

Do Deep Mitochondrial DNA Divergences within Intertidal Gastropods Reveal Phylogeographic Signals from Earlier Glacial Cycles?


Abstract
:1. Introduction
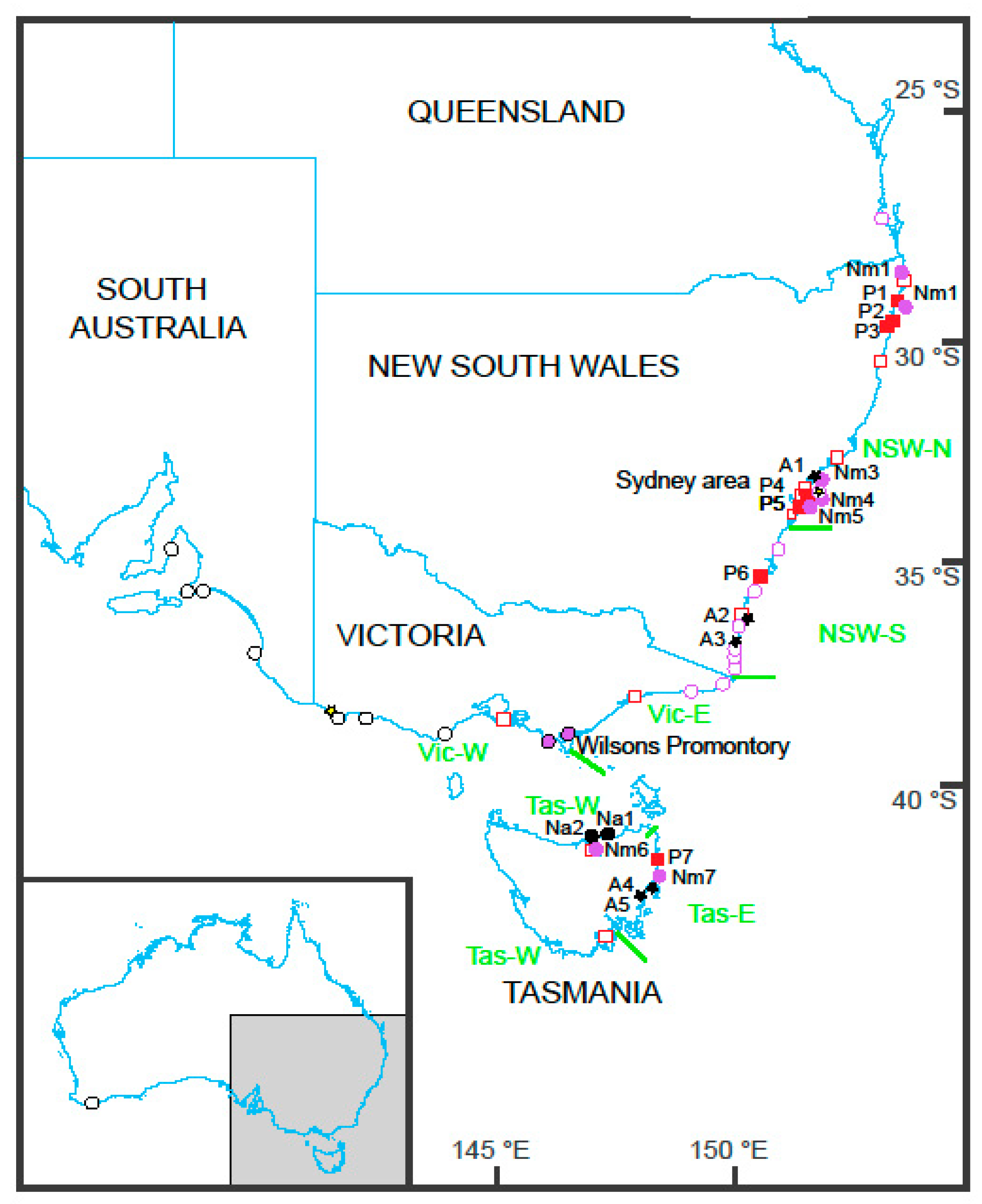
2. Materials and Methods
2.1. Collections and DNA Sequencing
2.2. Sequence Editing and Alignment
2.3. Analyses
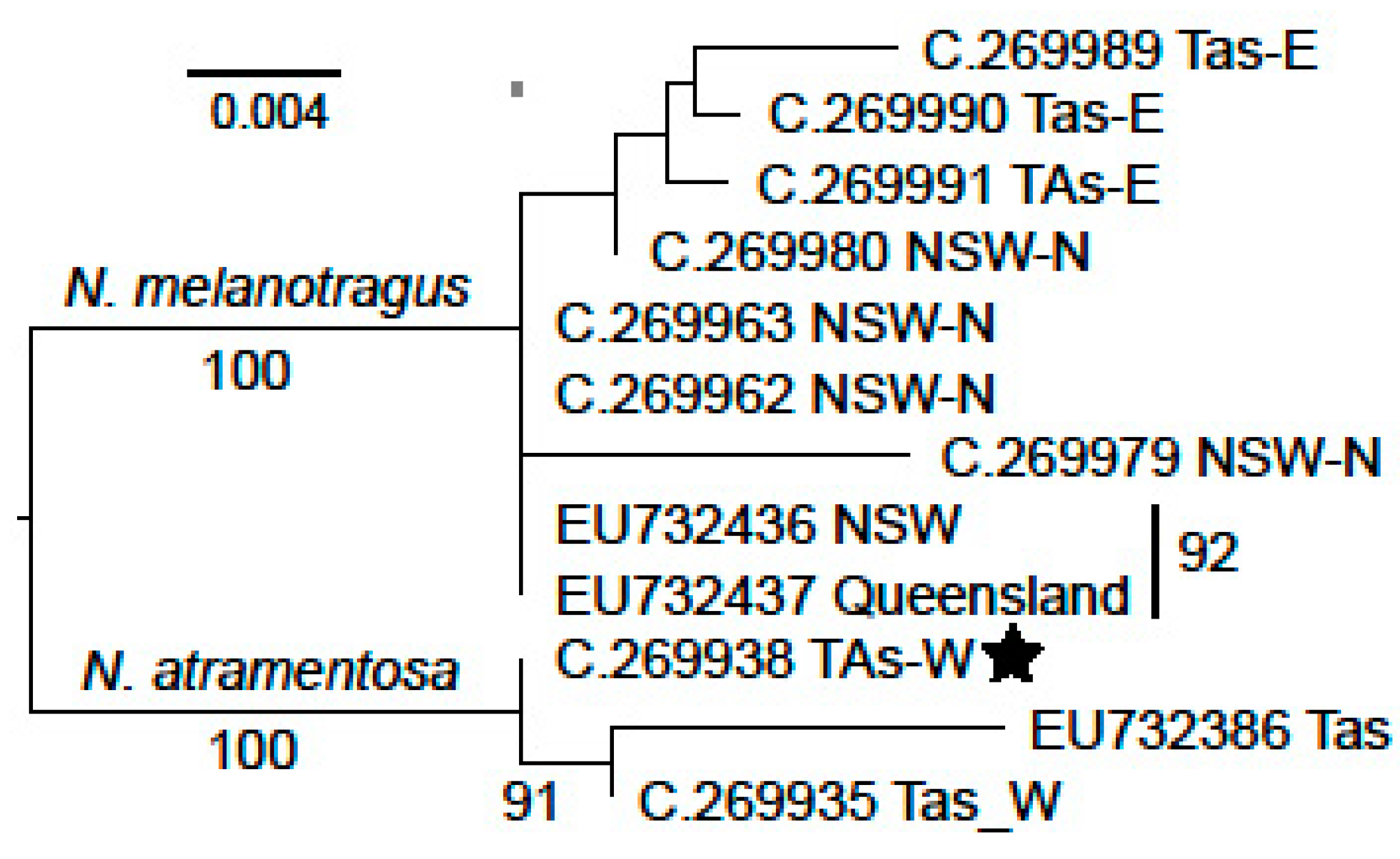
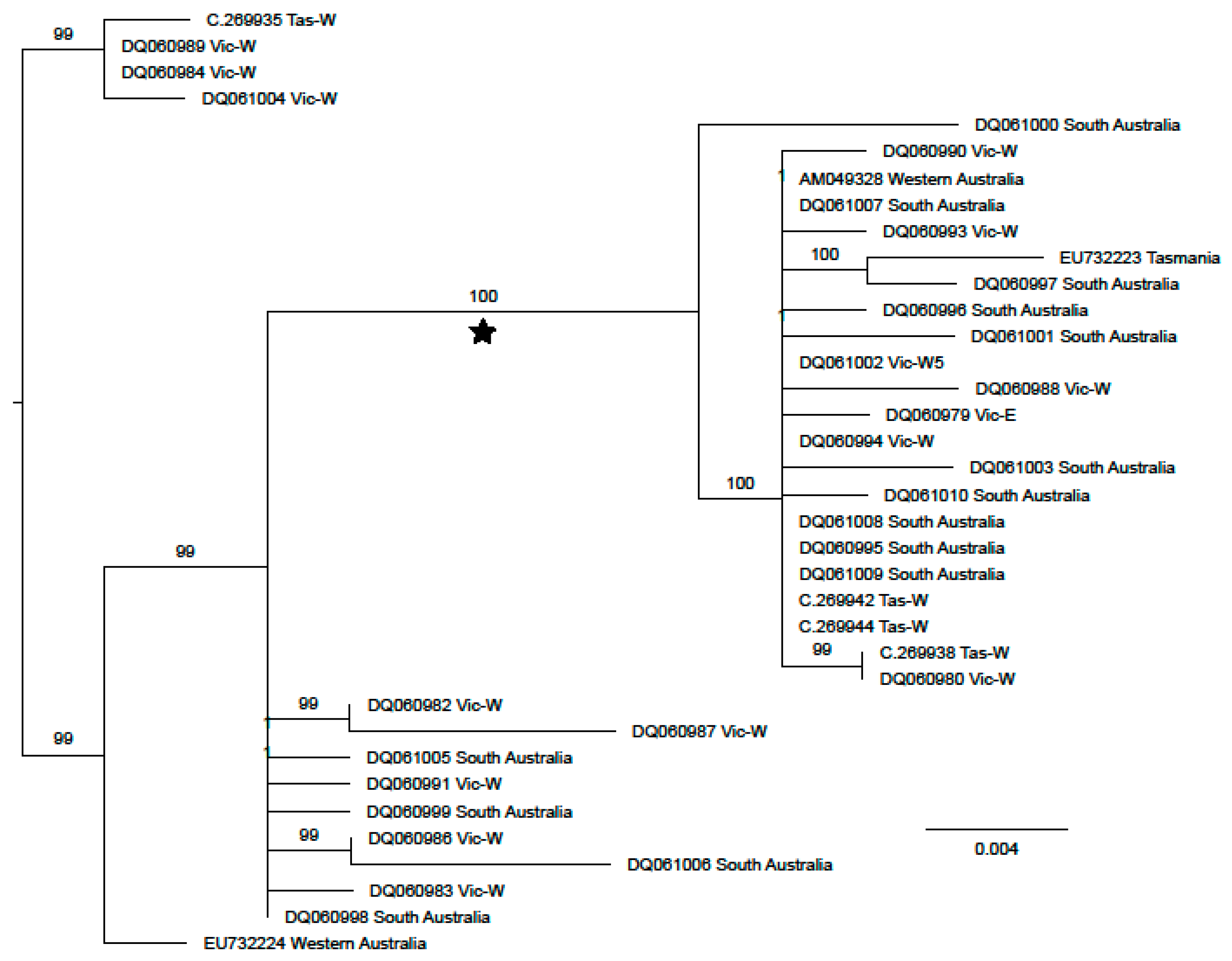
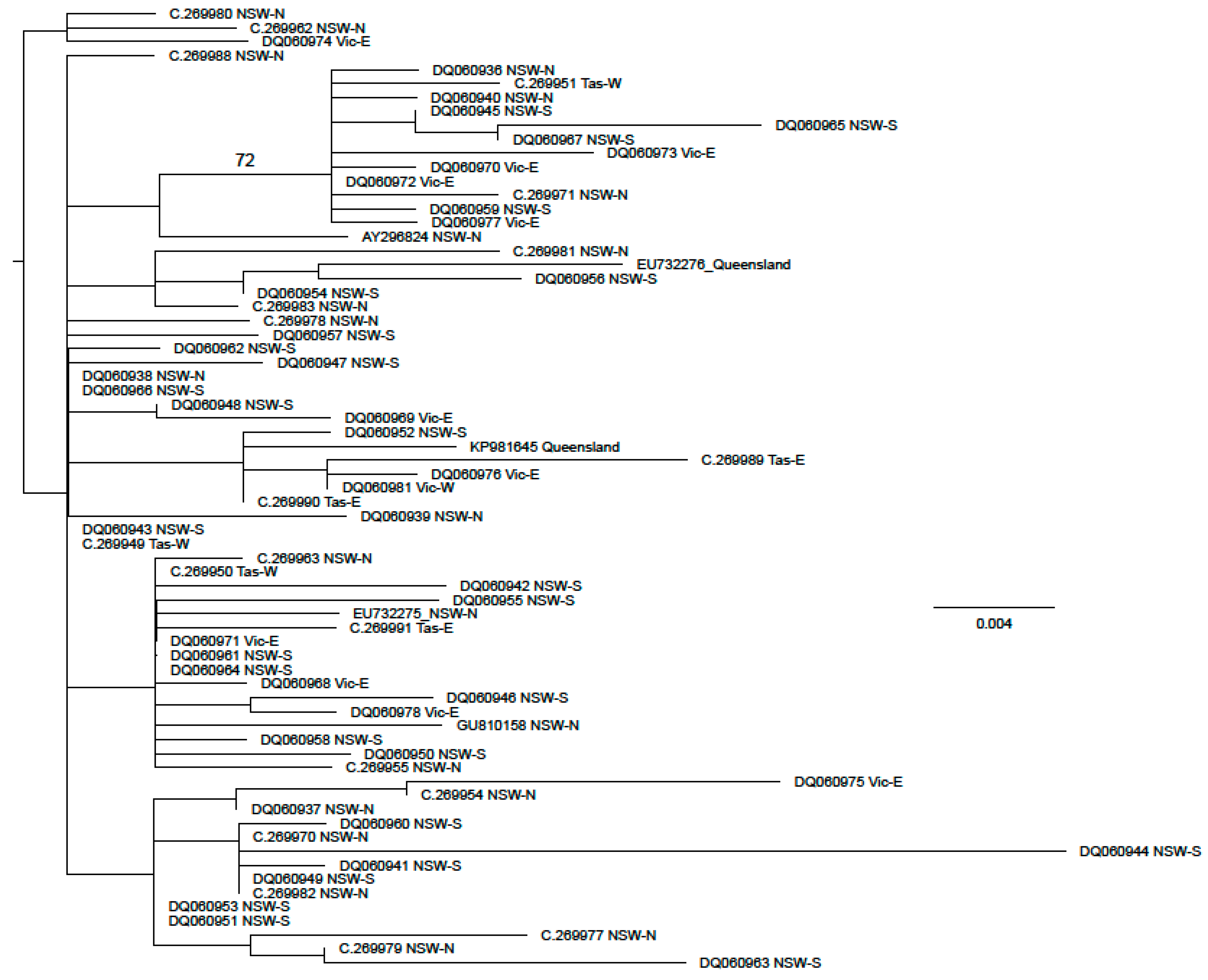
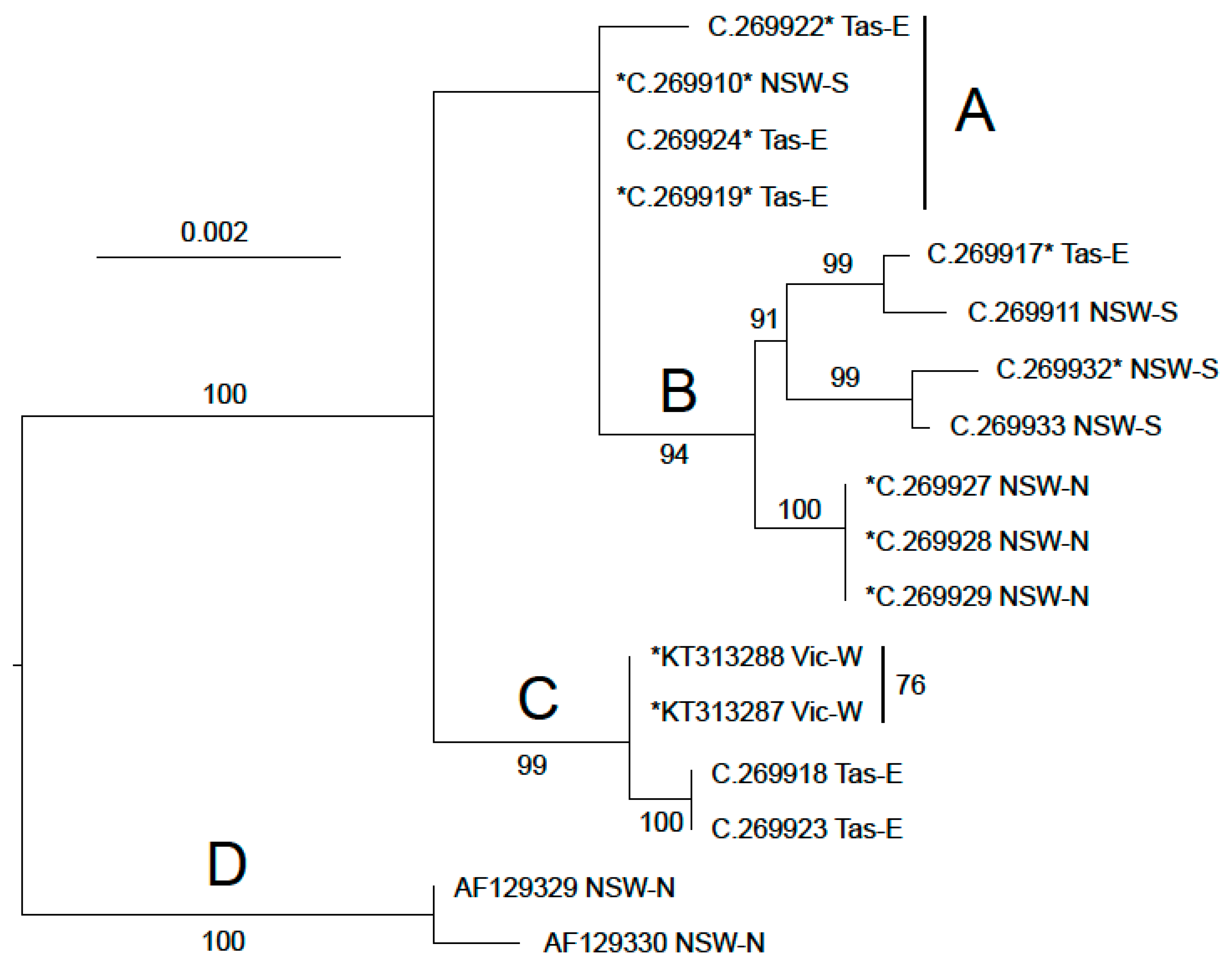
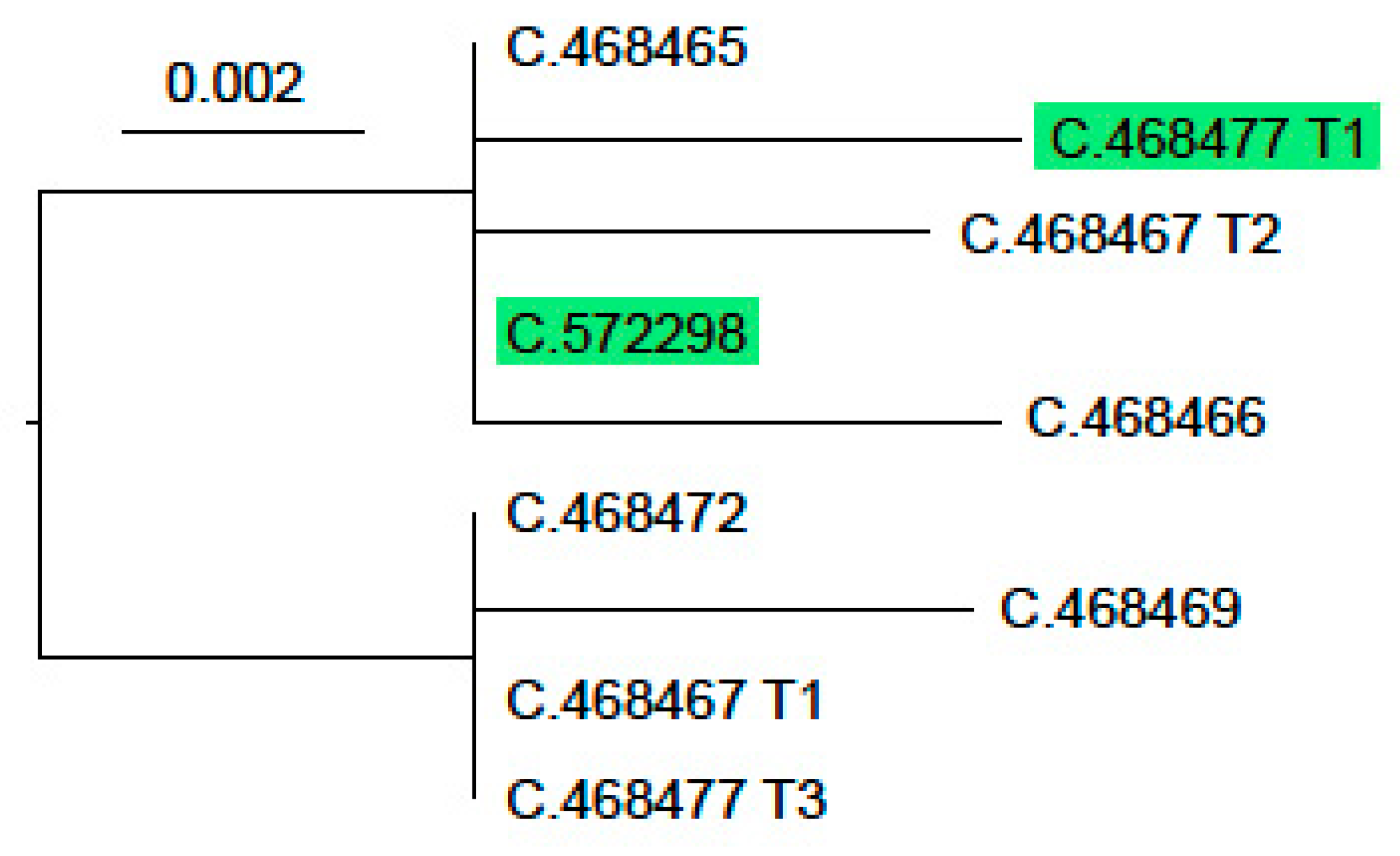
3. Results
3.1. Nerita
3.2. Ascorhis tasmanica
3.3. Phallomedusa solida
4. Discussion
Supplementary Materials
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Colgan, D.J. Marine and estuarine phylogeography of the coasts of south-eastern Australia. Mar. Freshw. Res. 2015, 67, 1597–1610. [Google Scholar] [CrossRef]
- Teske, P.R.; Sandoval-Castillo, J.; Waters, J.; Beheregaray, L.B. An overview of Australia’s temperate marine phylogeography, with new evidence from high-dispersal gastropods. J. Biogeogr. 2017, 44, 217–229. [Google Scholar] [CrossRef]
- Siddall, M.; Chappell, J.; Potter, E.K. Eustatic sea level during past interglacials. Dev. Quatern. Sci. 2007, 7, 75–92. [Google Scholar]
