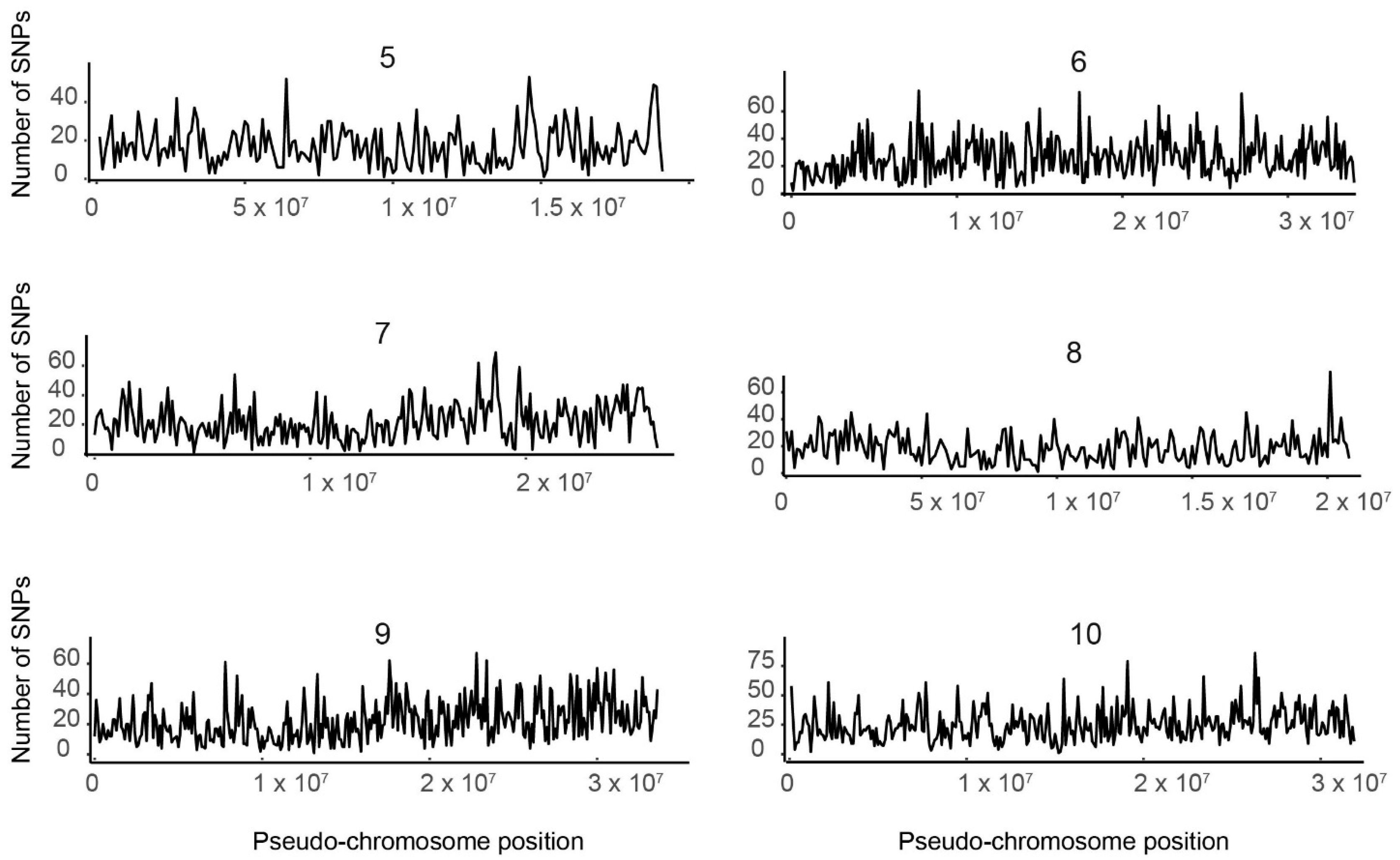
3.3. SNP Distribution along the 10 Tef Pseudomolecules
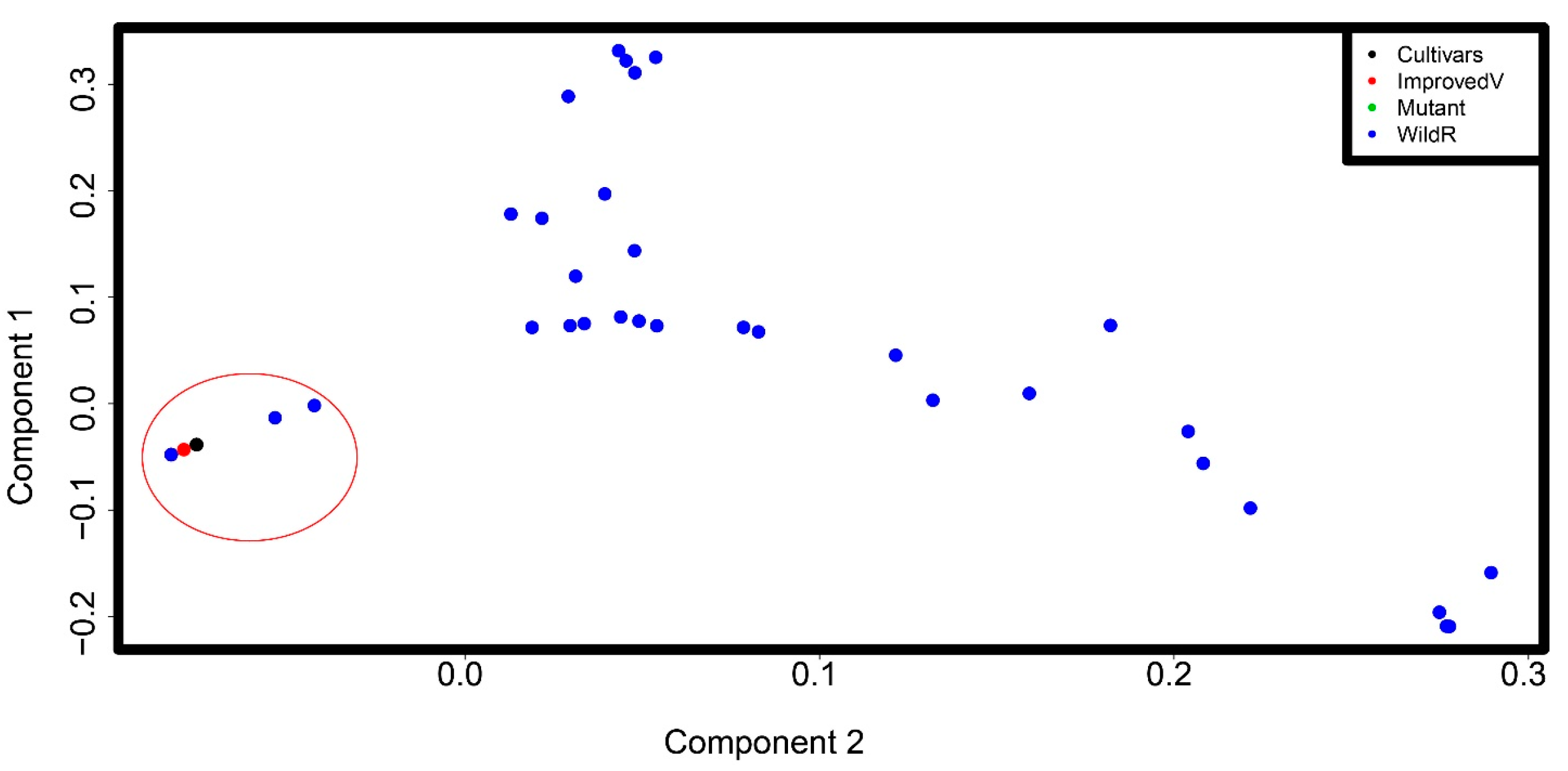
3.4. Principal Component Analysis Captures the Genetic Differentiation between Tef and Wild Eragrostis Species
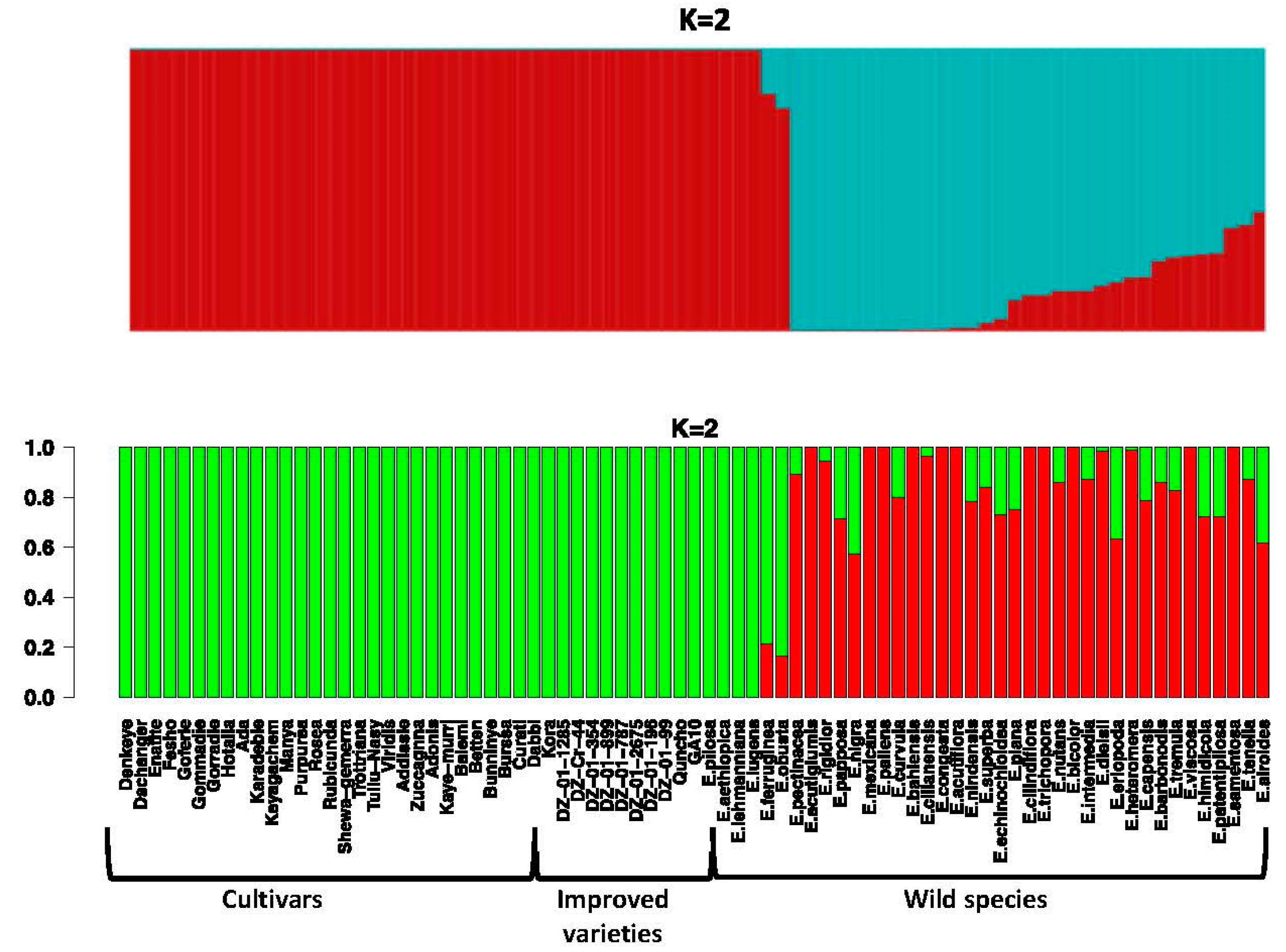
3.5. Population Structure in the Genus Eragrostis
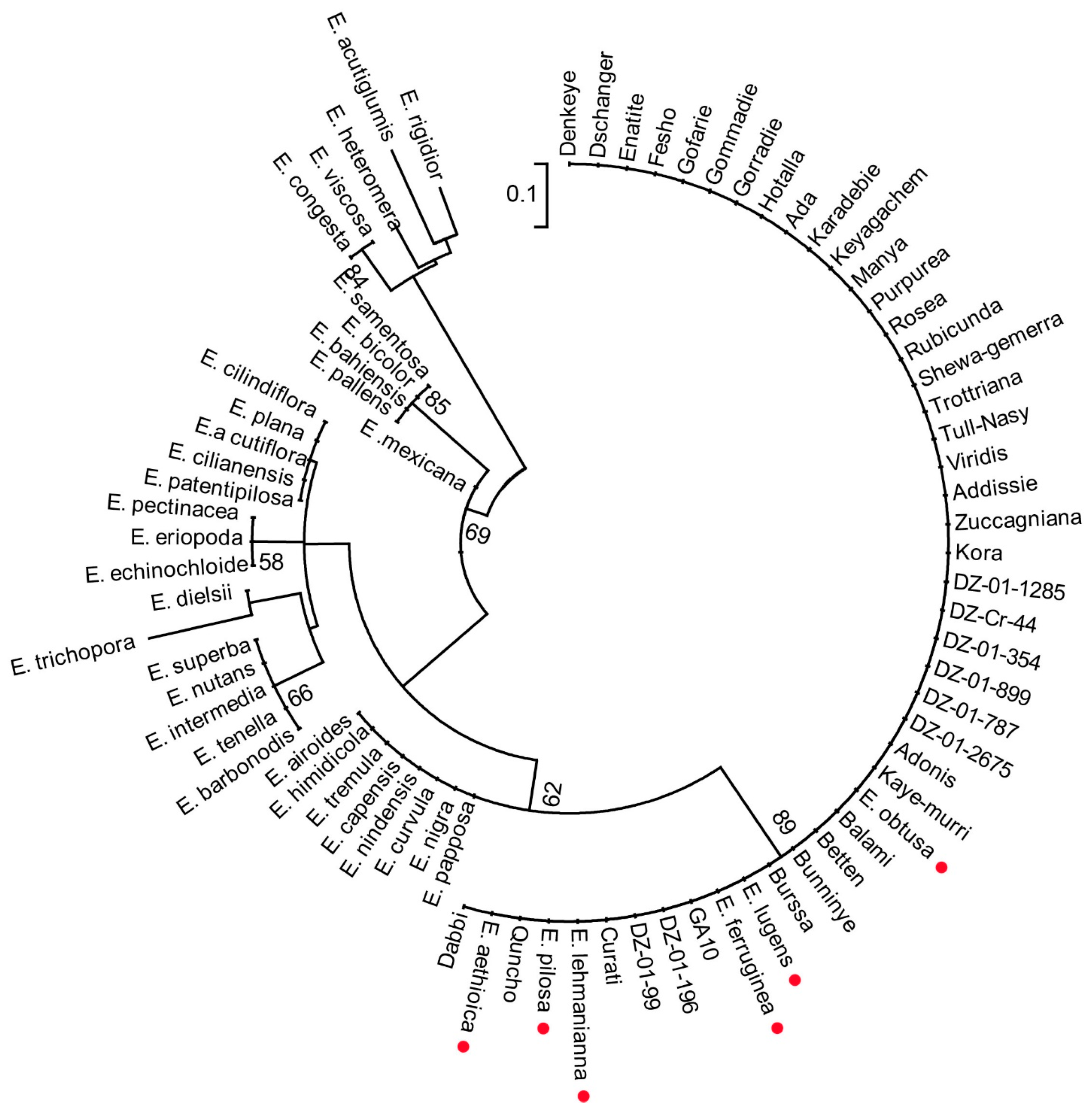
3.6. Molecular Phylogenetic Analysis Grouped Six Wild Species within the Tef Cultivars Clade
3.7. Wild Species Show High Level of Genetic Differentiation Compared to the Tef Cultivars Subpopulation
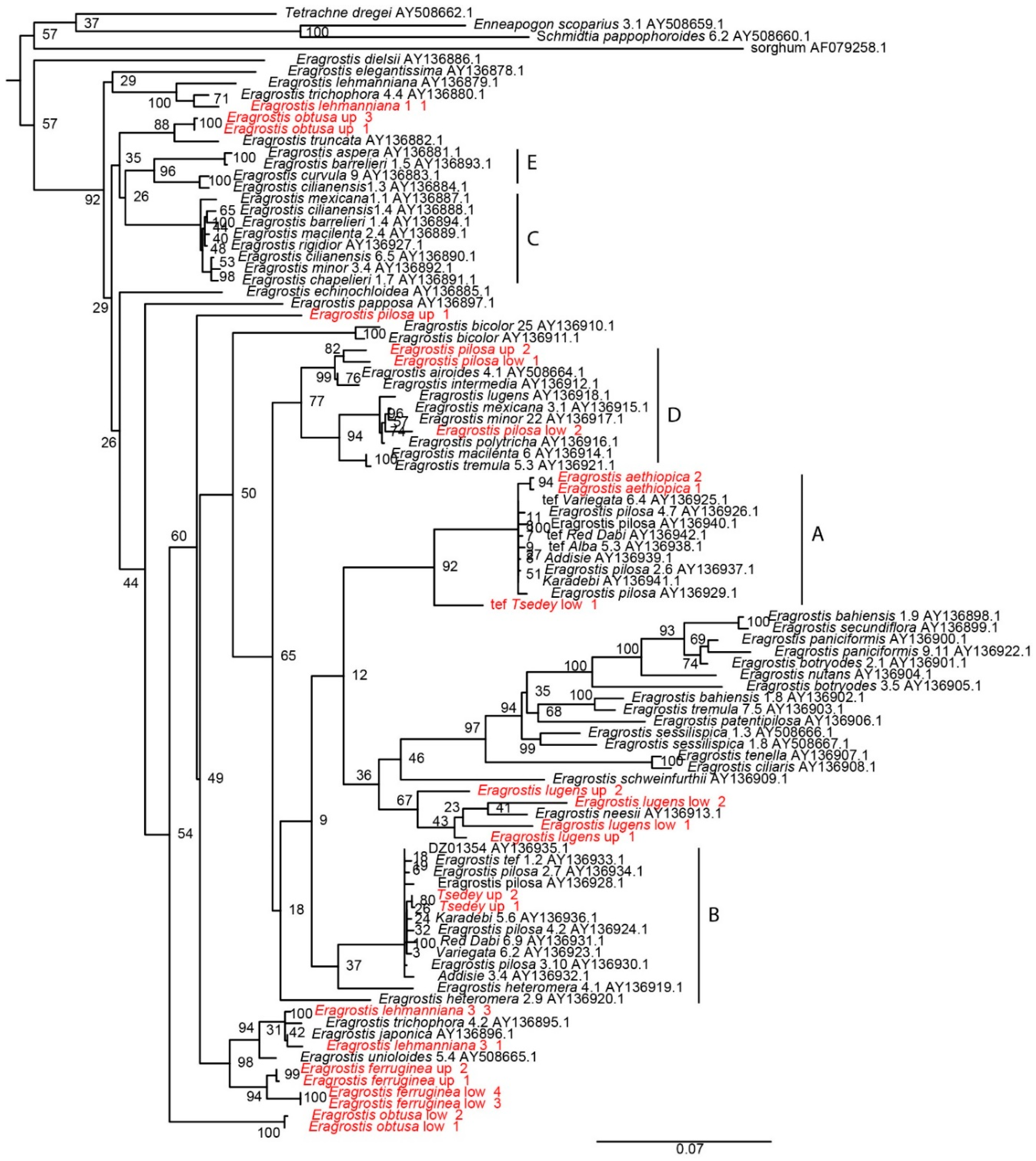
3.8. Phylogeny Tree from the Waxy Gene
4. Discussion
4.1. Genotyping by Sequencing Enabled Comprehensive Genomic Analysis of Eragrostis Species
4.2. Genomic Distribution of GBS-SNPs in the Tef Genome
4.3. Sequence Divergence between Tef Cultivars and Putative Wild Progenitors
4.4. Low Nucleotide Diversity in the Tef Species
4.5. Phylogenetic Analyses of Eragrostis Species Using Genome-Scale Data Reasserts Previously Reported Single-Gene–Based Analyses
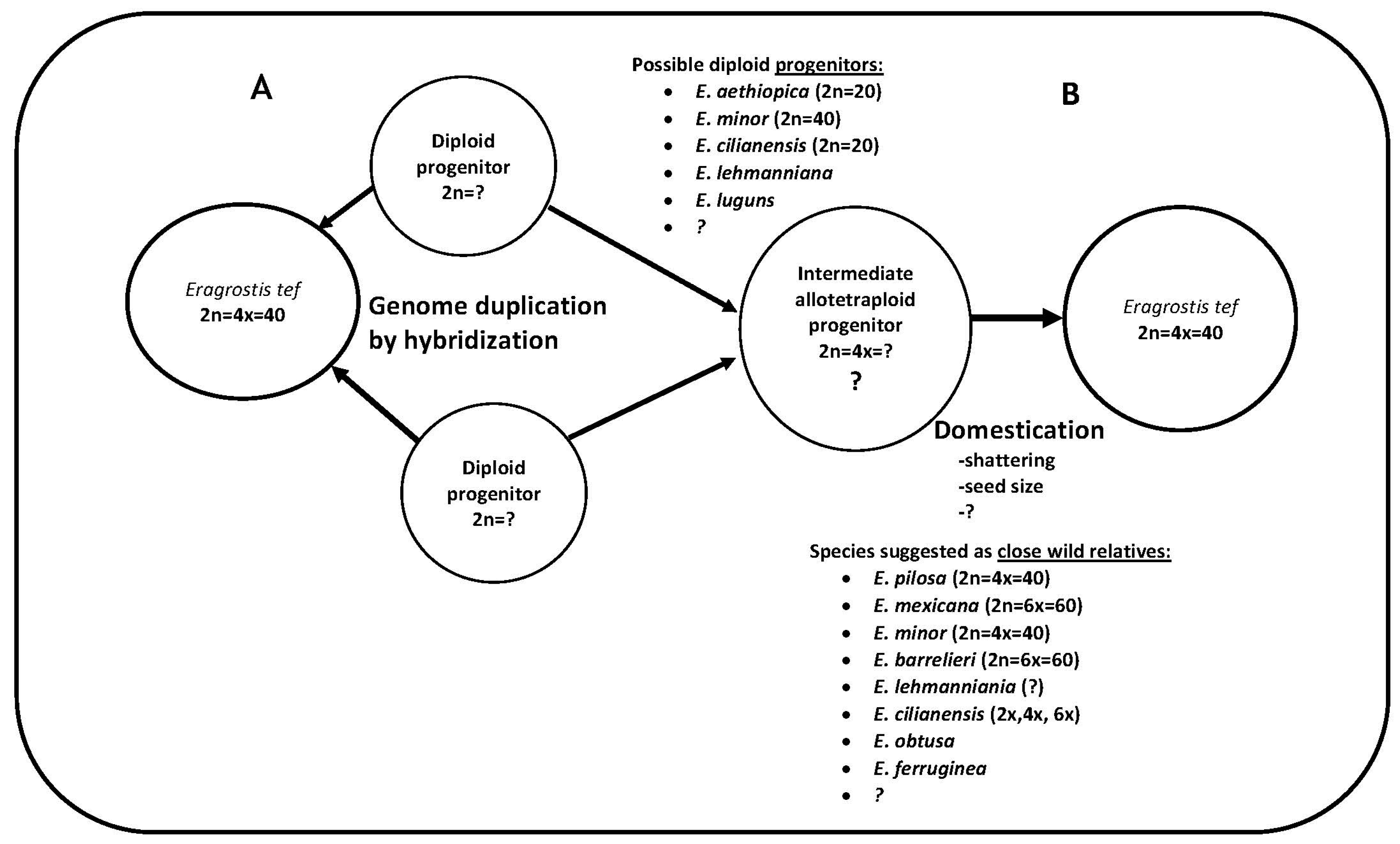
4.6. Coupling the Potential of the Wild Eragrostis Species with Tef Breeding
4.7. Deciphering the Diploid Pieces of the Allotetraploid Tef Genome
4.8. Phylogeny Tree from the Waxy Gene
