
Protein Structure, Models of Sequence Evolution, and Data Type Effects in Phylogenetic Analyses of Mitochondrial Data: A Case Study in Birds



Abstract
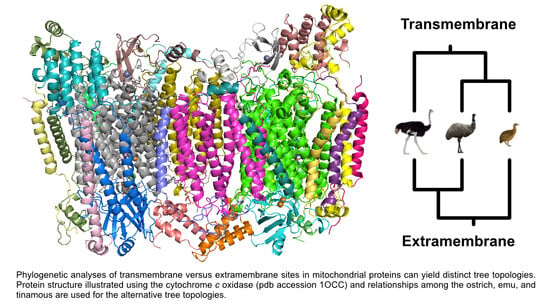
:
1. Introduction
2. Materials and Methods
2.1. Data Matrix Construction
2.2. Analyses of Molecular Evolution and Phylogeny
3. Results
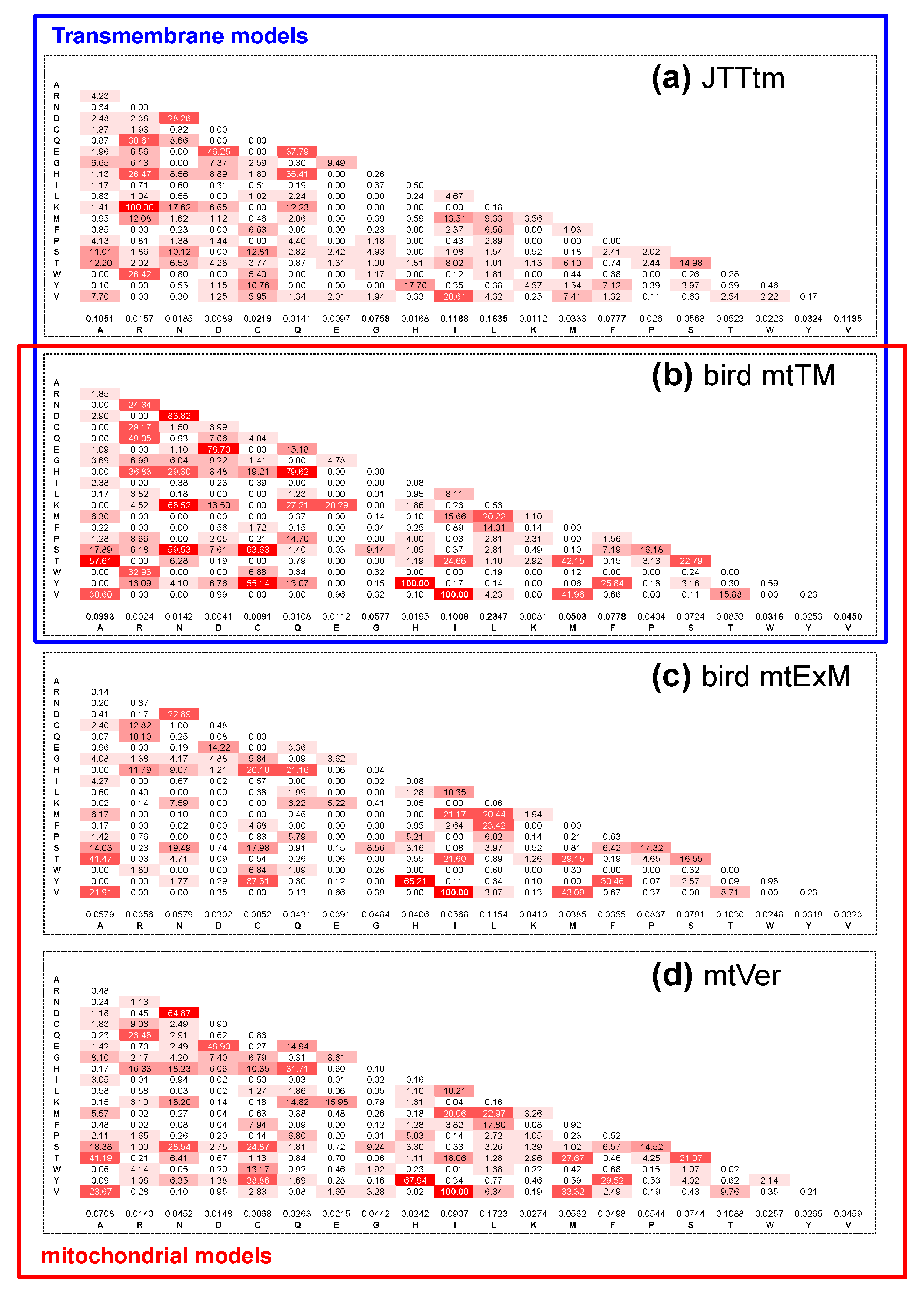
3.1. Do the mtTM (Transmembrane) and mtExM (Extramembrane) Models Differ?
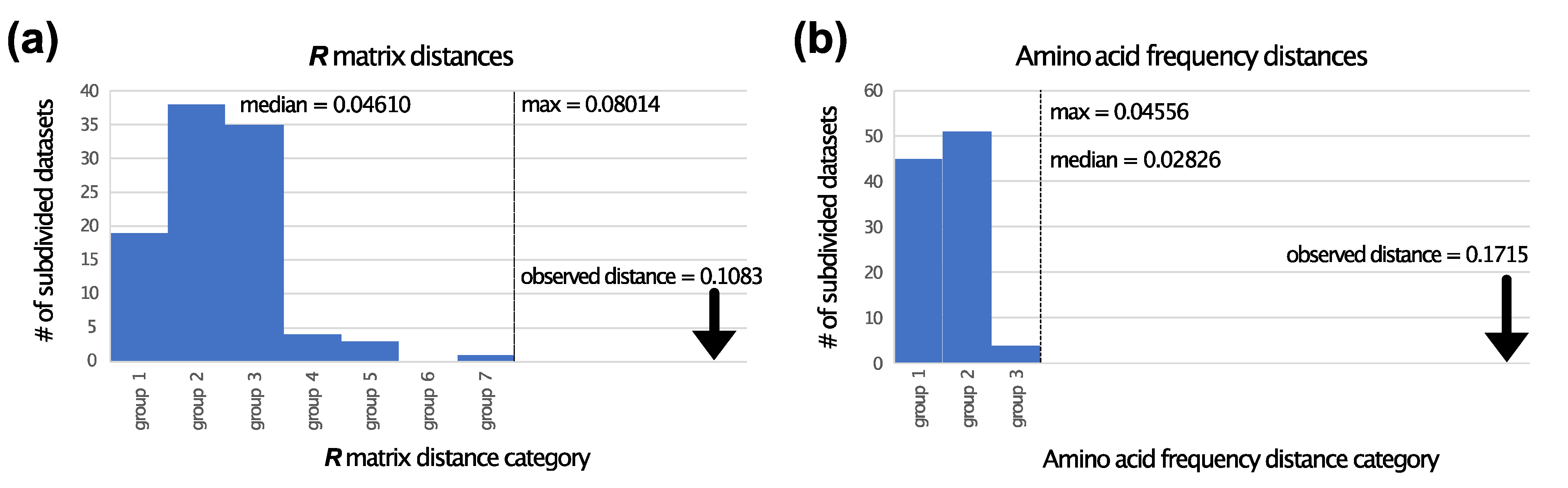
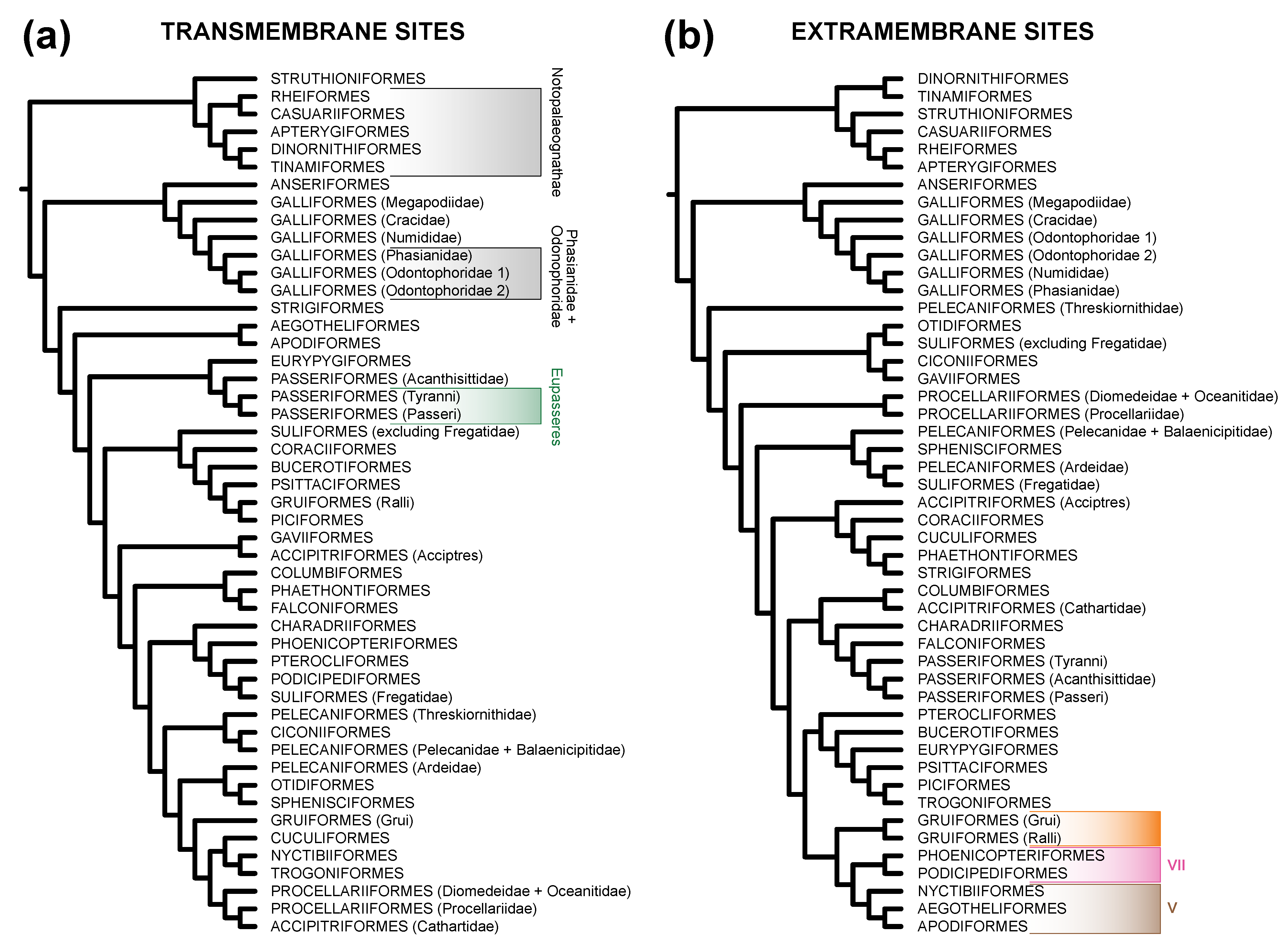
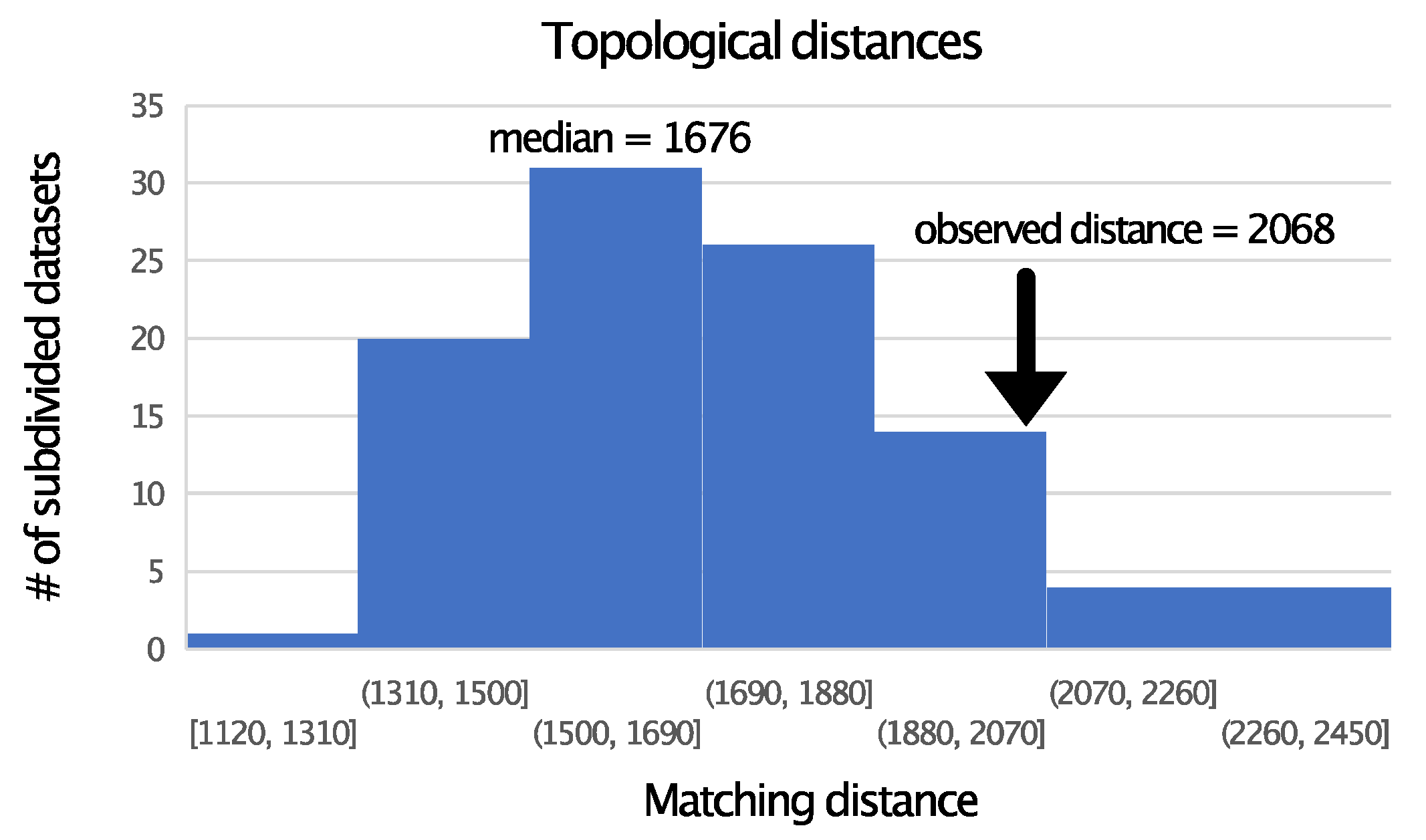
3.2. TM Helix and ExM Loops Tree Topologies: Stochastic Error, Not Data Type Effects
3.3. Is There Evidence for Heterogeneity within TM and ExM Sites?
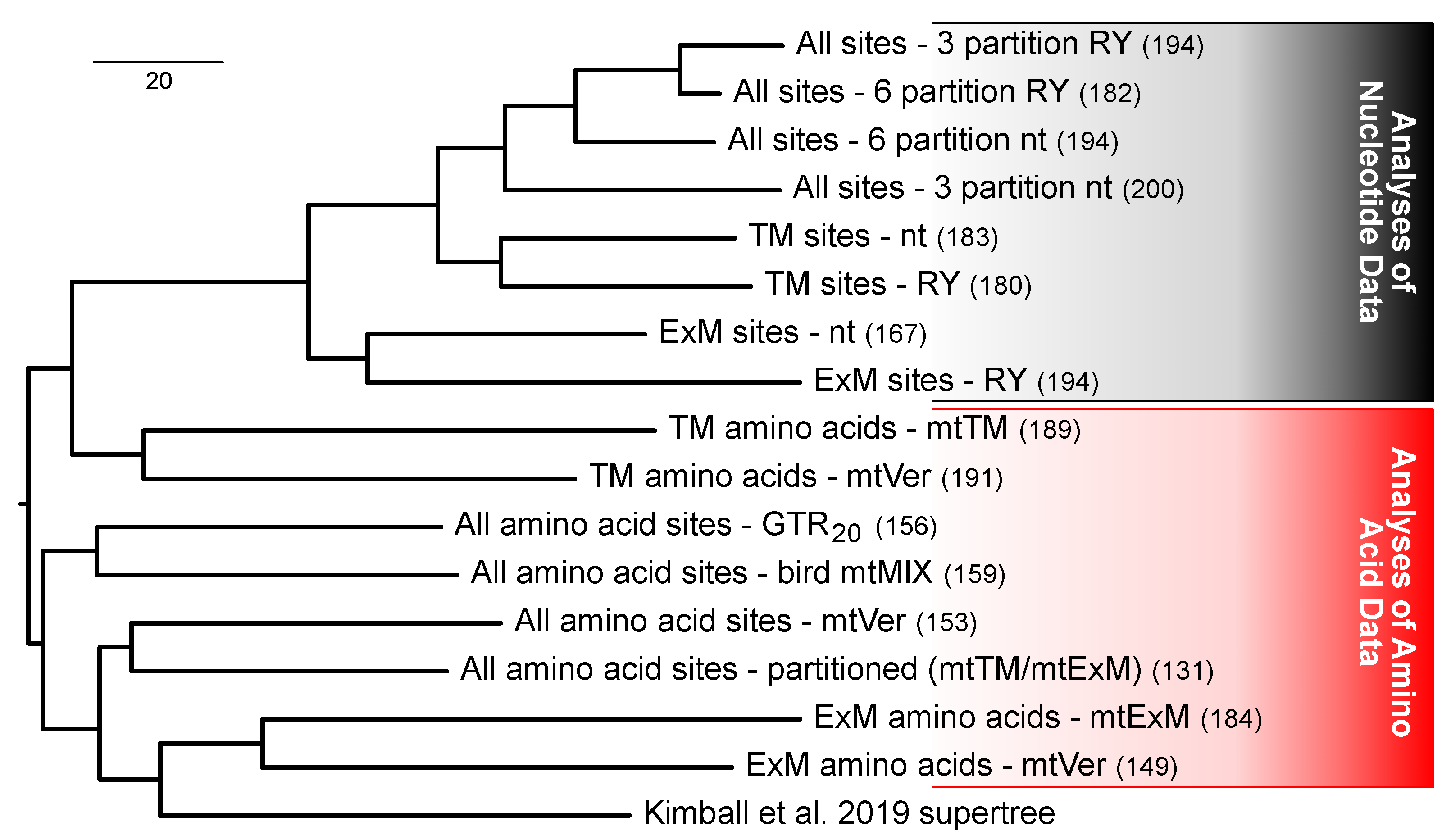
3.4. Protein Structure Has an Impact on Analyses of Nucleotide and Purine-Pyrimidine Data
3.5. Multiple Factors Shape the Tree Space for Analyses of Mitochondrial Proteins
4. Discussion
4.1. Data Type Effects and Process Partitions
4.2. Models of Transmembrane Protein Evolution and the NB Hypothesis
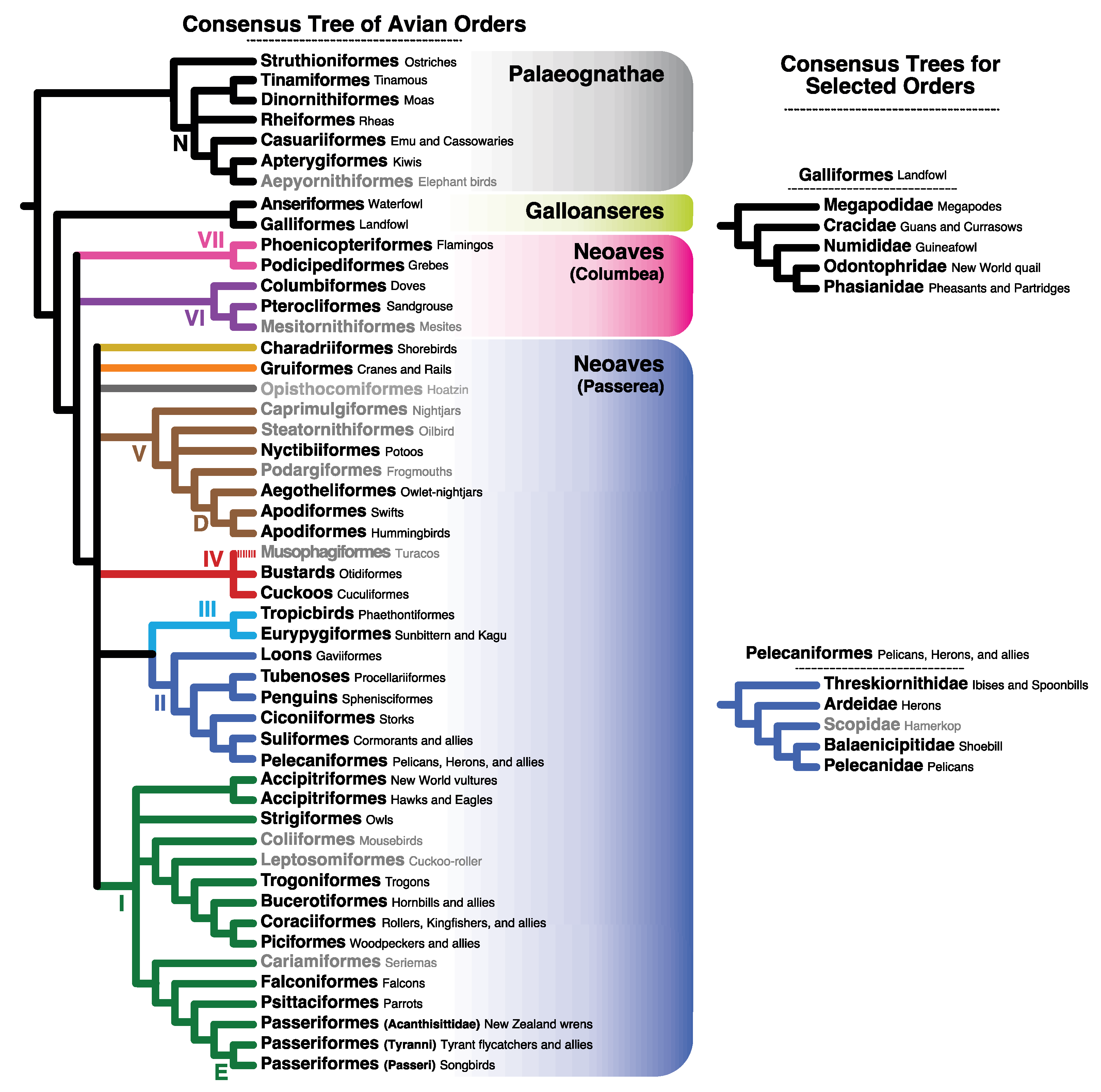
4.3. Implications for Avian Systematics and Evolution
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gee, H. Evolution: Ending incongruence. Nature 2003, 425, 782. [Google Scholar] [CrossRef]
- Rokas, A.; Williams, B.L.; King, N.; Carroll, S.B. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 2003, 425, 798–804. [Google Scholar] [CrossRef]
- Jeffroy, O.; Brinkmann, H.; Delsuc, F.; Philippe, H. Phylogenomics: The beginning of incongruence? Trends Genet. 2006, 22, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Pamilo, P.; Nei, M. Relationships between gene trees and species trees. Mol. Biol. Evol. 1988, 5, 568–583. [Google Scholar] [CrossRef]
- Maddison, W.P. Gene trees in species trees. Syst. Biol. 1997, 46, 523–536. [Google Scholar] [CrossRef]
- Edwards, S.V. Is a new and general theory of molecular systematics emerging? Evolution 2009, 63, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degnan, J.H.; Rosenberg, N.A. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 2009, 24, 332–340. [Google Scholar] [CrossRef]
- Edwards, S.V.; Xi, Z.; Janke, A.; Faircloth, B.C.; McCormack, J.E.; Glenn, T.C.; Zhong, B.; Wu, S.; Lemmon, E.M.; Lemmon, A.R.; et al. Implementing and testing the multispecies coalescent model: A valuable paradigm for phylogenomics. Mol. Phylogenet. Evol. 2016, 94, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Braun, E.L.; Kimball, R.T. Polytomies, the power of phylogenetic inference, and the stochastic nature of molecular evolution: A comment on Walsh (1999). Evolution 2001, 55, 1261–1263. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Kimball, R.T.; Pandey, A.; Hosner, P.A.; Braun, M.J.; Hackett, S.J.; Han, K.-L.; Harshman, J.; Huddleston, C.J.; Kingston, S.; et al. Why do phylogenomic data sets yield conflicting trees? Data type influences the avian tree of life more than taxon sampling. Syst. Biol. 2017, 66, 857–879. [Google Scholar] [CrossRef]
- Bull, J.J.; Huelsenbeck, J.P.; Cunningham, C.W.; Swofford, D.L.; Waddell, P.J. Partitioning and combining data in phylogenetic analysis. Syst. Biol. 1993, 42, 384. [Google Scholar] [CrossRef]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.W.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prum, R.O.; Berv, J.S.; Dornburg, A.; Field, D.J.; Townsend, J.P.; Lemmon, E.M.; Lemmon, A.R. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature 2015, 526, 569–573. [Google Scholar] [CrossRef]
- Braun, E.L.; Kimball, R.T. Data types and the phylogeny of Neoaves. Birds 2021, 2, 1. [Google Scholar] [CrossRef]
- Chen, M.-Y.; Liang, D.; Zhang, P. Phylogenomic resolution of the phylogeny of laurasiatherian mammals: Exploring phylogenetic signals within coding and noncoding sequences. Genome Biol. Evol. 2017, 9, 1998–2012. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.O.; Hutter, C.R.; Wood, P.L.; Grismer, L.L.; Brown, R.M. Larger, Unfiltered datasets are more effective at resolving phylogenetic conflict: Introns, exons, and UCEs resolve ambiguities in golden-backed frogs (Anura: Ranidae; Genus Hylarana). Mol. Phylogenet. Evol. 2020, 151, 106899. [Google Scholar] [CrossRef]