- Lawver, L.A.; Gahagan, L.M. Evolution of Cenozoic seaways in the circum-Antarctic region. Palaeogeog. Palaeoclimatol. Palaeoecol. 2003, 198, 11–37. [Google Scholar] [CrossRef]
- McKay, R.; Naish, T.; Carter, L.; Riesselman, C.; Dunbar, R.; Sjunneskog, C.; Winter, D.; Sangiorgi, F.; Warren, C.; Pagani, M.; et al. Antarctic and Southern Ocean influences on Late Pliocene global cooling. Proc. Natl. Acad. Sci. USA 2012, 109, 6423–6428. [Google Scholar] [CrossRef] [Green Version]
- Sikes, E.L.; Howard, W.R.; Samson, C.R.; Mahan, T.S.; Robertson, L.G.; Volkman, J.K. Southern Ocean seasonal temperature and Subtropical Front movement on the South Tasman Rise in the late Quaternary. Paleoceanography 2009, 24, PA2201. [Google Scholar] [CrossRef]
- Ayre, D.J.; Minchinton, T.E.; Perrin, C. Does life history predict past and current connectivity for rocky intertidal invertebrates across a marine biogeographic barrier? Mol. Ecol. 2009, 18, 1887–1903. [Google Scholar] [CrossRef] [PubMed]
- Waters, J.M.; McCulloch, G.A.; Eason, J.A. Marine biogeographical structure in two highly dispersive gastropods: Implications for trans-Tasman dispersal. J. Biogeog. 2007, 34, 678–687. [Google Scholar] [CrossRef]
