An Infrared Actin Probe for Deep-Cell Electroporation-Based Single-Molecule Speckle (eSiMS) Microscopy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. NIR-Labeled Actin
2.2. Plasmids
2.3. Cell Culture and Electroporation
2.4. Single-Molecule Speckle Imaging and Data Analysis
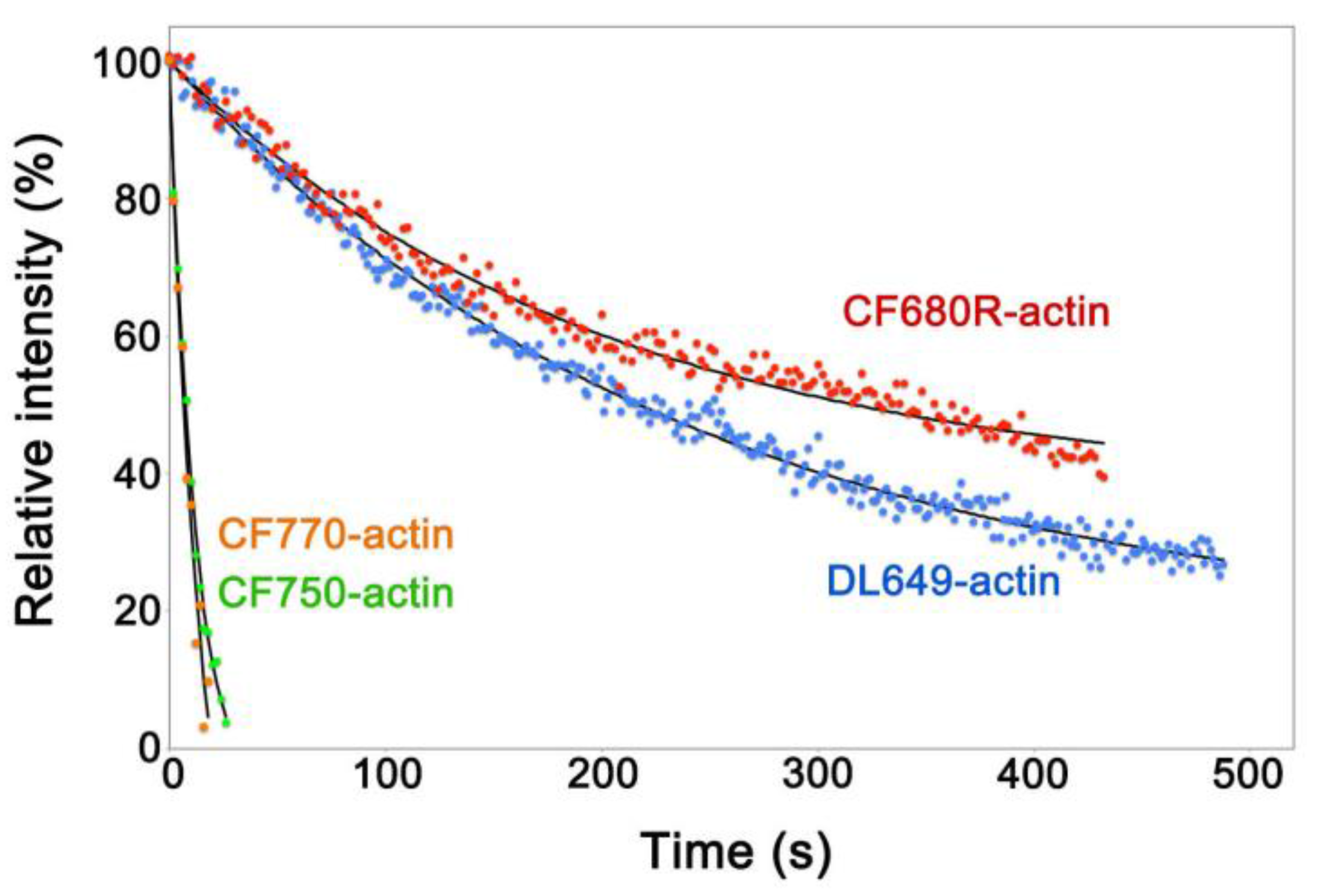
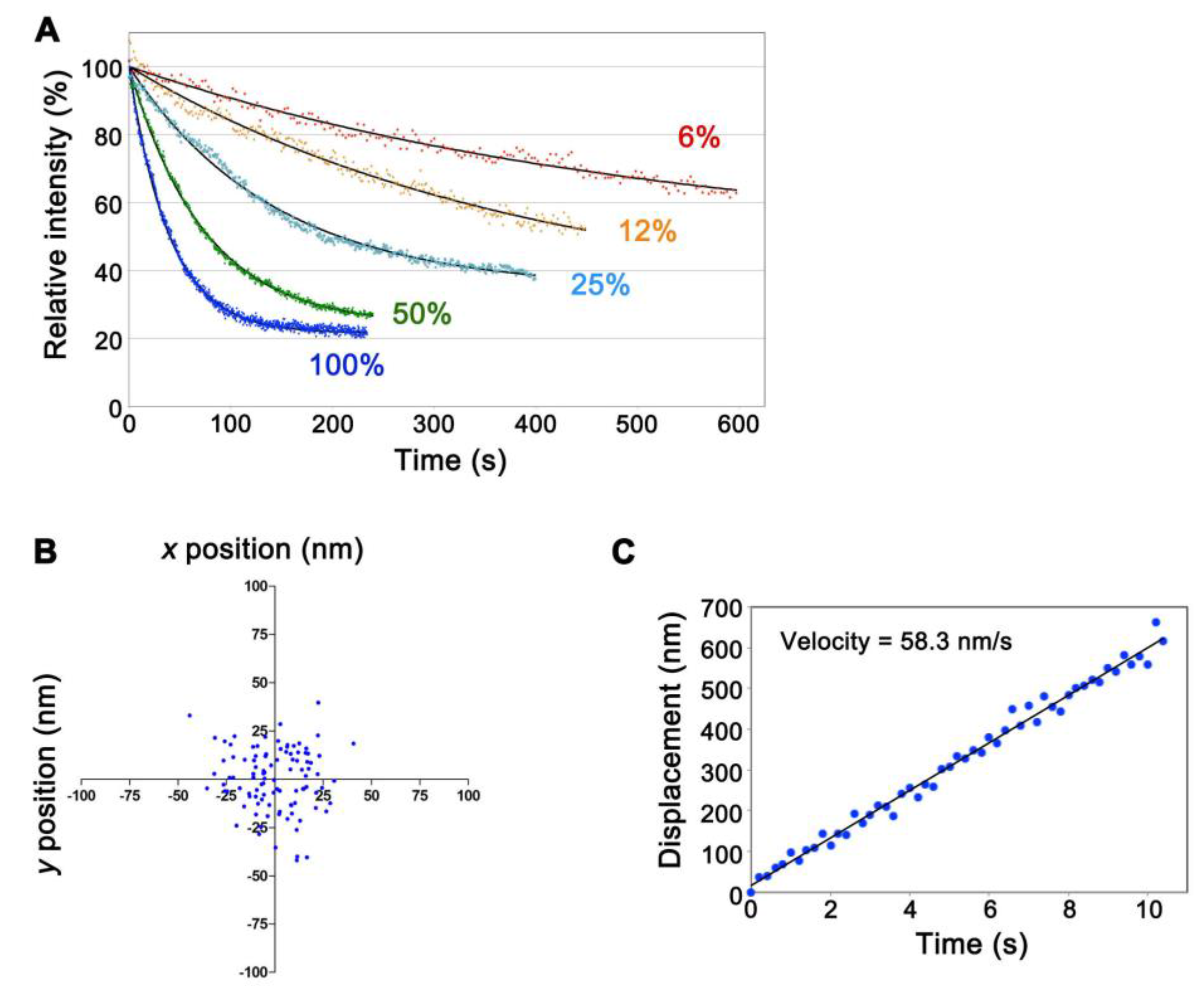
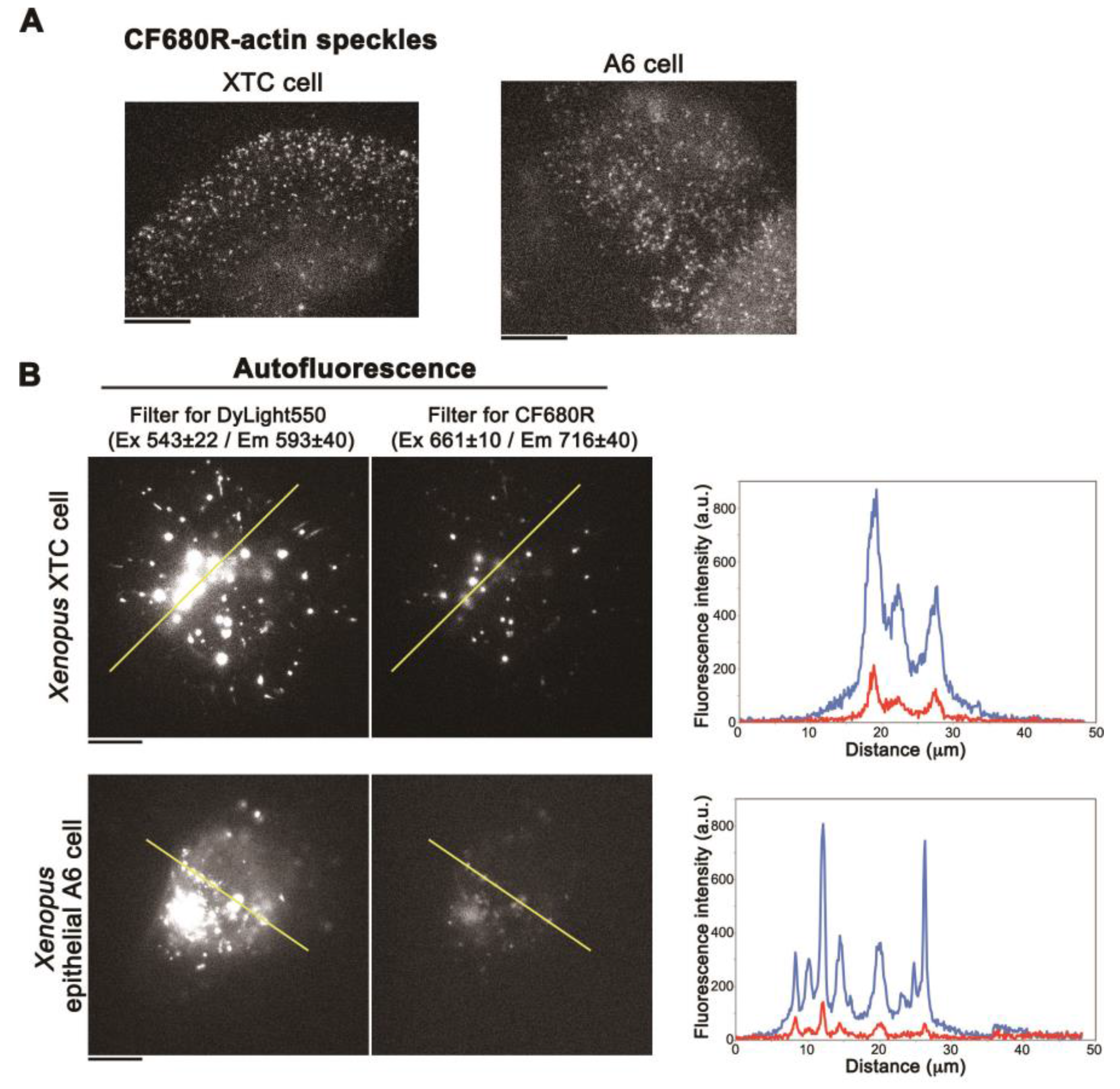
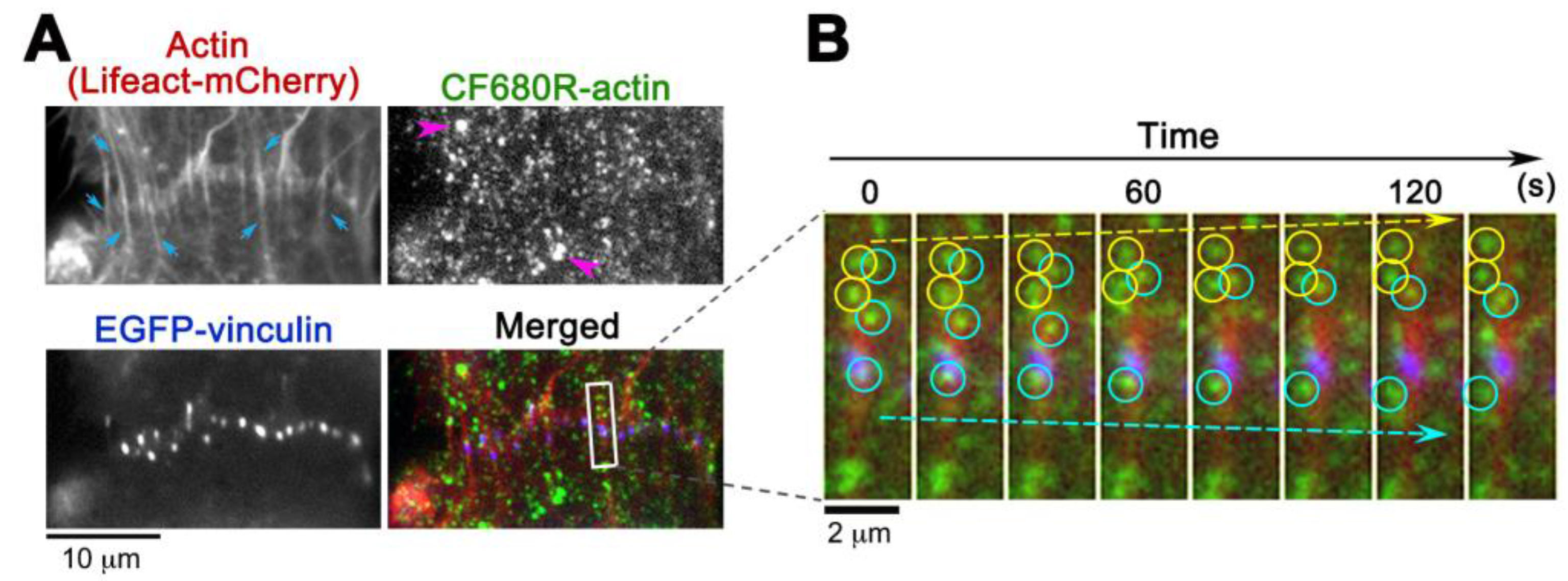
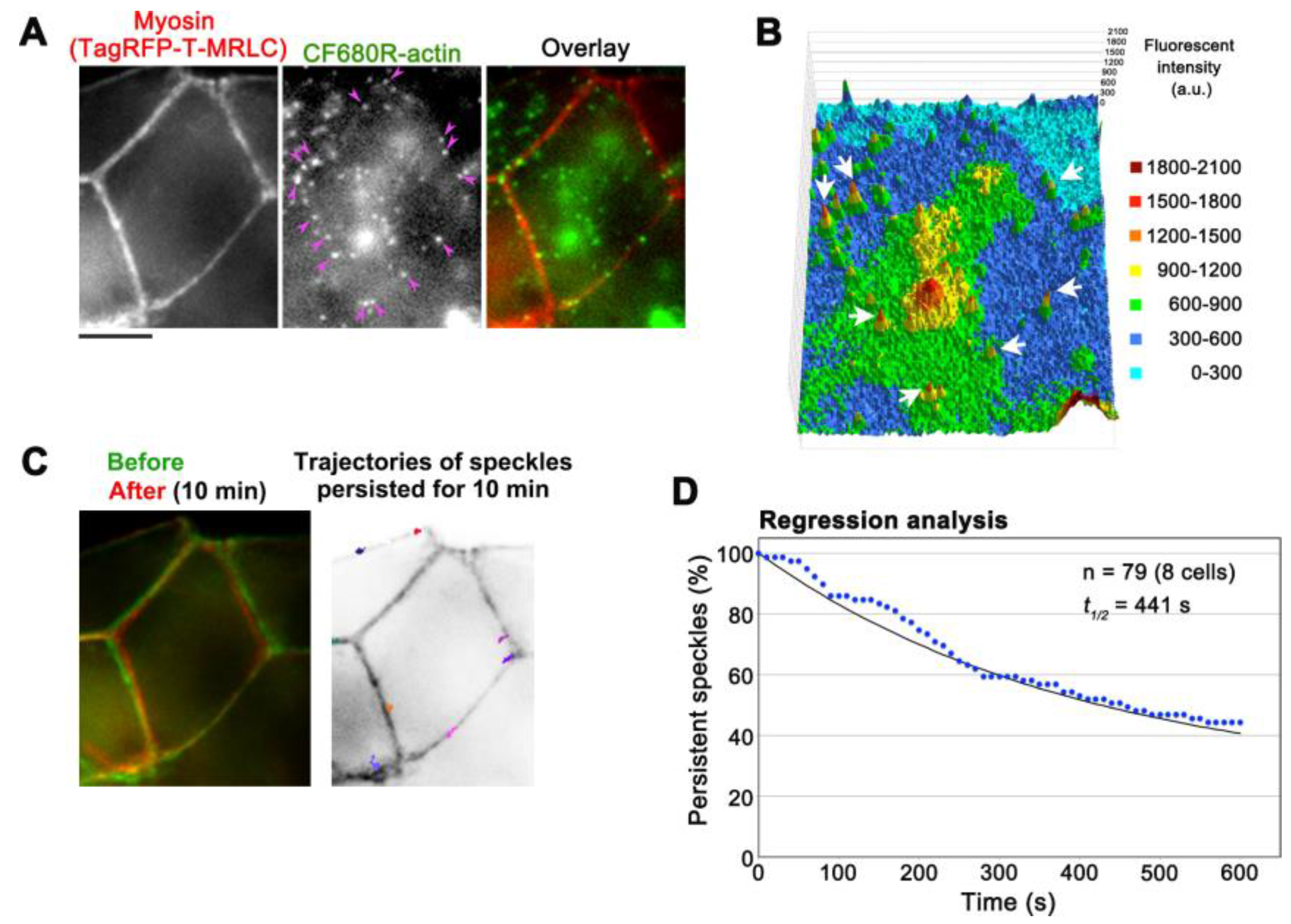
3. Results
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Watanabe, N. Inside view of cell locomotion through single-molecule: Fast F-/G-actin cycle and G-actin regulation of polymer restoration. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 62–83. [Google Scholar] [CrossRef] [PubMed]
- Higashida, C.; Miyoshi, T.; Fujita, A.; Oceguera-Yanez, F.; Monypenny, J.; Andou, Y.; Narumiya, S.; Watanabe, N. Actin polymerization-driven molecular movement of mDia1 in living cells. Science 2004, 303, 2007–2010. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, T.; Tsuji, T.; Higashida, C.; Hertzog, M.; Fujita, A.; Narumiya, S.; Scita, G.; Watanabe, N. Actin turnover-dependent fast dissociation of