- Pandey, A.; Braun, E.L. Phylogenetic analyses of sites in different protein structural environments result in distinct placements of the metazoan root. Biology 2020, 9, 64. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Lindsey, A.R.I.; Peters, R.S.; Heraty, J.M.; Hopper, K.R.; Werren, J.H.; Martinson, E.O.; Woolley, J.B.; Yoder, M.J.; Krogmann, L. Conflicting signal in transcriptomic markers leads to a poorly resolved backbone phylogeny of chalcidoid wasps. Syst. Entomol. 2020, 45, 783–802. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Y.-H.; Jin, J.-J.; Stull, G.W.; Bruneau, A.; Cardoso, D.; De Queiroz, L.P.; Moore, M.J.; Zhang, S.-D.; Chen, S.-Y.; et al. Exploration of plastid phylogenomic conflict yields new insights into the deep relationships of Leguminosae. Syst. Biol. 2020, 69, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Braun, E.L. The roles of protein structure, taxon sampling, and model complexity in phylogenomics: A case study focused on early animal divergences. Biophysica 2021, 1, 8. [Google Scholar] [CrossRef]
- Tiley, G.P.; Pandey, A.; Kimball, R.T.; Braun, E.L.; Burleigh, J.G. Whole genome phylogeny of Gallus: Introgression and data-type effects. Avian Res. 2020, 11, 7. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. Cases in which parsimony or compatibility methods will be positively misleading. Syst. Zool. 1978, 27, 401–410. [Google Scholar] [CrossRef]
- Hendy, M.D.; Penny, D. A framework for the quantitative study of evolutionary trees. Syst. Zool. 1989, 38, 297–309. [Google Scholar] [CrossRef]
- Conant, G.C.; Lewis, P.O. Effects of nucleotide composition bias on the success of the parsimony criterion in phylogenetic inference. Mol. Biol. Evol. 2001, 18, 1024–1033. [Google Scholar] [CrossRef] [Green Version]
- Katsu, Y.; Braun, E.L.; Guillette, L.J.; Iguchi, T. From reptilian phylogenomics to reptilian genomes: Analyses of c-Jun and DJ-1 proto-oncogenes. Cytogenet. Genome Res. 2009, 127, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Kubatko, L.S.; Degnan, J.H. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol. 2007, 56, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roch, S.; Steel, M. Likelihood-based tree reconstruction on a concatenation of aligned sequence data sets can be statistically inconsistent. Theor. Popul. Biol. 2015, 100C, 56–62. [Google Scholar] [CrossRef]
- Wang, N.; Braun, E.L.; Liang, B.; Cracraft, J.; Smith, S.A. Categorical edge-based analyses of phylogenomic data reveal conflicting signals for difficult relationships in the avian tree. BioRxiv 2021, 2021.05.17.444565. [Google Scholar] [CrossRef]
- Goldman, N.; Thorne, J.L.; Jones, D.T. Assessing the impact of secondary structure and solvent accessibility on protein evolution. Genetics 1998, 149, 445–458. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O. Accounting for solvent accessibility and secondary structure in protein phylogenetics is clearly beneficial. Syst. Biol. 2010, 59, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.; Braun, E.L. Protein evolution is structure dependent and non-homogeneous across the tree of life. In Proceedings of the 11th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Virtual Event, 21–24 September 2020; ACM: New York, NY, USA, 2020; pp. 1–11, Article 28. [Google Scholar] [CrossRef]
- Kessel, A.; Ben-Tal, N. Introduction to Proteins: Structure, Function, and Motion, 2nd ed.; Chapman & Hall/CRC Computational Biology Series; Chapman and Hall/CRC: Boca Raton, FL, USA, 2018; p. 988. ISBN 9781498747172. [Google Scholar]
- Naylor, G.J.; Brown, W.M. Structural biology and phylogenetic estimation. Nature 1997, 388, 527–528. [Google Scholar] [CrossRef]
- Naylor, G.J.; Brown, W.M. Amphioxus mitochondrial DNA, chordate phylogeny, and the limits of inference based on comparisons of sequences. Syst. Biol. 1998, 47, 61–76. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.-G. Maintenance and expression of mammalian mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef] [PubMed]
- Formenti, G.; Rhie, A.; Balacco, J.; Haase, B.; Mountcastle, J.; Fedrigo, O.; Brown, S.; Capodiferro, M.R.; Al-Ajli, F.O.; Ambrosini, R.; et al. Complete vertebrate mitogenomes reveal widespread repeats and gene duplications. Genome Biol. 2021, 22, 120. [Google Scholar] [CrossRef] [PubMed]