- Miller, A.D.; Versace, V.L.; Matthews, T.G.; Montgomery, S.; Bowie, K.C. Ocean currents influence the genetic structure of an intertidal mollusc in southeastern Australia–implications for predicting the movement of passive dispersers across a marine biogeographic barrier. Ecol. Evol. 2013, 3, 1248–1261. [Google Scholar] [CrossRef]
- Colgan, D.J.; Schreiter, S. Extrinsic and intrinsic influences on the phylogeography of the Austrocochlea constricta species group. J. Exp. Mar. Biol. Ecol. 2011, 397, 44–51. [Google Scholar] [CrossRef]
- Colgan, D.J. Inter-boundary comparison between Bass Strait and southeastern Tasmania reveals taxon-specific effects of phylogeographic determinants in marine Mollusca. Regional Stud. Mar. Sci. 2019, 25, 100449. [Google Scholar] [CrossRef]
- Waters, J.M.; King, T.M.; O’loughlin, P.M.; Spencer, H.G. Phylogeographical disjunction in abundant high-dispersal littoral gastropods. Mol. Ecol. 2005, 14, 2789–2802. [Google Scholar] [CrossRef] [PubMed]
- Golding, R.E.; Colgan, D.J.; Nelmes, G.; Reutelshöfer, T. Sympatry and allopatry in the deeply divergent mitochondrial DNA clades of the estuarine pulmonate gastropod genus Phallomedusa (Mollusca, Gastropoda). Mar. Biol. 2011, 158, 1259–1269. [Google Scholar] [CrossRef]
- York, K.L.; Blacket, M.J.; Appleton, B.R. The Bassian Isthmus and the major ocean currents of southeast Australia influence the phylogeography and population structure of a southern Australian intertidal barnacle Catomerus polymerus (Darwin). Mol. Ecol. 2008, 17, 1948–1961. [Google Scholar] [CrossRef]
- Hershler, R.; Liu, H.P.; Mulvey, M. Phylogenetic relationships within the aquatic snail genus Tryonia: Implications for biogeography of the North American Southwest. Mol. Phylogenet. Evol. 1999, 13, 377–391. [Google Scholar] [CrossRef] [Green Version]
- Zielske, S.; Ponder, W.F.; Haase, M. The enigmatic pattern of long-distance dispersal of minute freshwater gastropods (Caenogastropoda, Truncatelloidea, Tateidae) across the South Pacific. J. Biogeogr. 2017, 44, 195–206. [Google Scholar] [CrossRef]
- Waters, J.M. Competitive exclusion: Phylogeography’s ‘elephant in the room’? Mol. Ecol. 2011, 20, 4388–4394. [Google Scholar] [CrossRef]