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ketema, S. Tef, Eragrostis Tef (Zucc.) Trotter; Institute of Plant Genetics and Crop Plant Research, Gatersleben/International Plant Genetic Resources Institute: Rome, Italy, 1997; p. 52. [Google Scholar]
- Yemane, K.; Yilma, H. Food and grain losses in traditional storage facilities in three areas of Ethiopia. In Proceedings of the National Workshop on Food Strategies for Ethiopia, Alemaya University, Alemaya, Ethiopia, 8–12 December 1986; pp. 407–430. [Google Scholar]
- Central Statistical Agency (CSA). Agricultural Sample Survey for 2013/14; Central Statistical Agency: Addis Ababa, Ethiopia, 2014.
- Hamaker, B.R. Technology of Functional Cereal Products; Woodhead Publishing: Sawston, UK, 2007. [Google Scholar]
- Tome, D.; Bos, C. Lysine requirement through the human life cycle. J. Nutr. 2007, 137, 1642s–1645s. [Google Scholar] [CrossRef] [PubMed]
- Spaenij-Dekking, L.; Kooy-Winkelaar, Y.; Koning, F. The Ethiopian cereal tef in celiac disease. N. Engl. J. Med. 2005, 353, 1748–1749. [Google Scholar] [CrossRef] [PubMed]
- Cannarozzi, G.; Plaza-Wuthrich, S.; Esfeld, K.; Larti, S.; Wilson, Y.S.; Girma, D.; de Castro, E.; Chanyalew, S.; Blosch, R.; Farinelli, L.; et al. Genome and transcriptome sequencing identifies breeding targets in the orphan crop tef (Eragrostis tef). BMC Genom. 2014, 15, 581. [Google Scholar] [CrossRef] [PubMed]