- Takezaki, N.; Gojobori, T. Correct and incorrect vertebrate phylogenies obtained by the entire mitochondrial DNA sequences. Mol. Biol. Evol. 1999, 16, 590–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: Approximate methods. J. Mol. Evol. 1994, 39, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Kjer, K.M.; Honeycutt, R.L. Site specific rates of mitochondrial genomes and the phylogeny of Eutheria. BMC Evol. Biol. 2007, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamashiro, R.A.; White, N.D.; Braun, M.J.; Faircloth, B.C.; Braun, E.L.; Kimball, R.T. What are the roles of taxon sampling and model fit in tests of cyto-nuclear discordance using avian mitogenomic data? Mol. Phylogenet. Evol. 2019, 130, 132–142. [Google Scholar] [CrossRef]
- Meiklejohn, K.A.; Danielson, M.J.; Faircloth, B.C.; Glenn, T.C.; Braun, E.L.; Kimball, R.T. Incongruence among different mitochondrial regions: A case study using complete mitogenomes. Mol. Phylogenet. Evol. 2014, 78, 314–323. [Google Scholar] [CrossRef]
- Braun, E.L.; Kimball, R.T. Examining basal avian divergences with mitochondrial sequences: Model complexity, taxon sampling, and sequence length. Syst. Biol. 2002, 51, 614–625. [Google Scholar] [CrossRef] [Green Version]
- Delsuc, F.; Phillips, M.J.; Penny, D. Comment on “Hexapod origins: Monophyletic or paraphyletic? ” Science 2003, 301, 1482. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.J.; Penny, D. The root of the mammalian tree inferred from whole mitochondrial genomes. Mol. Phylogenet. Evol. 2003, 28, 171–185. [Google Scholar] [CrossRef]
- Gibson, A.; Gowri-Shankar, V.; Higgs, P.G.; Rattray, M. A comprehensive analysis of mammalian mitochondrial genome base composition and improved phylogenetic methods. Mol. Biol. Evol. 2005, 22, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Pratt, R.C.; Gibb, G.C.; Morgan-Richards, M.; Phillips, M.J.; Hendy, M.D.; Penny, D. Toward resolving deep Neoaves phylogeny: Data, signal enhancement, and priors. Mol. Biol. Evol. 2009, 26, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Nesnidal, M.P.; Helmkampf, M.; Bruchhaus, I.; Hausdorf, B. The complete mitochondrial genome of Flustra foliacea (Ectoprocta, Cheilostomata)—Compositional bias affects phylogenetic analyses of lophotrochozoan relationships. BMC Genom. 2011, 12, 572. [Google Scholar] [CrossRef]
- Song, F.; Li, H.; Jiang, P.; Zhou, X.; Liu, J.; Sun, C.; Vogler, A.P.; Cai, W. Capturing the phylogeny of Holometabola with mitochondrial genome data and Bayesian site-heterogeneous mixture models. Genome Biol. Evol. 2016, 8, 1411–1426. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. A mutation data matrix for transmembrane proteins. FEBS Lett. 1994, 339, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Liò, P.; Goldman, N. Using protein structural information in evolutionary inference: Transmembrane proteins. Mol. Biol. Evol. 1999, 16, 1696–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimball, R.T.; Oliveros, C.H.; Wang, N.; White, N.D.; Barker, F.K.; Field, D.J.; Ksepka, D.T.; Chesser, R.T.; Moyle, R.G.; Braun, M.J.; et al. A phylogenomic supertree of birds. Diversity 2019, 11, 109. [Google Scholar] [CrossRef] [Green Version]
- Kuhl, H.; Frankl-Vilches, C.; Bakker, A.; Mayr, G.; Nikolaus, G.; Boerno, S.T.; Klages, S.; Timmermann, B.; Gahr, M. An unbiased molecular approach using 3′UTRs resolves the avian family-level tree of life. Mol. Biol. Evol. 2020, 1, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; White, N.D.; Benson, R.B.J.; Braun, M.J.; Field, D.J. Total-evidence framework reveals complex morphological evolution in nightbirds (Strisores). Diversity 2019, 11, 143. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Field, D.J. Phylogenetic definitions for Caprimulgimorphae (Aves) and major constituent clades under the International Code of Phylogenetic Nomenclature. Vertebr. Zool. 2020, 70, 571–585. [Google Scholar] [CrossRef]
- Yuri, T.; Kimball, R.T.; Harshman, J.; Bowie, R.C.K.; Braun, M.J.; Chojnowski, J.L.; Han, K.-L.; Hackett, S.J.; Huddleston, C.J.; Moore, W.S.; et al. Parsimony and model-based analyses of indels in avian nuclear genes reveal congruent and incongruent phylogenetic signals. Biology 2013, 2, 419–444. [Google Scholar] [CrossRef] [PubMed]
- Sangster, G. A name for the clade formed by owlet-nightjars, swifts and hummingbirds (Aves). Zootaxa 2005, 799, 1. [Google Scholar] [CrossRef] [Green Version]
