
Phylogeography of Ramalina farinacea (Lichenized Fungi, Ascomycota) in the Mediterranean Basin, Europe, and Macaronesia

 ,
,  , and
, and 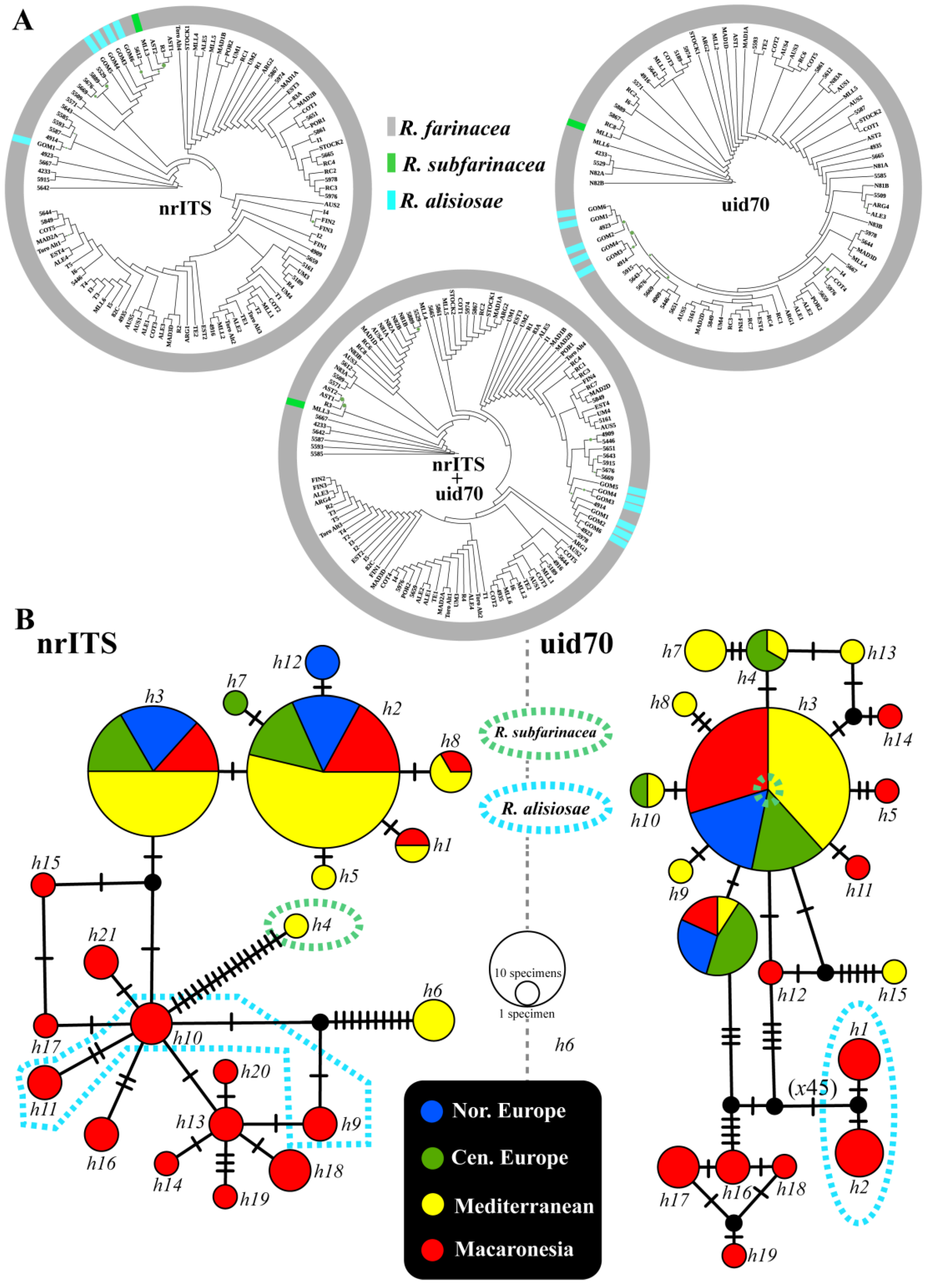{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling, Pretreatment of the Samples, and DNA Extraction
2.2. PCR Amplification and Sequencing
2.3. Phylogenetic Analyses
2.4. Inference of Genealogical Relationships among Haplotypes and Population Structure
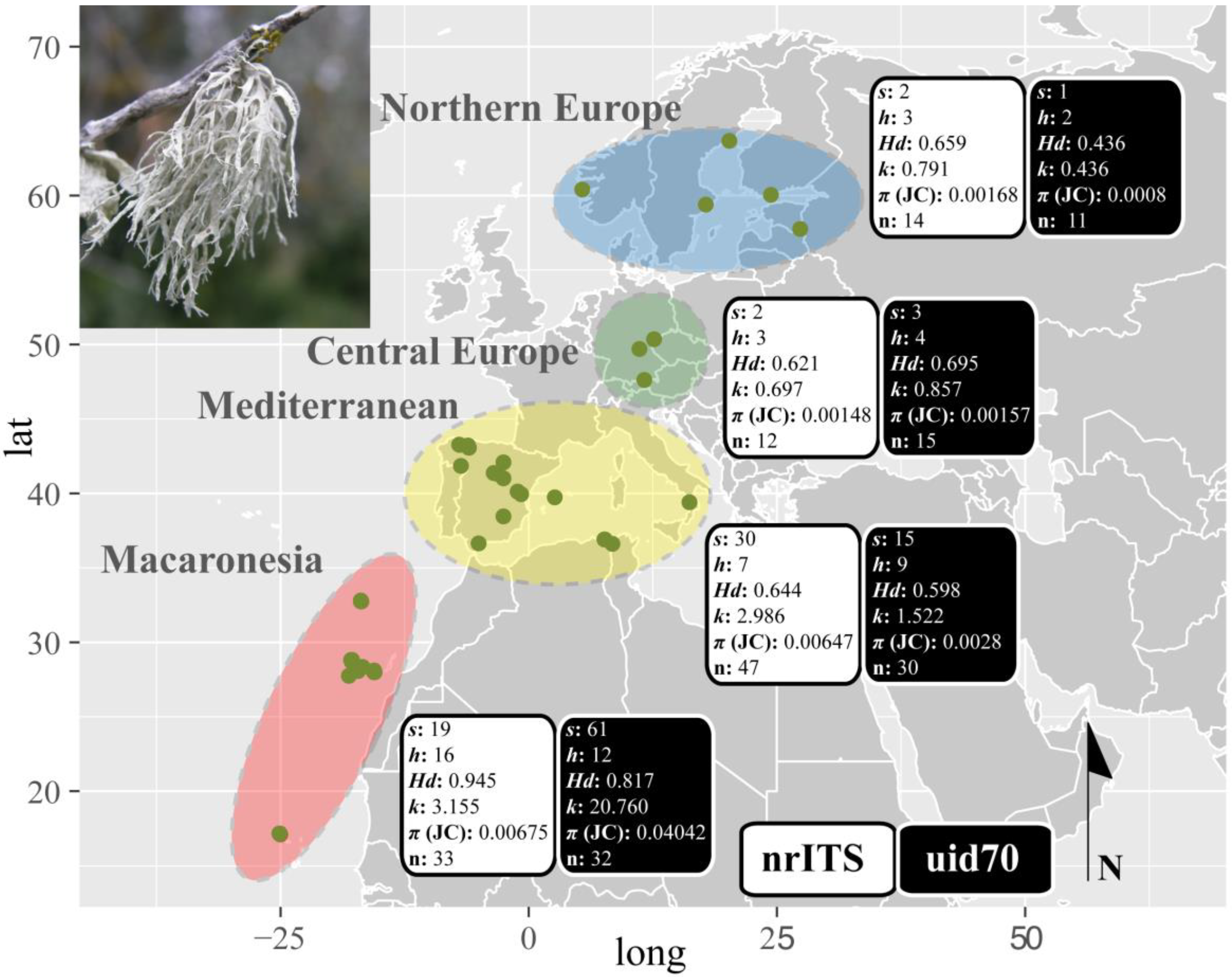
2.5. Polymorphism Analyses
2.6. Dating Analyses
3. Results
3.1. Molecular Datasets and Phylogenetic Analyses
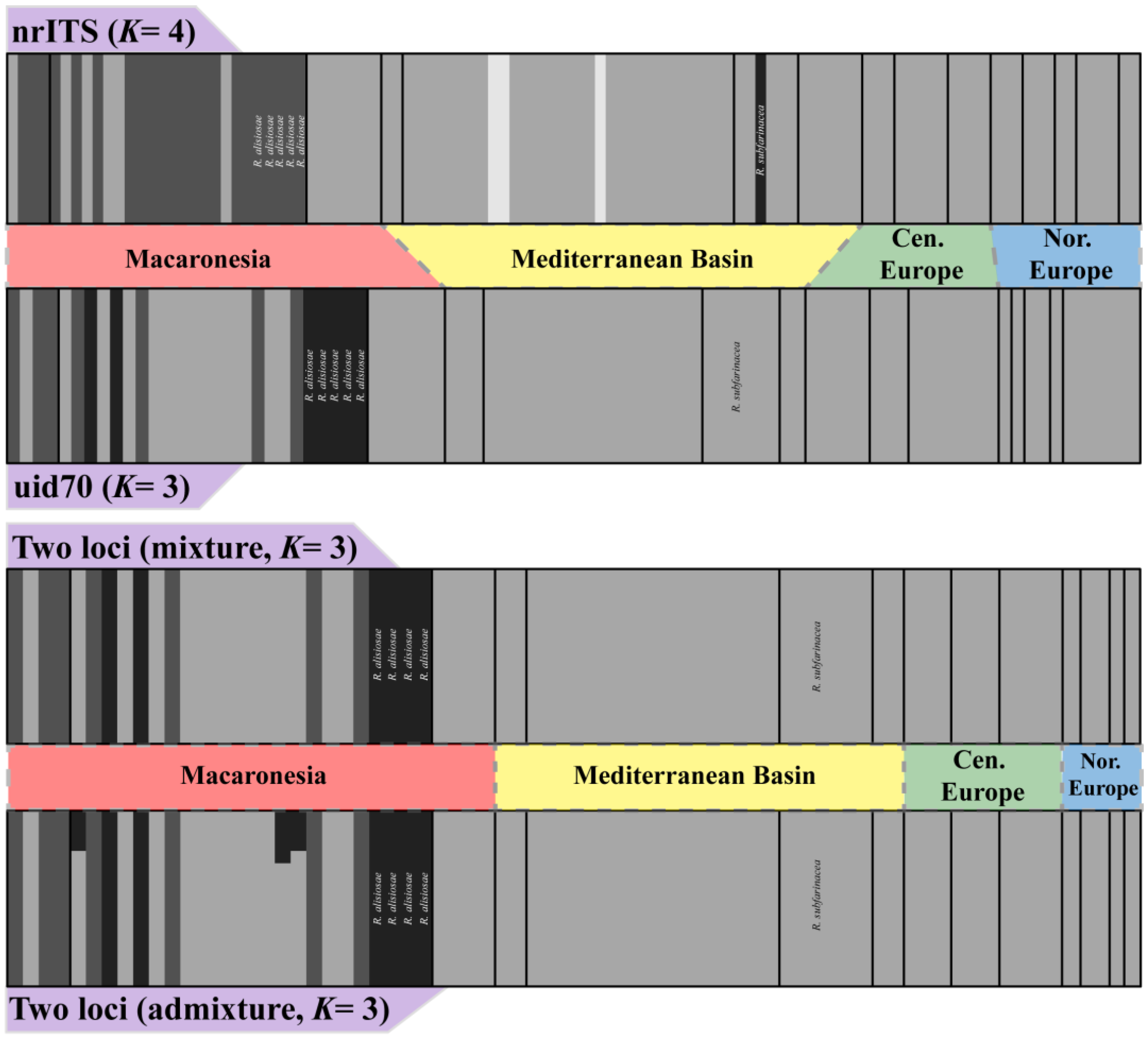
3.2. Haplotype Networks, BAPS Cluster Assignment and Polymorphism Statistics
3.3. Dating Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chapman, M.J.; Margulis, L. Morphogenesis by symbiogenesis. Int. Microbiol. 1998, 1, 319–326. [Google Scholar] [PubMed]
- Hawksworth, D.L.; Honegger, R. The lichen thallus: A symbiotic phenotype of nutritionally specialized fungi and its response to gall producers. Syst. Assoc. Spec. Vol. 1994, 49, 77. [Google Scholar]
- Feuerer, T.; Hawksworth, D.L. Biodiversity of lichens, including a world-wide analysis of checklist data based on Takhtajan’s floristic regions. Biodivers. Conser. 2007, 16, 85–98. [Google Scholar] [CrossRef]
- Kirk, P.M.; Cannon, P.F.; Minter, D.W.; Stalpers, J.A. Dictionary of the Fungi, 10th ed.; Cromwell Press: Townbridge, UK, 2008; p. 771. [Google Scholar]
- Honegger, R. The symbiotic phenotype of lichen-forming ascomycetes and their endo- and epibionts. In Fungal Associations. The Mycota IX, 2nd ed.; Hock, B., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 287–339. [Google Scholar]
- Aschenbrenner, I.A.; Cardinale, M.; Berg, G.; Grube, M. Microbial cargo: Do bacteria on symbiotic propagules reinforce the microbiome of lichens? Environ. Microbiol. 2014, 16, 3743–3752. [Google Scholar] [CrossRef] [PubMed]