- Clayton, S.D.; Renvoize, S.A. Genera Graminum: Grasses of the World; University of Chicago Press: Chicago, IL, USA, 1986. [Google Scholar]
- Vavilov, I. The Origin, Variation, Immunity and Breeding of Cultivated Plants; Translated from the Russian by Chester, K.S.; Ronald Press Co.: New York, NY, USA, 1951. [Google Scholar]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Hu, Y.; Mao, B.; Xiang, H.; Shao, Y.; Pan, Y.; Sheng, X.; Li, Y.; Ni, X.; Xia, Y.; et al. Genetic analysis for rice grain quality traits in the YVB stable variant line using RAD-seq. Mol. Genet. Genom. 2016, 291, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Begum, H.; Spindel, J.E.; Lalusin, A.; Borromeo, T.; Gregorio, G.; Hernandez, J.; Virk, P.; Collard, B.; McCouch, S.R. Genome-wide association mapping for yield and other agronomic traits in an elite breeding population of tropical rice (Oryza sativa). PLoS ONE 2015, 10, e0119873. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.J.; Wu, T.T.; Ye, J.; Sun, J.; Jiang, Y.; Yu, J.; Tang, J.P.; Chen, G.M.; Wang, C.M.; Wan, J.M. SNP-based analysis of genetic diversity reveals important alleles associated with seed size in rice. BMC Plant Biol. 2016, 16. [Google