- Ericson, P.G.P.; Irestedt, M.; Johansson, U.S. Evolution, biogeography, and patterns of diversification in passerine birds. J. Avian Biol. 2003, 34, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Cox, W.A.; Kimball, R.T.; Braun, E.L. Phylogenetic position of the New World quail (Odontophoridae): Eight nuclear loci and three mitochondrial regions contradict morphology and the Sibley-Ahlquist Tapestry. Auk 2007, 124, 71–84. [Google Scholar] [CrossRef]
- Gibb, G.C.; Kennedy, M.; Penny, D. Beyond phylogeny: Pelecaniform and Ciconiiform birds, and long-term niche stability. Mol. Phylogenet. Evol. 2013, 68, 229–238. [Google Scholar] [CrossRef]
- Braun, E.L.; Cracraft, J.; Houde, P. Resolving the avian tree of life from top to bottom: The promise and potential boundaries of the phylogenomic era. In Avian Genomics in Ecology and Evolution: From the Lab into the Wild; Kraus, R.H.S., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 151–210. ISBN 978-3-030-16476-8. [Google Scholar]
- Nabholz, B.; Uwimana, N.; Lartillot, N. Reconstructing the phylogenetic history of long-term effective population size and life-history traits using patterns of amino acid replacement in mitochondrial genomes of mammals and birds. Genome Biol. Evol. 2013, 5, 1273–1290. [Google Scholar] [CrossRef] [Green Version]
- Paton, T.A.; Baker, A.J. Sequences from 14 mitochondrial genes provide a well-supported phylogeny of the charadriiform birds congruent with the nuclear RAG-1 tree. Mol. Phylogenet. Evol. 2006, 39, 657–667. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Maddison, D.R.; Swofford, D.L.; Maddison, W.P. NEXUS: An extensible file format for systematic information. Syst. Biol. 1997, 46, 590–621. [Google Scholar] [CrossRef]
- Hildebrand, P.W.; Preissner, R.; Frömmel, C. Structural features of transmembrane helices. FEBS Lett. 2004, 559, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. Ufboot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Schwarz, G. Estimating the dimension of a model. Ann. Statist. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Le, V.S.; Dang, C.C.; Le, Q.S. Improved mitochondrial amino acid substitution models for metazoan evolutionary studies. BMC Evol. Biol. 2017, 17, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosiol, C.; Goldman, N. Different versions of the Dayhoff rate matrix. Mol. Biol. Evol. 2005, 22, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Bogdanowicz, D.; Giaro, K. Matching split distance for unrooted binary phylogenetic trees. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 9, 150–160. [Google Scholar] [CrossRef]
- Lin, Y.; Rajan, V.; Moret, B.M.E. A metric for phylogenetic trees based on matching. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1014–1022. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods); Sinauer Associates: Sunderland, UK, 2003. [Google Scholar]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Farris, J.S.; Kallersjo, M.; Kluge, A.G.; Bult, C. Testing significance of incongruence. Cladistics 1994, 10, 315–319. [Google Scholar] [CrossRef]
- Farris, J.S.; Kallersjo, M.; Kluge, A.G.; Bult, C. Constructing a significance test for incongruence. Syst. Biol. 1995, 44, 570. [Google Scholar] [CrossRef]
- Grantham, R. Amino acid difference formula to help explain protein evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef]
- Pacheco, M.A.; Battistuzzi, F.U.; Lentino, M.; Aguilar, R.F.; Kumar, S.; Escalante, A.A. Evolution of modern birds revealed by mitogenomics: Timing the radiation and origin of major orders. Mol. Biol. Evol. 2011, 28, 1927–1942. [Google Scholar] [CrossRef] [Green Version]
- Sangster, G. A name for the flamingo-grebe clade. Ibis 2005, 147, 612–615. [Google Scholar] [CrossRef]
- Houde, P.; Braun, E.L.; Narula, N.; Minjares, U.; Mirarab, S. Phylogenetic signal of indels and the neoavian radiation. Diversity 2019, 11, 108. [Google Scholar] [CrossRef] [Green Version]
- Gatesy, J.; O’Grady, P.; Baker, R.H. Corroboration among data sets in simultaneous analysis: Hidden support for phylogenetic relationships among higher level artiodactyl taxa. Cladistics 1999, 15, 271–313. [Google Scholar] [CrossRef]