- Cernava, T.; Berg, G.; Grube, M. High life expectancy of bacteria on lichens. Microbial. Ecol. 2016, 72, 510–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, M.; Grube, M.; Schiefelbein, U.; Zühlke, D.; Bernhardt, J.; Riedel, K. The lichens’ microbiota, still a mystery? Front. Microbiol. 2021, 12, 714. [Google Scholar] [CrossRef] [PubMed]
- Acharius, E. Lichenographia Universalis; Apud Iust. Frid. Danckwerts: Göttingen, Germany, 1810. [Google Scholar]
- Lücking, R.; Hodkinson, B.P.; Leavitt, S.D. The 2016 classification of lichenized fungi in the Ascomycota and Basidiomycota–Approaching one thousand genera. Bryologist 2017, 119, 361–416. [Google Scholar] [CrossRef]
- Krog, H.; Østhagen, H. The genus Ramalina in the Canary Islands. Norw. J. Bot. 1980, 27, 255–296. [Google Scholar]
- Stevens, G.N. The lichen genus Ramalina in Australia. Bull. Br. Mus. Nat. Hist. Bot. 1987, 16, 107–223. [Google Scholar]
- Krog, H. New Ramalina species from Porto Santo, Madeira. Lichenologist 1990, 22, 241–247. [Google Scholar] [CrossRef]
- Blanchon, D.J.; Braggins, J.E.; Stewart, A. The lichen genus Ramalina in New Zealand. J. Hattori bot. Lab. 1996, 79, 43–98. [Google Scholar]
- Aptroot, A.; Bungartz, F. The lichen genus Ramalina on the Galapagos. Lichenologist 2007, 39, 519–542. [Google Scholar] [CrossRef]
- Aptroot, A. Lichens of St Helena and Ascension Island. Bot. J. Linn. Soc. 2008, 158, 147–171. [Google Scholar] [CrossRef]
- Aptroot, A.; Schumm, F. Key to Ramalina species known from Atlantic islands, with two new species from the Azores. Sauteria 2008, 15, 21–57. [Google Scholar]
- Pérez-Vargas, I.; Pérez-Ortega, S. A new endemic Ramalina species from the Canary Islands (Ascomycota, Lecanorales). Phytotaxa 2014, 159, 269–278. [Google Scholar] [CrossRef] [Green Version]
- Sparrius, L.B.; Aptroot, A.; Sipman, H.J.M.; Pérez-Vargas, I.; Matos, P.; Gerlach, A.; Vervoort, M. Estimating the population size of the endemic lichens Anzia centrifuga (Parmeliaceae) and Ramalina species (Ramalinaceae) on Porto Santo (Madeira archipelago). Bryologist 2017, 120, 293–301. [Google Scholar] [CrossRef]