- Spencer, H.G.; Waters, J.M.; Eichhorst, T.E. Taxonomy and nomenclature of black nerites (Gastropoda: Neritimorpha: Nerita) from the South Pacific. Invert. Syst. 2007, 21, 229–237. [Google Scholar] [CrossRef]
- Frey, M.A.; Vermeij, G.J. Molecular phylogenies and historical biogeography of a circumtropical group of gastropods (Genus: Nerita): Implications for regional diversity patterns in the marine tropics. Mol. Phylogenett. Evol. 2008, 48, 1067–1086. [Google Scholar] [CrossRef]
- Waters, J.M. Marine biogeographical disjunction in temperate Australia: Historical landbridge, contemporary currents, or both? Div. Dist. 2008, 14, 692–700. [Google Scholar] [CrossRef]
- Grove, S.J. A Guide to the Seashells and other Marine Molluscs of Tasmania Web-Site. Available online: https://molluscsoftasmania.org.au/ (accessed on 22 December 2022).
- Hales, T.J. The family Neritidae in southern Australia. Mal. Soc. Australasia Newsl. 2021, 176, 9–10. [Google Scholar]
- Ponder, W.F.; Clark, G.A. A morphological and electrophoretic examination of Hydrobia buccinoides, a variable brackish-water gastropod from temperate Australia (Mollusca, Hydrobiidae). Aust. J. Zool. 1988, 36, 661–689. [Google Scholar] [CrossRef] [Green Version]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotech. 1994, 3, 294–299. [Google Scholar]
- Jarman, S.N.; Ward, R.D.; Elliott, N.G. Oligonucleotide primers for PCR amplification of coelomate introns. Mar. Biotech. 2002, 4, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Colgan, D.J.; Ponder, W.F.; Eggler, P.E. Gastropod evolutionary rates and phylogenetic relationships assessed using partial 28S rDNA and histone H3 sequences. Zool. Scr. 2000, 2, 29–63. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl. Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user–friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Stephens, M.; Smith, N.J.; Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Amin, S.; Prentis, P.J.; Gilding, E.K.; Collett, C.; Pavasovic, A. Comparative Transcriptome Analysis of Two Marine Gastropods (Nerita melanotragus and N. albicilla) Offers Novel Insights into Adaptation to Temperature Stress. Unpublished Reference in GenBank Accession Data 2016. Available online: https://www.ncbi.nlm.nih.gov/nuccore/KP981645.1 (accessed on 23 February 2023).