capping protein in the dendritic nucleation actin network: Evidence of frequent filament severing. J. Cell Biol. 2006, 175, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Douglass, A.D.; Vale, R.D. Single-molecule microscopy reveals plasma membrane microdomains created by protein-protein networks that exclude or trap signaling molecules in T cells. Cell 2005, 121, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Kasai, R.S.; Kusumi, A. Single-molecule imaging revealed dynamic GPCR dimerization. Curr. Opin. Cell Biol. 2014, 27, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Ananthanarayanan, V.; Schattat, M.; Vogel, S.K.; Krull, A.; Pavin, N.; Tolic-Norrelykke, I.M. Dynein motion switches from diffusive to directed upon cortical anchoring. Cell 2013, 153, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; McEwen, D.P.; Martens, J.R.; Meyhofer, E.; Verhey, K.J. Single molecule imaging reveals differences in microtubule track selection between Kinesin motors. PLoS Biol. 2009, 7, e1000216. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, M.; Imamoto, N.; Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods 2008, 5, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Zhen, C.Y.; Tatavosian, R.; Huynh, T.N.; Duc, H.N.; Das, R.; Kokotovic, M.; Grimm, J.B.; Lavis, L.D.; Lee, J.; Mejia, F.J.; et al. Live-cell single-molecule tracking reveals co-recognition of H3K27me3 and DNA targets polycomb Cbx7-PRC1 to chromatin. Elife 2016, 5, e17667. [Google Scholar] [CrossRef] [PubMed]
- Izeddin, I.; Recamier, V.; Bosanac, L.; Cisse, I.I.; Boudarene, L.; Dugast-Darzacq, C.; Proux, F.; Benichou, O.; Voituriez, R.; Bensaude, O.; et al. Single-molecule tracking in live cells reveals distinct target-search strategies of transcription factors in the nucleus. Elife 2014, 3, e02230. [Google Scholar] [CrossRef] [PubMed]
- Robin, F.B.; McFadden, W.M.; Yao, B.; Munro, E.M. Single-molecule analysis of cell surface dynamics in Caenorhabditis elegans embryos. Nat. Methods 2014, 11, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, J.C.; Suter, D.M.; Roy, R.; Zhao, Z.W.; Chapman, A.R.; Basu, S.; Maniatis, T.; Xie, X.S. Single-molecule imaging of transcription factor binding to DNA in live mammalian cells. Nat. Methods 2013, 10, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.C.; Legant, W.R.; Wang, K.; Shao, L.; Milkie, D.E.; Davidson, M.W.; Janetopoulos, C.; Wu, X.S.; Hammer, J.A., 3rd; Liu, Z.; et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 2014, 346, 1257998. [Google Scholar] [CrossRef] [PubMed]