- Arhoun, M.; Barreno, E.; Torres, J.R.; Ramis-Ramos, G. Releasing rates of inorganic ions from the lichen Ramalina farinacea by capillary zone electrophoresis (CZE) as an indicator of atmospheric pollution. Cryptogamie Bryol. L. 2000, 21, 275–289. [Google Scholar]
- Fadila, K.; Houria, D.; Rachid, R.; Reda, D.M. Cellular response of a pollution bioindicator model (Ramalina farinacea) following treatment with fertilizer (NPKs). Am.-Euras. J. Toxicol. Sci. 2009, 1, 69–73. [Google Scholar]
- Arroyo, R.; Manrique, E. Estudios químicos en Ramalina farinacea (L.) Ach. del centro de España. Anales Jard. Bot. Madr. 1988, 45, 53–59. [Google Scholar]
- Stocker-Wörgötter, E.; Elix, J.A.; Grube, M. Secondary chemistry of lichen-forming fungi: Chemosyndromic variation and DNA-analyses of cultures and chemotypes in the Ramalina farinacea complex. Bryologist 2004, 107, 152–162. [Google Scholar] [CrossRef]
- Armstrong, R.A. Soredial dispersal from individual soralia in the lichen Hypogymnia physodes (L.) Nyl. Env. Exp. Bot. 1992, 32, 55–63. [Google Scholar] [CrossRef]
- Armstrong, R.A. Dispersal of soredia from individual soralia of the lichen Hypogymnia physodes (L.) Nyl. in a simple wind tunnel. Env. Exp. Bot. 1994, 34, 39–45. [Google Scholar] [CrossRef]
- Walser, J.C.; Zoller, S.; Büchler, U.; Scheidegger, C. Species-specific detection of Lobaria pulmonaria (lichenized ascomycete) diaspores in litter samples trapped in snow cover. Mol. Ecol. 2001, 10, 2129–2138. [Google Scholar] [CrossRef] [PubMed]
- Casano, L.M.; del Campo, E.M.; García-Breijo, F.J.; Reig-Armiñana, J.; Gasulla, F.; Del Hoyo, A.; Guéra, A.; Barreno, E. Two Trebouxia algae with different physiological performances are ever-present in lichen thalli of Ramalina farinacea. Coexistence versus competition? Environ. Microbiol. 2011, 13, 806–818. [Google Scholar] [CrossRef]
- Moya, P.; Molins, A.; Chiva, S.; Bastida, J.; Barreno, E. Symbiotic microalgal diversity within lichenicolous lichens and crustose hosts on Iberian Peninsula gypsum biocrusts. Sci. Rep. UK 2020, 10, 14060. [Google Scholar] [CrossRef]