- Williams, S.T.; Ozawa, T. Molecular phylogeny suggests polyphyly of both the turban shells(family Turbinidae) and the superfamily Trochoidea (Mollusca: Vetigastropoda). Mol. Phylogenet. Evol. 2006, 39, 33–51. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web-servers. Syst. Biol. 2008, 75, 758–771. [Google Scholar] [CrossRef]
- Pattengale, N.D.; Alipour, M.; Bininda-Emonds, O.R.P.; Moret, B.M.E.; Stamatakis, A. How many bootstrap replicates are necessary? J. Computat. Biol. 2010, 17, 337–354. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1999, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Sánchez-DelBarrio, J.C.; Messeguer, X.; Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 2002, 19, 2496–2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Onsins, S.E.; Rozas, J. Statistical properties of new neutrality tests against population growth. Mol. Biol. Evol. 2002, 19, 2092–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Meth. Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Clement, M.; Posada, D.C.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1659. [Google Scholar] [CrossRef] [Green Version]
- Puillandre, N.; Brouillet, S.; Achaz, G. ASAP: Assemble species by automatic partitioning. Mol. Ecol. Resour. 2021, 21, 609–620. [Google Scholar] [CrossRef]
- Teske, P.R.; Sandoval-Castillo, J.; van Sebille, E.; Waters, J.; Beheregaray, L.B. On-shelf larval retention limits population connectivity in a coastal broadcast spawner. Mar. Ecol. Progr. Ser. 2015, 532, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Marko, P.B. Fossil calibration of molecular clocks and the divergence times of geminate species pairs separated by the Isthmus of Panama. Mol. Biol. Evol. 2002, 19, 2005–2021. [Google Scholar] [CrossRef] [Green Version]
- Donald, K.M.; Kennedy, M.; Spencer, H.G. Cladogenesis as the result of long-distance rafting events in South Pacific topshells (Gastropoda, Trochidae). Evolution 2005, 59, 1701–1711. [Google Scholar]
- Nürnberg, D.; Brughmans, N.; Schönfeld, J.; Ninnemann, U.; Dullo, C. Paleo–export production, terrigenous flux and sea surface temperatures around Tasmania—Implications for glacial/interglacial changes in the Subtropical Convergence Zone. In The cenozoic Southern Ocean: Tectonics, Sedimentation and Climate Change between Australia and Antarctica; Exon, N.F., Kennett, J.P., Malone, M.J., Eds.; Geophysical Monograph Series; American Geophysical Union: Washington, DC, USA, 2004; Volume 151, pp. 291–317. [Google Scholar]
- Colgan, D.J. Fine-scale spatial partitioning of genetic variation and evolutionary contestability in the invasive estuarine mussel Xenostrobus securis. Mar. Biol. Res. 2017, 13, 1059–1072. [Google Scholar] [CrossRef]
- Paradis, E. The distribution of branch lengths in phylogenetic trees. Mol. Phylogenet. Evol. 2016, 94, 136–145. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Location Number | Collection Event (MAL#) | Locality | Latitude | Longitude | Collection Date | No. of Sequences |
|---|---|---|---|---|---|---|---|
| Ascorhis tasmanica | |||||||
| A1 | MAL71148 | North Avoca | 33°27′27″ | 151°26′30″ | 11 May 2007 | 3, 3 | |
| A2 | MAL88537 | Lake Nangudga, | 36°17′ | 150°08′ | 13 June 2006 | 1, 2, 1 | |
| A3 | MAL72686 | Merimbula, Back Lagoon, | 36°53′08″ | 149°55′01″ | 1 March 2008 | 1, 2, 1 | |
| A4 | MAL72687 | Moulding Lagoon | 41°59′ | 148°14′ | 3 April 2007 | 2, 3 | |
| A5 | MAL72688 | Swansea, | 42°07′52″ | 148°04′35″ | 3 April 2007 | 2, 3, 1 | |
| Nerita atramentosa | |||||||
| Na1 | MAL72701 | Bridport | 40°58′54″ | 147°23′08″ | 3 April 2007 | 2, 1 | |
| Na2 | MAL71157 | Georgetown | 41°06′33″ | 146°48′39″ | 4 April 2007 | 2, 2, 1 | |
| Nerita melanotragus | |||||||