- Gatesy, J.; Baker, R.H. Hidden likelihood support in genomic data: Can forty-five wrongs make a right? Syst. Biol. 2005, 54, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Pagel, M.; Meade, A. A phylogenetic mixture model for detecting pattern-heterogeneity in gene sequence or character-state data. Syst. Biol. 2004, 53, 571–581. [Google Scholar] [CrossRef]
- Zink, R.M.; Barrowclough, G.F. Mitochondrial DNA under siege in avian phylogeography. Mol. Ecol. 2008, 17, 2107–2121. [Google Scholar] [CrossRef] [PubMed]
- Barrowclough, G.F.; Zink, R.M. Funds enough, and time: mtDNA, nuDNA and the discovery of divergence. Mol. Ecol. 2009, 18, 2934–2936. [Google Scholar] [CrossRef]
- Smith, B.T.; McCormack, J.E.; Cuervo, A.M.; Hickerson, M.J.; Aleixo, A.; Cadena, C.D.; Pérez-Emán, J.; Burney, C.W.; Xie, X.; Harvey, M.G.; et al. The drivers of tropical speciation. Nature 2014, 515, 406–409. [Google Scholar] [CrossRef]
- Berlin, S.; Ellegren, H. Evolutionary genetics. Clonal inheritance of avian mitochondrial DNA. Nature 2001, 413, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Berlin, S.; Smith, N.G.C.; Ellegren, H. Do avian mitochondria recombine? J. Mol. Evol. 2004, 58, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Dayhoff, M.O.; Schwartz, R.M.; Orcutt, B.C. A model of evolutionary change in proteins. In Atlas of Protein Sequence and Structure; Dayhoff, M.O., Ed.; National Biomedical Research Foundation: Silver Springs, MD, USA, 1978; Volume 5, pp. 345–352. [Google Scholar]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Bioinformatics 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [Green Version]
- Kosiol, C.; Holmes, I.; Goldman, N. An empirical codon model for protein sequence evolution. Mol. Biol. Evol. 2007, 24, 1464–1479. [Google Scholar] [CrossRef] [Green Version]
- Worth, C.L.; Gong, S.; Blundell, T.L. Structural and functional constraints in the evolution of protein families. Nat. Rev. Mol. Cell Biol. 2009, 10, 709–720. [Google Scholar] [CrossRef]
- Woese, C.R.; Achenbach, L.; Rouviere, P.; Mandelco, L. Archaeal phylogeny: Reexamination of the phylogenetic position of Archaeoglohus fulgidus in light of certain composition-induced artifacts. Syst. Appl. Microbiol. 1991, 14, 364–371. [Google Scholar] [CrossRef]
- Phillips, M.J.; Delsuc, F.; Penny, D. Genome-scale phylogeny and the detection of systematic niases. Mol. Biol. Evol. 2004, 21, 1455–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatesy, J. A tenth crucial question regarding model use in phylogenetics. Trends Ecol. Evol. 2007, 22, 509–510. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, D.A.; Klaere, S. How well does your phylogenetic model fit your data? Syst. Biol. 2019, 68, 157–167. [Google Scholar] [CrossRef]
- Sanderson, M.J.; Kim, J. Parametric phylogenetics? Syst. Biol. 2000, 49, 817–829. [Google Scholar] [CrossRef] [Green Version]
- Holder, M.T.; Lewis, P.O.; Swofford, D.L. The Akaike information criterion will not choose the no common mechanism model. Syst. Biol. 2010, 59, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Field, D.J.; Berv, J.S.; Hsiang, A.Y.; Lanfear, R.; Landis, M.J.; Dornburg, A. Timing the extant avian radiation: The rise of modern birds, and the importance of modeling molecular rate variation. PeerJ Preprints 2019, 7, e27521. [Google Scholar] [CrossRef]
- Kim, J. Slicing hyperdimensional oranges: The geometry of phylogenetic estimation. Mol. Phylogenet. Evol. 2000, 17, 58–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Stiller, J.; Deng, Y.; Armstrong, J.; Fang, Q.; Reeve, A.H.; Xie, D.; Chen, G.; Guo, C.; Faircloth, B.C.; et al. Dense sampling of bird diversity increases power of comparative genomics. Nature 2020, 587, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Bravo, G.A.; Schmitt, C.J.; Edwards, S.V. What have we learned from the first 500 avian genomes? Annu. Rev. Ecol. Evol. Syst. 2021, 52. early access online. [Google Scholar] [CrossRef]
- Suh, A. The phylogenomic forest of bird trees contains a hard polytomy at the root of Neoaves. Zool. Scr. 2016, 45, 50–62. [Google Scholar] [CrossRef]
- Houde, P.; Braun, E.L.; Zhou, L. Deep-time demographic inference suggests ecological release as driver of neoavian adaptive radiation. Diversity 2020, 12, 164. [Google Scholar] [CrossRef]
- Moore, W.S. Inferring phylogenies from mtDNA variation: Mitochondrial gene trees versus nuclear gene trees. Evolution 1995, 49, 718–726. [Google Scholar] [CrossRef]