- Blázquez, M.; Hernández-Moreno, L.S.; Gasulla, F.; Pérez-Vargas, I.; Pérez-Ortega, S. The role of photobionts as drivers of diversification in an island radiation of lichen-forming fungi. Front. Microbiol. 2021, 12, 4037. [Google Scholar] [CrossRef]
- De Carolis, R.; Cometto, A.; Moya, P.; Barreno, E.; Grube, M.; Tretiach, M.; Leavitt, S.D.; Muggia, L. Photobiont diversity in lichen symbioses from extreme environments. Front. Microbiol. 2022, 13, 809804. [Google Scholar] [CrossRef]
- Kosecka, M.; Kukwa, M.; Jabłonska, A.; Flakus, A.; Rodríguez-Flakus, P.; Ptach, Ł.; Guzow-Krzeminska, B. Phylogeny and ecology of Trebouxia photobionts from Bolivian lichens. Front. Microbiol 2022, 13, 779784. [Google Scholar] [CrossRef]
- Del Campo, E.M.; Català, S.; Casano, L.M.; Gimeno, J.; del Hoyo, A.; Martínez-Alberola, F.; Grube, M.; Barreno, E. The genetic structure of the cosmopolitan three-partner lichen Ramalina farinacea evidences the concerted diversification of symbionts. FEMS Microbiol. Ecol. 2013, 83, 310–323. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, R.J.; Ladle, R.J.; Araújo, M.B.; Fernández-Palacios, J.M.; Domingo Delgado, J.; Arévalo, J.R. The island immaturity–speciation pulse model of island evolution: An alternative to the “diversity begets diversity” model. Ecography 2007, 30, 321–327. [Google Scholar] [CrossRef]
- Molins, A.; Moya, P.; Muggia, L.; Barreno, E. Thallus growth stage and geographic origin shape microalgal diversity in Ramalina farinacea lichen holobionts. J. Phycol. 2021, 57, 975–987. [Google Scholar] [CrossRef]
- Wickham, H. Data analysis. In ggplot2; Springer: Cham, Switzerland, 2016; pp. 189–201. [Google Scholar]
- Ordynets, A.; Heilmann-Clausen, J.; Savchenko, A.; Bässler, C.; Volobuev, S.; Akulov, O.; Karadelev, M.; Kotiranta, H.; Saitia, A.; Langer, E.; et al. Do plant-based biogeographical regions shape aphyllophoroid fungal communities in Europe? J. Biogeogr. 2018, 45, 1182–1195. [Google Scholar] [CrossRef]