Scholar] [CrossRef]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Bayer, M.; Druka, A.; Russell, J.R.; Hackett, C.A.; Poland, J.; Ramsay, L.; Hedley, P.E.; Waugh, R. An evaluation of genotyping by sequencing (GBS) to map the Breviaristatum-e (ari-e) locus in cultivated barley. BMC Genom. 2014, 15, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.P.; Ramu, P.; Deshpande, S.P.; Hash, C.T.; Shah, T.; Upadhyaya, H.D.; Riera-Lizarazu, O.; Brown, P.J.; Acharya, C.B.; Mitchell, S.E.; et al. Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc. Natl. Acad. Sci. USA 2013, 110, 453–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipka, A.E.; Gore, M.A.; Magallanes-Lundback, M.; Mesberg, A.; Lin, H.; Tiede, T.; Chen, C.; Buell, C.R.; Buckler, E.S.; Rocheford, T.; et al. Genome-wide association study and pathway-level analysis of tocochromanol levels in maize grain. G3 2013, 3, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Romay, M.C.; Millard, M.J.; Glaubitz, J.C.; Peiffer, J.A.; Swarts, K.L.; Casstevens, T.M.; Elshire, R.J.; Acharya, C.B.; Mitchell, S.E.; Flint-Garcia, S.A.; et al. Comprehensive genotyping of the USA national maize inbred seed bank. Genome Biol. 2013, 14, R55. [Google Scholar] [CrossRef] [PubMed]