- Croce, A.C.; Bottiroli, G. Autofluorescence spectroscopy and imaging: A tool for biomedical research and diagnosis. Eur. J. Histochem. 2014, 58, 2461. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Mitchison, T.J. Single-molecule speckle analysis of actin filament turnover in lamellipodia. Science 2002, 295, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, S.; Mizuno, H.; Smith, M.B.; Ryan, G.L.; Kiuchi, T.; Vavylonis, D.; Watanabe, N. New single-molecule speckle microscopy reveals modification of the retrograde actin flow by focal adhesions at nanometer scales. Mol. Biol. Cell 2014, 25, 1010–1024. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, S.; Mizuno, H.; Watanabe, N. An easy-to-use single-molecule speckle microscopy enabling nanometer-scale flow and wide-range lifetime measurement of cellular actin filaments. Methods Cell Biol. 2015, 125, 43–59. [Google Scholar] [PubMed]
- Weissleder, R. A clearer vision for in vivo imaging. Nat. Biotechnol. 2001, 19, 316–317. [Google Scholar] [CrossRef] [PubMed]
- Frangioni, J.V. In vivo near-infrared fluorescence imaging. Curr. Opin. Chem. Biol. 2003, 7, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Ryan, G.L.; Petroccia, H.M.; Watanabe, N.; Vavylonis, D. Excitable actin dynamics in lamellipodial protrusion and retraction. Biophys. J. 2012, 102, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Uchimura, T.; Fumoto, K.; Yamamoto, Y.; Ueda, K.; Hosoya, H. Spatial localization of mono-and diphosphorylated myosin II regulatory light chain at the leading edge of motile HeLa cells. Cell Struct. Funct. 2002, 27, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N. Fluorescence single-molecule imaging of actin turnover and regulatory mechanisms. Methods Enzymol. 2012, 505, 219–232. [Google Scholar] [PubMed]
- Mimori-Kiyosue, Y.; Matsui, C.; Sasaki, H.; Tsukita, S. Adenomatous polyposis coli (APC) protein regulates epithelial cell migration and morphogenesis via PDZ domain-based interactions with plasma membranes. Genes Cells 2007, 12, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Maruthamuthu, V.; Aratyn-Schaus, Y.; Gardel, M.L. Conserved F-actin dynamics and force transmission at cell adhesion. Curr. Opin. Cell Biol. 2010, 22, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Vasioukhin, V.; Bauer, C.; Mei, Y.; Fuchs, E. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell 2000, 100, 209–219. [Google Scholar] [CrossRef]
- Kametani, Y.; Takeichi, M. Basal-to-apical cadherin flow at cell junctions. Nat. Cell Biol. 2007, 9, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Yonemura, S. Cadherin-actin interactions at adherens junctions. Curr. Opin. Cell Biol. 2011, 23, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Breitsprecher, D.; Goode, B.L. Formins at a glance. J. Cell Sci. 2013, 126, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.S.; Pollard, T.D. Review of the mechanism of processive actin filament elongation by formins. Cell Motil. Cytoskeleton 2009, 66, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Grikscheit, K.; Grosse, R. Formins at the Junction. Trends Biochem. Sci. 2016, 41, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Nag, S.; Pollard, T.D. Formins filter modified actin subunits during processive elongation. J. Struct. Biol. 2012, 177, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Grikscheit, K.; Frank, T.; Wang, Y.; Grosse, R. Junctional actin assembly is mediated by Formin-like 2 downstream of Rac1. J. Cell Biol. 2015, 209, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, E.M.; Verma, S.; Ali, R.G.; Ratheesh, A.; Hamilton, N.A.; Akhmanova, A.; Yap, A.S. N-WASP regulates the epithelial junctional actin cytoskeleton through a non-canonical post-nucleation pathway. Nat. Cell Biol. 2011, 13, 934–943. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamashiro, S.; Watanabe, N. An Infrared Actin Probe for Deep-Cell Electroporation-Based Single-Molecule Speckle (eSiMS) Microscopy. Sensors 2017, 17, 1545. https://doi.org/10.3390/s17071545
Yamashiro S, Watanabe N. An Infrared Actin Probe for Deep-Cell Electroporation-Based Single-Molecule Speckle (eSiMS) Microscopy. Sensors. 2017; 17(7):1545. https://doi.org/10.3390/s17071545
Chicago/Turabian StyleYamashiro, Sawako, and Naoki Watanabe. 2017. "An Infrared Actin Probe for Deep-Cell Electroporation-Based Single-Molecule Speckle (eSiMS) Microscopy" Sensors 17, no. 7: 1545. https://doi.org/10.3390/s17071545
APA StyleYamashiro, S., & Watanabe, N. (2017). An Infrared Actin Probe for Deep-Cell Electroporation-Based Single-Molecule Speckle (eSiMS) Microscopy. Sensors, 17(7), 1545. https://doi.org/10.3390/s17071545