- Rivas-Martínez, S.; Penas, Á.; del Río, S.; Díaz González, T.E.; Rivas-Sáenz, S. Bioclimatology of the Iberian Peninsula and the Balearic Islands. In The Vegetation of the Iberian Peninsula; Loidi, J., Ed.; Springer: Cham, Switzerland, 2017; Volume 12, pp. 29–80. [Google Scholar]
- Blázquez, M. Evolution of the Ramalina decipiens Group (Lichenized Ascomycota) in Macaronesia: Comparative Study of Its Symbionts and Ecophysiological Traits. Ph.D. Thesis, Universidad Rey Juan Carlos, Madrid, Spain, 2023. [Google Scholar]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Academic Press: Cambridge, MA, USA, 1990; pp. 315–322. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. Mafft multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid boostrap algorithm for the RAxML web server. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. The CIPRES science gateway: A community resource for phylogenetic analyses. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010. [Google Scholar]
- Mason-Gamer, R.J.; Kellogg, E.A. Testing for phylogenetic conflict among molecular data sets in the tribe Triticeae (Gramineae). Syst. Biol. 1996, 45, 524–545. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanfear, R.; Calcott, B.; Ho, S.Y.; Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, J.W.; Bryant, D. PopART: Full feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Clement, M.; Snell, Q.; Walker, P.; Posada, D.; Crandall, K. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Templeton, A.R.; Crandall, K.A.; Sing, C.F. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 1992, 132, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Corander, J.; Marttinen, P. Bayesian identification of admixture events using multilocus molecular markers. Mol. Ecol. 2006, 15, 2833–2843. [Google Scholar] [CrossRef]
- Corander, J.; Marttinen, P.; Sirén, J.; Tang, J. Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinform. 2008, 9, 539. [Google Scholar] [CrossRef] [Green Version]
- Maddison, W.P.; Maddison, D.R. Mesquite: A Modular System for Evolutionary Analysis. Version 3.02. 2015. Available online: http://mesquiteproject.org (accessed on 20 November 2022).