- Takuno, S.; Ralph, P.; Swarts, K.; Elshire, R.J.; Glaubitz, J.C.; Buckler, E.S.; Hufford, M.B.; Ross-Ibarra, J. Independent molecular basis of convergent highland adaptation in Maize. Genetics 2015, 200, 1297–1312. [Google Scholar] [CrossRef] [PubMed]
- Punnuri, S.M.; Wallace, J.G.; Knoll, J.E.; Hyma, K.E.; Mitchell, S.E.; Buckler, E.S.; Varshney, R.K.; Singh, B.P. Development of a high-density linkage map and tagging leaf spot resistance in Pearl Millet using genotyping-by-sequencing markers. Plant Genome 2016, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, D.; Das, S.; Upadhyaya, H.D.; Ranjan, R.; Badoni, S.; Kumar, V.; Tripathi, S.; Gowda, C.L.; Sharma, S.; Singh, S.; et al. A Genome-wide combinatorial strategy dissects complex genetic architecture of seed coat color in Chickpea. Front. Plant Sci. 2015, 6, 979. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-F.; Poland, J.A.; Wight, C.P.; Jackson, E.W.; Tinker, N.A. Using genotyping-by-sequencing (GBS) for genomic discovery in cultivated oat. PLoS ONE 2014, 9, e102448–10. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.; Upadhyaya, H.; Vetriventhan, M.; Buckler, E.; Hash, T.; Ramu, P. The genetic makeup of a global barnyard millet germplasm collection. Plant Genome 2015, 1–39. [Google Scholar] [CrossRef]