- Ballard, J.W.O.; Whitlock, M.C. The incomplete natural history of mitochondria. Mol. Ecol. 2004, 13, 729–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlin, S.; Tomaras, D.; Charlesworth, B. Low mitochondrial variability in birds may indicate Hill-Robertson effects on the W chromosome. Heredity 2007, 99, 389–396. [Google Scholar] [CrossRef] [Green Version]
- Hill, W.G.; Robertson, A. The effect of linkage on limits to artificial selection. Genet. Res. 1966, 8, 269. [Google Scholar] [CrossRef]
- Hickey, A.J.R. Avian mtDNA diversity?: An alternate explanation for low mtDNA diversity in birds: An age-old solution? Heredity 2008, 100, 443. [Google Scholar] [CrossRef]
- Lane, N. Mitochondria and the W chromosome: Low variability on the W chromosome in birds is more likely to indicate selection on mitochondrial genes. Heredity 2008, 100, 444–445. [Google Scholar] [CrossRef]
- Persons, N.W.; Hosner, P.A.; Meiklejohn, K.A.; Braun, E.L.; Kimball, R.T. Sorting out relationships among the grouse and ptarmigan using intron, mitochondrial, and ultra-conserved element sequences. Mol. Phylogenet. Evol. 2016, 98, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, M.J.; McCullough, J.M.; Gyllenhaal, E.F.; Mapel, X.M.; Haryoko, T.; Jønsson, K.A.; Joseph, L. Complex histories of gene flow and a mitochondrial capture event in a nonsister pair of birds. Mol. Ecol. 2021, 30, 2087–2103. [Google Scholar] [CrossRef]
- Kimball, R.T.; Guido, M.; Hosner, P.A.; Braun, E.L. When good mitochondria go bad: Cyto-nuclear discordance in landfowl (Aves: Galliformes). Gene 2021, 801, 145841. [Google Scholar] [CrossRef] [PubMed]
- Hill, G.E. Reconciling the mitonuclear compatibility species concept with rampant mitochondrial introgression. Integr. Comp. Biol. 2019, 59, 912–924. [Google Scholar] [CrossRef]
- Springer, M.S.; Gatesy, J. Retroposon insertions within a multispecies coalescent framework suggest that ratite phylogeny is not in the ‘Anomaly Zone’. BioRxiv 2019, 643296. [Google Scholar] [CrossRef]
- Smith, J.V.; Braun, E.L.; Kimball, R.T. Ratite nonmonophyly: Independent evidence from 40 novel loci. Syst. Biol. 2013, 62, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Hackett, S.J.; Kimball, R.T.; Reddy, S.; Bowie, R.C.K.; Braun, E.L.; Braun, M.J.; Chojnowski, J.L.; Cox, W.A.; Han, K.-L.; Harshman, J.; et al. A phylogenomic study of birds reveals their evolutionary history. Science 2008, 320, 1763–1768. [Google Scholar] [CrossRef]
- Castoe, T.A.; de Koning, A.P.J.; Kim, H.-M.; Gu, W.; Noonan, B.P.; Naylor, G.; Jiang, Z.J.; Parkinson, C.L.; Pollock, D.D. Evidence for an ancient adaptive episode of convergent molecular evolution. Proc. Natl. Acad. Sci. USA 2009, 106, 8986–8991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.-Y.; Liang, L.; Zhu, Z.-H.; Zhou, W.-P.; Irwin, D.M.; Zhang, Y.-P. Adaptive evolution of energy metabolism genes and the origin of flight in nats. Proc. Natl. Acad. Sci. USA 2010, 107, 8666–8671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.; Shen, X.; Irwin, D.M.; Shen, Y.; Zhang, Y. Mitogenomic analyses propose positive selection in mitochondrial henes for high-altitude adaptation in galliform nirds. Mitochondrion 2014, 18, 70–75. [Google Scholar] [CrossRef]
- Zhang, C.; Sayyari, E.; Mirarab, S. ASTRAL-III: Increased scalability and impacts of contracting low support nranches. In Comparative Genomics; Meidanis, J., Nakhleh, L., Eds.; Lecture Notes in Computer Science; Springer International Publishing: Cham, Switzerland, 2017; Volume 10562, pp. 53–75. ISBN 978-3-319-67978-5. [Google Scholar]
- Roch, S.; Warnow, T. On the robustness to gene tree estimation error (or lack thereof) of coalescent-nased species tree methods. Syst. Biol. 2015, 64, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.; Kimball, R.T.; Braun, E.L. Error in phylogenetic estimation for nushes in the tree of life. J. Phylogenet. Evol. Biol. 2013, 1, 110. [Google Scholar] [CrossRef] [Green Version]
- Gatesy, J.; Springer, M.S. Phylogenetic analysis at deep timescales: Unreliable gene trees, bypassed hidden support, and the coalescence/concatalescence conundrum. Mol. Phylogenet. Evol. 2014, 80, 231–266. [Google Scholar] [CrossRef]