- Corander, J.; Tang, J. Bayesian analysis of population structure based on linked molecular information. Math. Biosci. 2007, 205, 19–31. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Jukes, T.H.; Cantor, C.R. Evolution of protein molecules. Mam. Prot. Metab. 1969, 3, 21–132. [Google Scholar]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leavitt, S.D.; Esslinger, T.L.; Lumbsch, H.T. Neogene-dominated diversification in neotropical montane lichens: Dating divergence events in the lichen-forming fungal genus Oropogon (Parmeliaceae). Am. J. Bot. 2012, 99, 1764–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leavitt, S.D.; Esslinger, T.L.; Divakar, P.K.; Lumbsch, H.T. Miocene and Pliocene dominated diversification of the lichen-forming fungal genus Melanohalea (Parmeliaceae, Ascomycota) and Pleistocene population expansions. BMC Evol. Biol. 2012, 12, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marthinsen, G.; Rui, S.; Timdal, E. OLICH: A reference library of DNA barcodes for Nordic lichens. Biodivers. Data J. 2019, 7, e36252. [Google Scholar] [CrossRef] [Green Version]
- Maddison, W.P. Gene Trees in Species Trees. Syst. Biol. 1997, 46, 523–536. [Google Scholar] [CrossRef]
- Jeffroy, O.; Brinkmann, H.; Delsuc, F.; Philippe, H. Phylogenomics: The beginning of incongruence? Trends Genet. 2006, 22, 225–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinová, J.; Stenroos, S.; Grube, M.; Škaloud, P. Genetic diversity and species delimitation of the zeorin-containing red-fruited Cladonia species (lichenized Ascomycota) assessed with ITS rDNA and β-tubulin data. Lichenologist 2013, 45, 665–684. [Google Scholar] [CrossRef]
- Saag, L.; Mark, K.; Saag, A.; Randlane, T. Species delimitation in the lichenized fungal genus Vulpicida (Parmeliaceae, Ascomycota) using gene concatenation and coalescent-based species tree approaches. Am. J. Bot. 2014, 101, 2169–2182. [Google Scholar] [CrossRef] [Green Version]
- Leavitt, S.D.; Grewe, F.D.; Widhelm, T.; Muggia, L.; Wray, B.; Lumbsch, H.T. Resolving evolutionary relationships in lichen-forming fungi using diverse phylogenomic datasets and analytical approaches. Sci. Rep. 2016, 6, 22262. [Google Scholar] [CrossRef]
- Blanco-Pastor, J.L.; Vargas, P.; Pfeil, B.E. Coalescent simulations reveal hybridization and incomplete lineage sorting in Mediterranean Linaria. PLoS ONE 2012, 7, e39089. [Google Scholar] [CrossRef] [PubMed]
- Morando, M.; Avila, L.J.; Baker, J.; Sites, J.W., Jr. Phylogeny and phylogeography of the Liolaemus darwinii complex (Squamata: Liolaemidae): Evidence for introgression and incomplete lineage sorting. Evolution 2004, 58, 842–859. [Google Scholar] [PubMed]
- Jakob, S.S.; Blattner, F.R. A chloroplast genealogy of Hordeum (Poaceae): Long-term persisting haplotypes, incomplete lineage sorting, regional extinction, and the consequences for phylogenetic inference. Mol. Biol. Evol. 2006, 23, 1602–1612. [Google Scholar] [CrossRef] [PubMed]
- Pollard, D.A.; Iyer, V.N.; Moses, A.M.; Eisen, M.B. Widespread discordance of gene trees with species tree in Drosophila: Evidence for incomplete lineage sorting. PLoS Genet. 2006, 2, e173. [Google Scholar] [CrossRef] [Green Version]
- Mark, K.; Saag, L.; Leavitt, S.D.; Will-Wolf, S.; Nelsen, M.P.; Tõrra, T.; Saag, A.; Randlane, T.; Lumbsch, H.T. Evaluation of traditionally circumscribed species in the lichen-forming genus Usnea, section Usnea (Parmeliaceae, Ascomycota) using a six-locus dataset. Org. Divers. Evol. 2016, 16, 497–524. [Google Scholar] [CrossRef]
- Clerc, P.; Naciri, Y. Usnea dasopoga (Ach.) Nyl. and U. barbata (L.) FH Wigg. (Ascomycetes, Parmeliaceae) are two different species: A plea for reliable identifications in molecular studies. Lichenologist 2021, 53, 221–230. [Google Scholar] [CrossRef]
- Boluda, C.G.; Rico, V.J.; Naciri, Y.; Hawksworth, D.L.; Scheidegger, C. Phylogeographic reconstructions can be biased by ancestral shared alleles: The case of the polymorphic lichen Bryoria fuscescens in Europe and North Africa. Mol. Ecol. 2021, 30, 4845–4865. [Google Scholar] [CrossRef]
- Athukorala, S.N.; Raquel, P.B.; Stenroos, S.; Teuvo, A.H.T.I.; Piercey-Normore, M.D. Phylogenetic relationships among reindeer lichens of North America. Lichenologist 2016, 48, 209–227. [Google Scholar] [CrossRef]