- Wang, S.; Chen, J.D.; Zhang, W.P.; Hu, Y.; Chang, L.J.; Fang, L.; Wang, Q.; Lv, F.N.; Wu, H.T.; Si, Z.F.; et al. Sequence-based ultra-dense genetic and physical maps reveal structural variations of allopolyploid cotton genomes. Genome Biol. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Hyma, K.E.; Barba, P.; Wang, M.; Londo, J.P.; Acharya, C.B.; Mitchell, S.E.; Sun, Q.; Reisch, B.; Cadle-Davidson, L. Heterozygous mapping strategy (HetMappS) for high resolution genotyping-by-sequencing markers: A case study in Grapevine. PLoS ONE 2015, 10, e0134880. [Google Scholar] [CrossRef] [PubMed]
- Hart, J.P.; Griffiths, P.D. Genotyping-by-sequencing enabled mapping and marker development for the by-2 potyvirus resistance allele in common bean. Plant Genome 2015, 8. [Google Scholar] [CrossRef]
- Lipka, A.E.; Lu, F.; Cherney, J.H.; Buckler, E.S.; Casler, M.D.; Costich, D.E. Accelerating the switchgrass (Panicum virgatum L.) breeding cycle using genomic selection approaches. PLoS ONE 2014, 9, e112227. [Google Scholar] [CrossRef] [PubMed]
- Ebba, T. Tef Cultivars: Morphology and Classification; Addis Ababa University, College of Agriculture: Dire Dawa, Ethiopia, 1975. [Google Scholar]
- Ministry of Agriculture (MoA). Crop Variety Register Issue No. 15; Ministry of Agriculture, Animal and Plant Health Regulatory Directorate: Addis Ababa, Ethiopia, 2014.