- Meiklejohn, K.A.; Faircloth, B.C.; Glenn, T.C.; Kimball, R.T.; Braun, E.L. Analysis of a rapid evolutionary radiation using ultraconserved elements: Evidence for a bias in some multispecies coalescent methods. Syst. Biol. 2016, 65, 612–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molloy, E.K.; Warnow, T. To include or not to include: The impact of gene filtering on species tree estimation methods. Syst. Biol. 2018, 67, 285–303. [Google Scholar] [CrossRef] [PubMed]
- Forthman, M.; Braun, E.L.; Kimball, R.T. Gene tree quality affects empirical coalescent branch length estimation. Zool. Scr. 2021. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clade 2 | TM Sites | ExM Sites | All Sites |
|---|---|---|---|
| PALAEOGNATHAE | 100 | 100 | 100 |
| Notopalaeognathae | 72 | – | 57 |
| (-) “Ratites”—Dinornithiformes 3 | – | 42 | – |
| Dinornithiformes + Tinamiformes | 87 | 92 | 98 |
| GALLOANSERES | 100 | 100 | 100 |
| Galliformes | 100 | 100 | 100 |
| (-) Numididae + Phasianidae | – | 74 | 57 |
| Odontophoridae + Phasianidae | 75 | – | – |
| Odontophoridae | 84 | – | 71 |
| NEOAVES | 95 | 99 | 100 |
| VII. Mirandornithes | – | 78 | 93 |
| VI. Columbimorphae | – | – | – |
| “Orphan Orders” 4 | n/a | n/a | n/a |
| Charadriiformes | 89 | 76 | 98 |
| Gruiformes | – | 90 | – |
| V. Strisores | – | 59 | 75 |
| Daedalornithes | 82 | 35 | 80 |
| Apodiformes | 97 | 92 | 99 |
| IV. Otidimorphae | – | – | – |
| III. Phaethontimorphae | – | – | – |
| II. Aequornithes | – | – | – |
| Procellariiformes | – | 96 | 96 |
| Suliformes | – | – | 92 |
| Sulidae + Phalacrocoracidae + Anhingidae | 99 | 100 | 100 |
| Pelecaniformes | – | – | – |
| (-) Ardeidae + Threskiornithidae | – | – | 64 |
| Balaenicipitidae + Pelecanidae | 72 | 81 | 95 |
| I. Telluraves | – | – | – |
| Accipitriformes | – | – | – |
| Accipitres (Acciptriformes—Cathartidae) | 96 | 49 | 93 |
| Strigiformes | 99 | 100 | 100 |
| Coraciiformes | 36 | 84 | 79 |
| Passeriformes | 94 | 100 | 100 |
| Eupasseres | 94 | – | 87 |
| Site Type 1 | ML Estimate of Weight | Proportion of Sites |
|---|---|---|
| TM | 0.5943 | 0.5103 |
| ExM | 0.4057 | 0.4897 |
| lnL mtTM—lnL mtExM2 | TM Sites | ExM Sites |
| Lower Quartile | 0.3038 | −1.6424 |
| Median | 1.2827 | 0.0894 |
| Upper Quartile | 2.16975 | 1.23255 |
| Clade | Partitioned | birdMIX |
|---|---|---|
| PALAEOGNATHAE | 100 | 100 |
| Notopalaeognathae | 57 | — |
| (-) “Ratites”—Dinornithiformes | — | 23 |
| Dinornithiformes + Tinamiformes | 98 | 99 |
| GALLOANSERES | 100 | 100 |
| Galliformes | 100 | 100 |
| (-) Numididae + Phasianidae | 57 | 61 |
| Odontophoridae + Phasianidae | — | — |
| Odontophoridae | 67 | 71 |
| NEOAVES | 97 | 99 |
| VII. Mirandornithes | 94 | 93 |
| VI. Columbimorphae | — | — |
| “Orphan Orders” | n/a | n/a |
| Charadriiformes | 99 | 100 |
| Gruiformes | — | 97 |
| V. Strisores | 71 | 86 |
| Daedalornithes | 77 | 81 |
| Apodiformes | 99 | 99 |
| IV. Otidimorphae | — | — |
| III. Phaethontimorphae | — | — |
| II. Aequornithes | — | — |
| Procellariiformes | 97 | 97 |
| Suliformes | 94 | 65 |
| Sulidae + Phalacrocoracidae + Anhingidae | 100 | 100 |
| Pelecaniformes | — | — |
| (-) Ardeidae + Threskiornithidae | — | 35 |
| Balaenicipitidae + Pelecanidae | 98 | 98 |
| I. Telluraves | — | — |
| Accipitriformes | — | — |
| Accipitres (Acciptriformes—Cathartidae) | 97 | 96 |
| Strigiformes | 100 | 100 |
| Coraciiformes | 80 | 89 |
| Passeriformes | 100 | 100 |
| Eupasseres | 72 | 80 |
| Clade | TM Sites | ExM Sites | All Sites (3) | All Sites (6) |
|---|---|---|---|---|
| PALAEOGNATHAE | 100 | 100 | 100 | 100 |
| Notopalaeognathae | — | — | — | — |
| (-) PALAEOGNATHAE—Rheiformes 2 | — | 62 | — | 34 |
| (-) “Ratites”—Dinornithiformes 2 | 59 | — | — | — |
| (-) “Ratites” 2 | 79 | — | 48 | — |
| Dinornithiformes + Tinamiformes | — | 84 | — | 55 |
| GALLOANSERES | 100 | 100 | 100 | 100 |
| Galliformes | 100 | 100 | 100 | 100 |
| (-) Numididae + Phasianidae | — | 64 | — | — |
| Odontophoridae + Phasianidae | 65 | — | 69 | 70 |
| Odontophoridae | 99 | 76 | 100 | 100 |
| NEOAVES | 100 | 99 | 100 | 100 |
| VII. Mirandornithes | 88 | 98 | 100 | 100 |
| VI. Columbimorphae | — | — | — | — |
| “Orphan Orders” | n/a | n/a | n/a | n/a |
| Charadriiformes | 99 | 100 | — 3 | 100 |
| Gruiformes | 79 | 97 | — | 99 |
| V. Strisores | — | 84 | — | — |
| Daedalornithes | 97 | 79 | 95 | 100 |
| Apodiformes | 99 | 99 | 100 | 100 |
| IV. Otidimorphae | — | 46 | — | — |