- Joly, S.; McLenachan, P.A.; Lockhart, P.J. A statistical approach for distinguishing hybridization and incomplete lineage sorting. Am. Nat. 2009, 174, 54–70. [Google Scholar] [CrossRef] [Green Version]
- Reddy, S.; Kimball, R.T.; Pandey, A.; Hosner, P.A.; Braun, M.J.; Hackett, S.J.; Han, K.L.; Harshman, J.; Huddleston, C.J.; Kingston, S. Why do phylogenomic data sets yield conflicting trees? Data type influences the avian tree of life more than taxon sampling. Syst. Biol. 2017, 66, 857–879. [Google Scholar] [CrossRef] [Green Version]
- Holland, B.R.; Benthin, S.; Lockhart, P.J.; Moulton, V.; Huber, K.T. Using supernetworks to distinguish hybridization from lineage-sorting. BMC Evol. Biol. 2008, 8, 202. [Google Scholar] [CrossRef] [Green Version]
- Bloomquist, E.W.; Suchard, M.A. Unifying vertical and nonvertical evolution: A stochastic ARG-based framework. Syst. Biol. 2010, 59, 27–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molloy, E.K.; Warnow, T. To include or not to include: The impact of gene filtering on species tree estimation methods. Syst. Biol. 2018, 67, 285–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiva, S.; Garrido-Benavent, I.; Moya, P.; Molins, A.; Barreno, E. How did terricolous fungi originate in the Mediterranean region? A case study with a gypsicolous lichenized species. J. Biogeogr. 2019, 46, 515–525. [Google Scholar] [CrossRef]
- Fernández-Palacios, J.M.; De Nascimento, L.; Otto, R.; Delgado, J.D.; García-del-Rey, E.; Arévalo, J.R.; Whittaker, R.J. A reconstruction of Palaeo-Macaronesia, with particular reference to the long-term biogeography of the Atlantic Island laurel forests. J. Biogeogr. 2011, 38, 226–246. [Google Scholar] [CrossRef]
- Schmitt, T. Molecular biogeography of Europe: Pleistocene cycles and postglacial trends. Front. Zool. 2007, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svenning, J.C.; Normand, S.; Kageyama, M. Glacial refugia of temperate trees in Europe: Insights from species distribution modelling. J. Ecol. 2008, 96, 1117–1127. [Google Scholar] [CrossRef]
- Weiss, S.; Ferrand, N. Phylogeography of Southern European Refugia; Springer: Dordretch, The Netherlands, 2008; pp. 341–357. [Google Scholar]
- Hewitt, G.M. Quaternary phylogeography: The roots to hybrid zones. Genetics 2011, 139, 617–638. [Google Scholar] [CrossRef]
- Zachos, J.; Pagani, M.; Sloan, L.; Thomas, E.; Billups, K. Trends, rhythms, and aberrations in global climate 65Ma to present. Science 2001, 292, 686–693. [Google Scholar] [CrossRef]
- Clark, P.U.; Dyke, A.S.; Shakun, J.D.; Carlson, A.E.; Clark, J.; Wohlfarth, B.; Mitrovica, J.X.; Hostetler, S.W.; McCabe, A.M. The last glacial maximum. Science 2009, 325, 710–714. [Google Scholar] [CrossRef] [Green Version]
- Parducci, L.; Jorgensen, T.; Tollefsrud, M.M.; Elverland, E.; Alm, T.; Fontana, S.L.; Bennett, K.D.; Haile, J.; Matetovici, I.; Suyama, Y.; et al. Glacial survival of boreal trees in northern Scandinavia. Science 2012, 335, 1083–1086. [Google Scholar] [CrossRef]
- Tzedakis, P.C.; Emerson, B.C.; Hewitt, G.M. Cryptic or mystic? Glacial tree refugia in northern Europe. Trends Ecol. Evol. 2013, 28, 696–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennet, K.D.; Tzedakis, P.C.; Willis, K.J. Quaternary refugia of North European trees. J. Biogeogr. 1991, 18, 103–115. [Google Scholar] [CrossRef]
- Petit, J.M.; Aguinagalde, I.; Beaulieu, J.L.; Bittkau, C.; Brewer, S.; Cheddadi, R.; Ennos, R.; Fineschi, S.; Grivet, D.; Lascoux, M.; et al. Glacial refugia: Hotspots but not melting pots of genetic diversity. Science 2003, 300, 1563–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Médail, F.; Diadema, K. Glacial refugia influence plant diversity patterns in the Mediterranean Basin. J. Biogeogr. 2009, 36, 1333–1345. [Google Scholar] [CrossRef]
- Habel, J.C.; Drees, C.; Schmitt, T.; Assmann, T. Review of refugial areas and postglacial colonizations in the Western Palearctic. In Relict Species. Phylogeography and Conservation Biology; Habel, J.C., Assmann, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 189–197. [Google Scholar]
- Feliner, G.N. Southern European glacial refugia: A tale of tales. Taxon 2011, 60, 365–372. [Google Scholar] [CrossRef]