- Tadele, Z. Tef Improvement Project: Harnessing genetic and genomic tools to boost productivity. In Achievements and Prospects of Tef Improvement; Assefa, K., Chanyalew, S., Tadele, Z., Eds.; EIAR-University of Bern: Bern, Switzerland, 2013; pp. 333–342. [Google Scholar]
- Chua, K.Y.; Doyle, C.R.; Simpson, R.J.; Turner, K.J.; Stewart, G.A.; Thomas, W.R. Isolation of cDNA coding for the major mite allergen Der p II by IgE plaque immunoassay. Int. Arch. Allergy Appl. Immunol. 1990, 91, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 2014, 9, e90346. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; Stephens, M.; Pritchard, J.K. fastSTRUCTURE: Variational inference of population structure in large SNP data sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML Version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Richter, D.C.; Rausch, C.; Dezulian, T.; Franz, M.; Rupp, R. Dendroscope: An interactive viewer for large phylogenetic trees. BMC Bioinform. 2007, 8, 460. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Nei, M.; Li, W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef] [PubMed]
- Ingram, A.L.; Doyle, J.J. The origin and evolution of Eragrostis tef (Poaceae) and related polyploids: Evidence from nuclear waxy and plastid rps16. Am. J. Bot. 2003, 90, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ning, Z.; Hu, Y.; Chen, J.; Zhao, R.; Chen, H.; Ai, N.; Guo, W.; Zhang, T. Molecular mapping of restriction-site associated DNA markers in Allotetraploid Upland Cotton. PLoS ONE 2015, 10, e0124781. [Google Scholar] [CrossRef] [PubMed]
- Arai-Kichise, Y.; Shiwa, Y.; Nagasaki, H.; Ebana, K.; Yoshikawa, H.; Yano, M.; Wakasa, K. Discovery of genome-wide DNA polymorphisms in a landrace cultivar of Japonica rice by whole-genome sequencing. Plant Cell Physiol. 2011, 52, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Nagasaki, H.; Yonemaru, J.; Ebana, K.; Nakajima, M.; Shibaya, T.; Yano, M. Fine definition of the pedigree haplotypes of closely related rice cultivars by means of genome-wide discovery of single-nucleotide polymorphisms. BMC Genom. 2010, 11, 267. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.; Lorenc, M.T.; Lee, H.C.; Berkman, P.J.; Bayer, P.E.; Visendi, P.; Ruperao, P.; Fitzgerald, T.L.; Zander, M.; Chan, C.K.; et al. Identification and characterization of more than 4 million intervarietal SNPs across the group 7 chromosomes of bread wheat. Plant Biotechnol. J. 2015, 13, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Hao, D.; Chao, M.; Yin, Z.; Yu, D. Genome-wide association analysis detecting significant single nucleotide polymorphisms for chlorophyll and chlorophyll fluorescence parameters in soybean (Glycine max) landraces. Euphytica 2012, 186, 919–931. [Google Scholar] [CrossRef]
- Yadav, C.B.; Bhareti, P.; Muthamilarasan, M.; Mukherjee, M.; Khan, Y.; Rathi, P.; Prasad, M. Genome-wide SNP identification and characterization in two soybean cultivars with contrasting Mungbean Yellow Mosaic India Virus disease resistance traits. PLoS ONE 2015, 10, e0123897. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Izzah, N.K.; Jayakodi, M.; Perumal, S.; Joh, H.J.; Lee, H.J.; Lee, S.C.; Park, J.Y.; Yang, K.W.; Nou, I.S.; et al. Genome-wide SNP identification and QTL mapping for black rot resistance in cabbage. BMC Plant Biol. 2015, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Kujur, A.; Upadhyaya, H.D.; Shree, T.; Bajaj, D.; Das, S.; Saxena, M.S.; Badoni, S.; Kumar, V.; Tripathi, S.; Gowda, C.L.; et al. Ultra-high density intra-specific genetic linkage maps accelerate identification of functionally relevant molecular tags governing important agronomic traits in chickpea. Sci. Rep. 2015, 5, 9468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingram, A.; Doyle, J. Eragrostis (Poaceae): Monophyly and infrageneric classification. J. Syst. Evolut. Bot. 2007, 23. [Google Scholar] [CrossRef]
- Buckler, E.S.T.; Thornsberry, J.M.; Kresovich, S. Molecular diversity, structure and domestication of grasses. Genet. Res. 2001, 77, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Berhe, T.; Nelson, L.A.; Morris, M.R.; Schmidt, J.W. The genetics of qualitative traits in tef. In Proceedings of the International Workshop on Tef Genetics and Improvement, Debre Zeit, Ethiopia, 16–19 October 2000; pp. 79–85. [Google Scholar]
- Assefa, K.; Yu, J.K.; Zeid, M.; Belay, G.; Tefera, H.; Sorrells, M.E. Breeding tef [Eragrostis tef (Zucc.) trotter]: Conventional and molecular approaches. Plant Breed. 2011, 130, 1–9. [Google Scholar] [CrossRef]
- Smith, S.M.; Yuan, Y.; Doust, A.N.; Bennetzen, J.L. Haplotype analysis and linkage disequilibrium at five loci in Eragrostis tef. G3 2012, 2, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Zheng, Z.; Wang, L.; Liu, X.; Zhu, G.; Fang, W.; Cheng, S.; Zeng, P.; Chen, C.; Wang, X.; et al. Comparative population genomics reveals the domestication history of the peach, Prunus persica, and human influences on perennial fruit crops. Genome Biol. 2014, 15, 415. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Burke, J.M. Patterns of nucleotide diversity in wild and cultivated sunflower. Genetics 2006, 173, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.A.; Gaut, B.S.; Stec, A.O.; Fuerstenberg, S.I.; Goodman, M.M.; Coe, E.H.; Doebley, J.F. Evolution of anthocyanin biosynthesis in maize kernels: The role of regulatory and enzymatic loci. Genetics 1996, 143, 1395–1407. [Google Scholar] [PubMed]