| Nm1 | MAL71145 | Hastings Point | 28°21′46″ | 153°34′46″ | 1 April 2008 | 1 | |
| Nm2 | MAL71144 | Skennars Head | 28°49′20″ | 153°36′26″ | 1 April 2008 | 2, 2, 2 | |
| Nm3 | MAL71148 | Norh Avoca Rock Platform | 33°27′27″ | 151°26′30″ | 11 May 2007 | 2 | |
| Nm4 | MAL71138 | Bottle and Glass Rocks | 33°50′51″ | 151°16′12″ | 28 March 2007 | 2, 2, 2 | |
| Nm5 | MAL71152 | Shelly Beach, Cronulla | 33°04′01″ | 159°09′31″ | 12 May 2009 | 3, 6, 3 | |
| Nm6 | MAL71140 | Bicheno | 41°52′18″ | 148°17′54″ | 3 April 2007 | 3 | |
| Nm7 | MAL71151 | Deviot | 41°15′ | 146°56′ | 4 April 2007 | 3 | |
| Phallomedusa solida | |||||||
| P1 | MAL76554 | Clarence | 29°25′59″ | 153°14′17″ | 31 March 2008 | 1 | |
| P2 | MAL72691 | Ballina, North Creek | 29°28′44″ | 153°21′50″ | 1 April 2008 | 1 | |
| P3 | MAL71151 | Angourie | 28°10′34″ | 153°32′27″ | 2 April 2008 | 1 | |
| P4 | MAL76555 | Roseville, | 33°46′07″ | 151°11′57″ | 28 March 2007 | 2 | |
| P5 | MAL76552 | Little Salt Pan Creek | 33°58′17″ | 151°02′24″ | 3 February 2006 | 1 | |
| P7 | MAL71160 | Narooma, at highway bridge | 36°12′35″ | 150°07′20″ | 13 June 2006 | 2 | |
| P8 | MAL76559 | Falmouth | 41°30′ | 148°16′ | 3 April 2007 | 1 | |
| Species/Gene | Number of Sequences | Length | No. Hap. | Hap. Div. | π | Av. Diff. | Tajima’s D | Popn Growth | R2 |
|---|---|---|---|---|---|---|---|---|---|
| Ascorhis tasmanica | |||||||||
| ATPSα | 10 | 365 | 2 | 0.356 | 0.009 +/− 0.004 | 3.2 | 0.026, p > 0.1 | Θi 13.973 τ 0.000 | 0.178 |
| COI | 17 | 577 | 11 | 0.941 | 0.024 +/− 0.003 | 13.618 | −0.252, p > 0.1 | Θi 6.506 τ 7.112 | 0.125 |
| Nerita atramentosa | |||||||||
| ATPSα | 6 | 393 | 3 | 0.800 | 0.007 +/− 0.002 | 2.667 | 1.219, p > 0.1 | Θi 1.309, τ 1.788 | 0.267 |
| COI | 36 | 571 | 22 | 0.917 | 0.008 +/− 0.001 | 4.724 | −1.673, p > 0.05 | Θi 2.2267 τ 2.4971 | 0.051 |
| Nerita melanotragus | |||||||||
| ATPSα | 18 | 393 | 10 | 0.85 | 0.007 +/− 0.002 | 2.817 | −1.783, p > 0.05 | Θi 2.2260 τ 0.557 | 0.119 |
| COI | 69 | 512 | 48 | 0.974 | 0.008 +/− 0.001 | 4.179 | −2.066, p < 0.05 | Θi 0.525 τ 0.654 | 0.034 |
| Phallomedusa solida | |||||||||
| ATPSα−overall | 18 | 225 | 9 | 0.869 | 0.007 +/− 0.0001 | 1.601 | −0.723, p > 0.1 | Θi 0.000 τ 1.601 | 0.119 |
| COI−overall | 72 | 593 | 52 | 0.960 | 0.018 +/− 0.002 | 10.967 | −1.133, p > 0.1 | Θi 9.105 τ 1.862 | 0.066 |
| COI-clade A | 48 | 613 | 36 | 0.957 | 0.005 +/− 0.001 | 3.124 | −2.591, p < 0.001 | Θi 0.512 τ 2.612 | 0.024 |
| COI-clade B | 20 | 637 | 16 | 0.983 | 0.007 +/− 0.001 | 4.579 | −1.797, p > 0.05 | Θi 0.780 τ 0.799 | 0.058 |
| Group | Group A | Group B | Group C | Group D |
|---|---|---|---|---|
| Group A | 0.004 | 0.008 | 0.005 | |
| Group B | 0.017 | 0.008 | 0.006 | |
| Group C | 0.041 | 0.047 | 0.008 | |
| Group D | 0.019 | 0.030 | 0.041 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colgan, D.J. Do Deep Mitochondrial DNA Divergences within Intertidal Gastropods Reveal Phylogeographic Signals from Earlier Glacial Cycles? Diversity 2023, 15, 346. https://doi.org/10.3390/d15030346
Colgan DJ. Do Deep Mitochondrial DNA Divergences within Intertidal Gastropods Reveal Phylogeographic Signals from Earlier Glacial Cycles? Diversity. 2023; 15(3):346. https://doi.org/10.3390/d15030346
Chicago/Turabian StyleColgan, Donald James. 2023. "Do Deep Mitochondrial DNA Divergences within Intertidal Gastropods Reveal Phylogeographic Signals from Earlier Glacial Cycles?" Diversity 15, no. 3: 346. https://doi.org/10.3390/d15030346
APA StyleColgan, D. J. (2023). Do Deep Mitochondrial DNA Divergences within Intertidal Gastropods Reveal Phylogeographic Signals from Earlier Glacial Cycles? Diversity, 15(3), 346. https://doi.org/10.3390/d15030346