| III. Phaethontimorphae | — | — | — | — |
| II. Aequornithes | — | 73 | — | — |
| Procellariiformes | 100 | 100 | 100 | 100 |
| Suliformes | 100 | 86 | 100 | 100 |
| Sulidae + Phalacrocoracidae + Anhingidae | 100 | 100 | 100 | 100 |
| Pelecaniformes | 40 | — | — | — |
| (-) Ardeidae + Threskiornithidae | 60 | 84 | 94 | 97 |
| Balaenicipitidae + Pelecanidae | 93 | 100 | 100 | 100 |
| I. Telluraves | — | — | — | — |
| Accipitriformes | — | — | — | 22 |
| Accipitres (Acciptriformes—Cathartidae) | 98 | 100 | 100 | 100 |
| Strigiformes | 100 | 99 | 100 | 100 |
| Coraciiformes | — | — | — | — |
| Passeriformes | 100 | 100 | 100 | 100 |
| Eupasseres | 100 | — | 76 | 72 |
| Clade | Rate | A | C | G | T | A + G 1 |
|---|---|---|---|---|---|---|
| All sites (3 partition analysis) | ||||||
| 1st codon positions | 0.2806 | 0.292628 | 0.294518 | 0.212506 | 0.200348 | 0.505134 |
| 2nd codon positions | 0.1578 | 0.185234 | 0.295701 | 0.121601 | 0.397464 | 0.306835 |
| 3rd codon positions | 2.5616 | 0.399664 | 0.422178 | 0.0456232 | 0.132535 | 0.4452872 |
| TM sites (6 partition analysis) | ||||||
| 1st codon positions | 0.2730 | 0.274286 | 0.284418 | 0.216439 | 0.224857 | 0.490725 |
| 2nd codon positions | 0.1253 | 0.0871239 | 0.280307 | 0.116706 | 0.515864 | 0.2038299 |
| 3rd codon positions | 2.7629 | 0.386619 | 0.433748 | 0.0439558 | 0.135678 | 0.4305748 |
| ExM sites (6 partition analysis) | ||||||
| 1st codon positions | 0.2540 | 0.311341 | 0.304823 | 0.208493 | 0.175343 | 0.519834 |
| 2nd codon positions | 0.1611 | 0.285325 | 0.311407 | 0.126596 | 0.276672 | 0.411921 |
| 3rd codon positions | 2.4235 | 0.412972 | 0.410375 | 0.0473243 | 0.129329 | 0.4602963 |
| Clade | TM Sites | ExM Sites | All Sites (3) | All Sites (6) |
|---|---|---|---|---|
| PALAEOGNATHAE | 100 | 100 | 100 | 100 |
| Notopalaeognathae | 83 | — | — | — |
| (-) PALAEOGNATHAE—Rheiformes | — | 56 | 42 | 60 |
| Dinornithiformes + Tinamiformes | 75 | 95 | 95 | 94 |
| GALLOANSERES | 100 | 100 | 100 | 100 |
| Galliformes | 100 | 100 | 100 | 100 |
| (-) Numididae + Phasianidae | — | 77 | — | — |
| Odontophoridae + Phasianidae | 57 | — | 50 | 54 |
| Odontophoridae | 97 | 81 | 89 | 99 |
| NEOAVES | 100 | 100 | 100 | 100 |
| VII. Mirandornithes | 100 | 98 | 100 | 100 |
| VI. Columbimorphae | — | — | — | 58 |
| “Orphan Orders” | n/a | n/a | n/a | n/a |
| Charadriiformes | 100 | 98 | 100 | 100 |
| Gruiformes | 92 | 98 | 100 | 100 |
| V. Strisores | — | 72 | — | — |
| Daedalornithes | 95 | 80 | 100 | 99 |
| Apodiformes | 100 | 98 | 100 | 100 |
| IV. Otidimorphae | — | — | — | — |
| III. Phaethontimorphae | — | — | — | — |
| II. Aequornithes | — | — | — | — |
| Procellariiformes | 100 | 97 | 100 | 100 |
| Suliformes | 99 | 35 | 100 | 100 |
| Sulidae + Phalacrocoracidae + Anhingidae | 100 | 100 | 100 | 100 |
| Pelecaniformes | 54 | — | — | — |
| (-) Ardeidae + Threskiornithidae | 71 | — | 75 | 78 |
| Balaenicipitidae + Pelecanidae | 93 | 100 | 100 | 100 |
| I. Telluraves | — | — | — | — |
| Accipitriformes | — | — | — | — |
| Accipitres (Acciptriformes—Cathartidae) | 100 | 100 | 100 | 100 |
| Strigiformes | — | 94 | 98 | 97 |
| Coraciiformes | — | — | — | — |
| Passeriformes | 100 | 100 | 100 | 100 |
| Eupasseres | 79 | — | 70 | 69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gordon, E.L.; Kimball, R.T.; Braun, E.L. Protein Structure, Models of Sequence Evolution, and Data Type Effects in Phylogenetic Analyses of Mitochondrial Data: A Case Study in Birds. Diversity 2021, 13, 555. https://doi.org/10.3390/d13110555
Gordon EL, Kimball RT, Braun EL. Protein Structure, Models of Sequence Evolution, and Data Type Effects in Phylogenetic Analyses of Mitochondrial Data: A Case Study in Birds. Diversity. 2021; 13(11):555. https://doi.org/10.3390/d13110555
Chicago/Turabian StyleGordon, Emily L., Rebecca T. Kimball, and Edward L. Braun. 2021. "Protein Structure, Models of Sequence Evolution, and Data Type Effects in Phylogenetic Analyses of Mitochondrial Data: A Case Study in Birds" Diversity 13, no. 11: 555. https://doi.org/10.3390/d13110555
APA StyleGordon, E. L., Kimball, R. T., & Braun, E. L. (2021). Protein Structure, Models of Sequence Evolution, and Data Type Effects in Phylogenetic Analyses of Mitochondrial Data: A Case Study in Birds. Diversity, 13(11), 555. https://doi.org/10.3390/d13110555