- Garrido-Benavent, I.; Ballarà, J.; Liimatainen, K.; Dima, B.; Brandrud, T.E.; Mahiques, R. Cortinarius ochrolamellatus (Agaricales, Basidiomycota): A new species in C. sect. Laeti, with comments on the origin of its European–Hyrcanian distribution. Phytotaxa 2020, 460, 185–200. [Google Scholar] [CrossRef]
- Harmata, K.; Olech, M. Transect for aerobiological studies from Antarctica to Poland. Grana 1991, 30, 458–463. [Google Scholar] [CrossRef] [Green Version]
- Molins, A.; Chiva, S.; Calatayud, Á.; Marco, F.; García-Breijo, F.; Reig-Armiñana, J.; Carrasco, P.; Moya, P. Multidisciplinary approach to describe Trebouxia diversity within lichenized fungi Buellia zoharyi from the Canary Islands. Symbiosis 2020, 82, 19–34. [Google Scholar] [CrossRef]
- Næsborg, R.R.; Ekman, S.; Tibell, L. Molecular phylogeny of the genus Lecania (Ramalinaceae, lichenized Ascomycota). Mycol. Res. 2007, 111, 581–591. [Google Scholar] [CrossRef]
- Hur, J.S.; Wang, L.S.; Oh, S.O.; Kim, G.H.; Lim, K.M.; Jung, J.S.; Koh, Y.J. Highland macrolichen flora of northwestern Yunnan, China. J. Microbiol. 2005, 43, 228–236. [Google Scholar]
- Kelly, L.J.; Hollingsworth, P.M.; Coppins, B.J.; Ellis, C.J.; Harrold, P.; Tosh, J.; Yahr, R. DNA barcoding of lichenized fungi demonstrates high identification success in a floristic context. New Phytol. 2011, 191, 288–300. [Google Scholar] [CrossRef]
- Sérusiaux, E.; Van den Boom, P.; Ertz, D. A two-gene phylogeny shows the lichen genus Niebla (Lecanorales) is endemic to the New World and does not occur in Macaronesia nor in the Mediterranean basin. Fungal Biol. 2010, 114, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, R.; Del Hoyo, A.; Díaz-Rodríguez, C.; Coello, A.J.; Del Campo, E.M.; Barreno, E. Casano, L.M. Lichen rehydration in heavy metal-polluted environments: Pb modulates the oxidative response of both Ramalina farinacea thalli and its isolated microalgae. Microbial Ecol. 2015, 69, 698–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, H.L.; Rafat, A.; Ridden, J.D.; Cruickshank, R.H.; Ridgway, H.J.; Paterson, A.M. Phylogenetic congruence of lichenised fungi and algae is affected by spatial scale and taxonomic diversity. Peer J. 2014, 2, e573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, G.; Kukwa, M.; Dal Grande, F.; Łubek, A.; Otte, J.; Schmitt, I. A glimpse into genetic diversity and symbiont interaction patterns in lichen communities from areas with different disturbance histories in Białowieża forest, Poland. Microorganisms 2019, 7, 335. [Google Scholar] [CrossRef] [Green Version]
- Spjut, R.; Simon, A.; Guissard, M.; Magain, N.; Sérusiaux, E. The fruticose genera in the Ramalinaceae (Ascomycota, Lecanoromycetes): Their diversity and evolutionary history. MycoKeys 2020, 73, 1. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moya, P.; Garrido-Benavent, I.; Chiva, S.; Pérez-Ortega, S.; Blázquez, M.; Pazos, T.; Hamel, T.; Myllys, L.; Tønsberg, T.; Esseen, P.-A.; et al. Phylogeography of Ramalina farinacea (Lichenized Fungi, Ascomycota) in the Mediterranean Basin, Europe, and Macaronesia. Diversity 2023, 15, 310. https://doi.org/10.3390/d15030310
Moya P, Garrido-Benavent I, Chiva S, Pérez-Ortega S, Blázquez M, Pazos T, Hamel T, Myllys L, Tønsberg T, Esseen P-A, et al. Phylogeography of Ramalina farinacea (Lichenized Fungi, Ascomycota) in the Mediterranean Basin, Europe, and Macaronesia. Diversity. 2023; 15(3):310. https://doi.org/10.3390/d15030310
Chicago/Turabian StyleMoya, Patricia, Isaac Garrido-Benavent, Salvador Chiva, Sergio Pérez-Ortega, Miguel Blázquez, Tamara Pazos, Tarek Hamel, Leena Myllys, Tor Tønsberg, Per-Anders Esseen, and et al. 2023. "Phylogeography of Ramalina farinacea (Lichenized Fungi, Ascomycota) in the Mediterranean Basin, Europe, and Macaronesia" Diversity 15, no. 3: 310. https://doi.org/10.3390/d15030310
APA StyleMoya, P., Garrido-Benavent, I., Chiva, S., Pérez-Ortega, S., Blázquez, M., Pazos, T., Hamel, T., Myllys, L., Tønsberg, T., Esseen, P. -A., Carrasco, P., & Barreno, E. (2023). Phylogeography of Ramalina farinacea (Lichenized Fungi, Ascomycota) in the Mediterranean Basin, Europe, and Macaronesia. Diversity, 15(3), 310. https://doi.org/10.3390/d15030310