- Tanksley, S.D.; McCouch, S.R. Seed banks and molecular maps: Unlocking genetic potential from the wild. Science 1997, 277, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Tenaillon, M.I.; U’Ren, J.; Tenaillon, O.; Gaut, B.S. Selection versus demography: A multilocus investigation of the domestication process in maize. Mol. Biol. Evol. 2004, 21, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Willing, E.M.; Hoffmann, M.; Klein, J.D.; Weigel, D.; Dreyer, C. Paired-end RAD-seq for de novo assembly and marker design without available reference. Bioinformatics 2011, 27, 2187–2193. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. The genetical structure of populations. Ann. Eugen. 1951, 15, 323–354. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.K.; Kantety, R.V.; Graznak, E.; Benscher, D.; Tefera, H.; Sorrells, M.E. A genetic linkage map for tef [Eragrostis tef (Zucc.) Trotter]. Theor. Appl. Genet. 2006, 113, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, W.F.; Logsdon, J.M., Jr. Archaeal genomics: Do archaea have a mixed heritage? Curr. Biol. 1998, 8, R209–R211. [Google Scholar] [CrossRef]
- Huynen, M.A.; Bork, P. Measuring genome evolution. Proc. Natl. Acad. Sci. USA 1998, 95, 5849–5856. [Google Scholar] [CrossRef] [PubMed]
- Snel, B.; Bork, P.; Huynen, M.A. Genome phylogeny based on gene content. Nat. Genet. 1999, 21, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.T.; Beckstrom-Sternberg, S.M.; Pearson, T.; Beckstrom-Sternberg, J.S.; Chain, P.S.; Roberto, F.F.; Hnath, J.; Brettin, T.; Keim, P. Whole-genome-based phylogeny and divergence of the genus Brucella. J. Bacteriol. 2009, 191, 2864–2870. [Google Scholar] [CrossRef] [PubMed]
- Bekele, E.; Lester, R.N. Biochemical Assessment of the Relationships of Eragrostis tef (Zucc.) Trotter with some Wild Eragrostis Species (Gramineae). Ann. Bot. 1981, 48, 717–725. [Google Scholar] [CrossRef]
- Costanza, S.H.; deWet, J.M.J.; Harlan, J.R. Literature review and numerical taxonomy of Eragrostis tef (T’ef). Econ. Bot. 1979, 33, 413–424. [Google Scholar] [CrossRef]
- Jones, B.M.G.; Ponti, J.; Tavassoli, A.; Dixon, P.A. Relationships of the Ethiopian Cereal T′ef (Eragrostis tef (Zucc.) Trotter): Evidence from morphology and chromosome number. Ann. Bot. 1978, 42, 1369–1373. [Google Scholar] [CrossRef]
- Prescott-Allen, R.; Prescott-Allen, C. Using Wild Genetic Resources for Food and Raw Materials; Earthscan Publications: London, UK, 1988. [Google Scholar]
- Hajjar, R.; Hodgkin, T. The use of wild relatives in crop improvement: A survey of developments over the last 20 years. Euphytica 2007, 156, 1–13. [Google Scholar] [CrossRef]
- Dempewolf, H.; Eastwood, R.J.; Guarino, L.; Khoury, C.K.; Müller, J.V.; Toll, J. Adapting agriculture to climate change: A global initiative to collect, conserve, and use crop wild relatives. Agroecol. Sustain. Food Syst. 2014, 38, 369–377. [Google Scholar] [CrossRef]
- Berhe, T. A break-through in tef breeding techniques. FAO Int. Bull. Cereal Improv. Prod. 1975, 12, 11–13. [Google Scholar]
- Zhang, D.; Ayele, M.; Tefera, H.; Nguyen, H.T. RFLP linkage map of the Ethiopian cereal tef [Eragrostis tef (Zucc) Trotter]. Theor. Appl. Genet. 2001, 102, 957–964. [Google Scholar] [CrossRef]
- Gottlieb, L.D. Plant polyploidy: Gene expression and genetic redundancy. Heredity 2003, 91, 91–92. [Google Scholar] [CrossRef] [PubMed]
- Ranwez, V.; Holtz, Y.; Sarah, G.; Ardisson, M.; Santoni, S.; Glémin, S.; Tavaud-Pirra, M.; David, J. Disentangling homeologous contigs in allo-tetraploid assembly: Application to durum wheat. BMC Bioinform. 2013, 14, S15–S11. [Google Scholar] [CrossRef] [PubMed]
- Tennessen, J.A.; Govindarajulu, R.; Ashman, T.-L.; Liston, A. Evolutionary origins and dynamics of octoploid strawberry subgenomes revealed by dense targeted capture linkage maps. Genome Biol. Evolut. 2014, 6, 3295–3313. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | [73] | [72] | [71] | [45] | This Study |
|---|---|---|---|---|---|
| Method | Morphological and cytological analysis | Morphometric analysis | Biochemical analysis | Nuclear gene waxy Platid rps16 loci | GBS |
| Suggested ancestor(s) of tef | E. aethiopica 2 | E. aethiopica 2 | E. aethiopica 2 | E. pilosa 1 | E. aethiopica 2 |
| E. pilosa 1 | E. pilosa 1 | E. pilosa 1 | E. longifolia | E. pilosa 1 | |
| E. bicolor | E. macilenta | E. barrelieri | E. lehmanniana | ||
| E. cilianensis | E. bicolor | E. lugens | |||
| E. heteromera | E. cilianensis | E. obtusa | |||
| E. mexicana | E. curvula | E. ferruginea | |||
| E. minor | E. diploachnoides | ||||
| E. papposa | E. heteromera | ||||
| E. barrelieri | E. mexicana | ||||
| E. minor | |||||
| E. papposa | |||||
| E. viscosa |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girma, D.; Cannarozzi, G.; Weichert, A.; Tadele, Z. Genotyping by Sequencing Reasserts the Close Relationship between Tef and Its Putative Wild Eragrostis Progenitors. Diversity 2018, 10, 17. https://doi.org/10.3390/d10020017
Girma D, Cannarozzi G, Weichert A, Tadele Z. Genotyping by Sequencing Reasserts the Close Relationship between Tef and Its Putative Wild Eragrostis Progenitors. Diversity. 2018; 10(2):17. https://doi.org/10.3390/d10020017
Chicago/Turabian StyleGirma, Dejene, Gina Cannarozzi, Annett Weichert, and Zerihun Tadele. 2018. "Genotyping by Sequencing Reasserts the Close Relationship between Tef and Its Putative Wild Eragrostis Progenitors" Diversity 10, no. 2: 17. https://doi.org/10.3390/d10020017
APA StyleGirma, D., Cannarozzi, G., Weichert, A., & Tadele, Z. (2018). Genotyping by Sequencing Reasserts the Close Relationship between Tef and Its Putative Wild Eragrostis Progenitors. Diversity, 10(2), 17. https://doi.org/10.3390/d10020017