1. Introduction
The increasing importance of biosensors in everyday life is the driving force behind a merging of the microelectronic and biomedical communities. The common effort is the production of devices ready for mass production that will perform accurate analyses. Among microelectronic materials, silicon (Si) has the most mature and low cost technology, hence, several research groups are approaching Si compatible technology as innovative platform for biosensors. Moreover, Si based matrixes have been proved to be a very useful support for the immobilization of enzymes thanks to their capability of retaining biological activity. To develop a useful device, particular care must be used for biological molecule immobilization on Si-based surfaces. There is a body of data in the literature [
1,
2] regarding immobilization mechanisms of biological molecules on solid surfaces. The most used approach is the formation of covalent bonds with the solid surface [
1-
7], often using bifunctional reagents to bridge the biological molecule and the functionalized sample surface.
Si-based biosensors, as well as conventional microelectronic devices, must be fully characterized using standard microelectronics techniques allowing biological molecule monitoring. In this way, the new technology costs are contained, since no new equipment is needed. Among the different techniques, X-ray Photoelectron Spectroscopy (XPS) and Energy Dispersive X-rays (EDX) coupled to Scanning Electron Microscopy (SEM) are very intriguing. The first one provides information on chemical bonds and molecular composition of the material surfaces, combined with a high surface specificity, while the second one, when operated on cross-sections, allows one the investigation of thick layers of material without, if coupled to SEM, complex sample preparation.
The above mentioned techniques were used to study the immobilization of glucose oxidase (GOx) enzyme from
Aspergillus niger, on an SiO
2 surface. GOx is a dimeric globular protein having overall dimensions of 6.0×5.2×7.7 nm
3. It catalyzes the oxidation of β-
d-glucose to δ-gluconolactone by a reaction that can be summarized in two steps: i) glucose oxidation with the enzyme reduction, ii) re-oxidation of the enzyme with consumption of molecular oxide (O
2) and production of hydrogen peroxide (H
2O
2) [
8]. For this reason GOx is commonly used to monitor the glucose concentration in the blood; hence, a GOx based micro-biosensor [
9] would have immediate applications in monitoring diabetes [
10].
In order to visualize the enzyme with XPS, it was labelled with gold nanoparticles having a diameter of 1.4 nm. Au is regularly used to label the amino groups of biological molecules in solution when transmission electron microscopy measurements are performed [
11]. GOx molecules contain two disulphide bonds and two accessible thiol groups [
12]. Since GOx amino groups were used to immobilize the protein on the solid surface, the free sulfydryl groups were labelled with functionalized gold nanoparticles [
13]. Gold labelling in this study had a twofold purpose: to provide conclusive proof of the presence of the enzyme and as internal reference for XPS measurements. To verify the preservation of enzyme activity after immobilization, a spectrophotometric technique was used. It was based on the use of peroxidase to monitor hydrogen peroxide production.
The aims of this work were the definition of an optimized immobilization protocol that would allow the maximum surface coverage without any loss in the GOx enzyme performances and the characterization of the deposited organic layer using standard microelectronic techniques.
2. Results and Discussion
The most promising immobilization protocol reported in literature (see ref. [
2]) was tested on bulk SiO
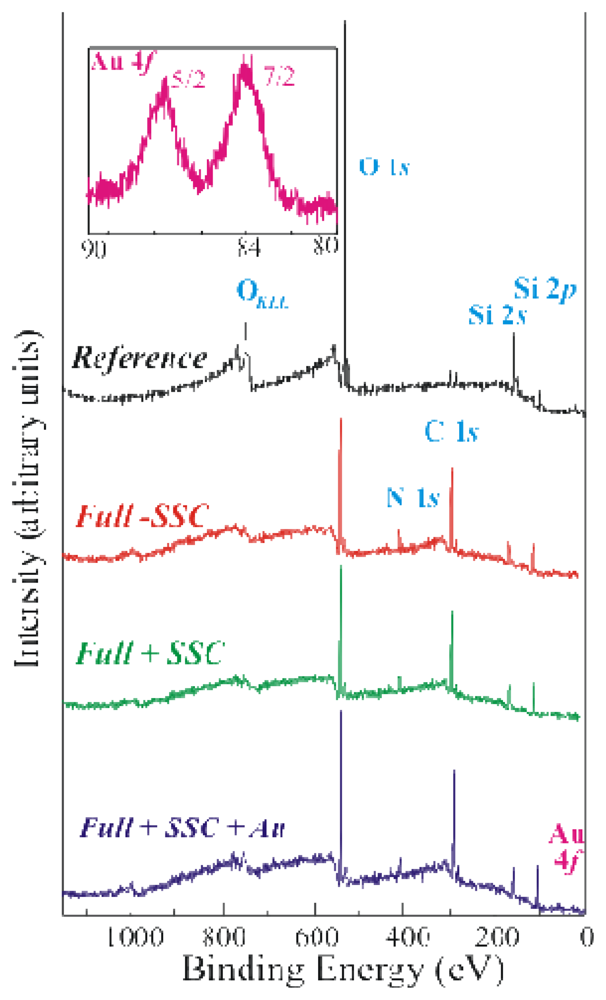
2 samples, mainly using XPS measurements. The results were compared with those obtained from a sample where a modified protocol was applied. The main difference with respect to the fist protocol was the addition of an oxide activation step at the beginning of the processing (see the experimental section for details). The XPS survey spectra of the reference (top spectrum, in black) and three fully processed samples (differently prepared) are shown in
Figure 1.
The second spectrum from the top was acquired from a sample obtained after the three step procedure reported in the literature [
2] (Full without SSC, Full-SSC, in red), while the other two spectra were obtained from samples where the modified protocol was applied (full +SSC, in green and in blue). GOx was labelled with Au nanoparticles before immobilization during the last sample preparation (Full+SSC+Au, in blue).
The reference sample exhibited the XPS Si peaks centred at about 155 eV (Si 2
s) and 104 eV (Si 2
p), a very small C peak (due to adventitious contamination) centred at about 285 eV and the O 1
s XPS signal centred at a binding energy of about 533eV. All the fully processed samples show the same signature already observed for the reference sample, even if their relative concentrations are quite different. As an example the C 1
s peak was well visible for all these samples, thanks to the presence of organic material. Moreover the N 1
s peak at about 400eV was detected in these spectra to demonstrate the enzyme presence in the fully processed samples [
14]. Finally, the Au 4
f peak was detected in the Full+SSC+Au sample. The expanded spectral region of Au
4f for this sample is shown in the inset of
Figure 1. The doublet Au
4f7/2 and Au
4f5/2 (used as reference) exhibited binding energies of 84.0 and 87.7eV, respectively. Au presence provided a conclusive proof of GOx immobilization on the sample. It should be mentioned that the Au peaks observed in XPS are a direct experimental evidence of the GOx presence on the sample. Literature results provide only indirect evidences of GOx immobilization, obtained using its enzymatic activity (as an example see refs. [
4,
5]).
The three fully processed samples, prepared using three different methods (Full+SSC, Full-SSC and Full+SSC+Au) allowed us to answer many open questions on the goodness of our protocol. First of all, the comparison with a protocol widely used in literature allowed us to directly measure any improvement in the sample preparation due to the introduction of a further step (SSC treatment). Moreover, gold labelling provided an experimental direct proof of the GOx presence, not found, to our knowledge, in literature.
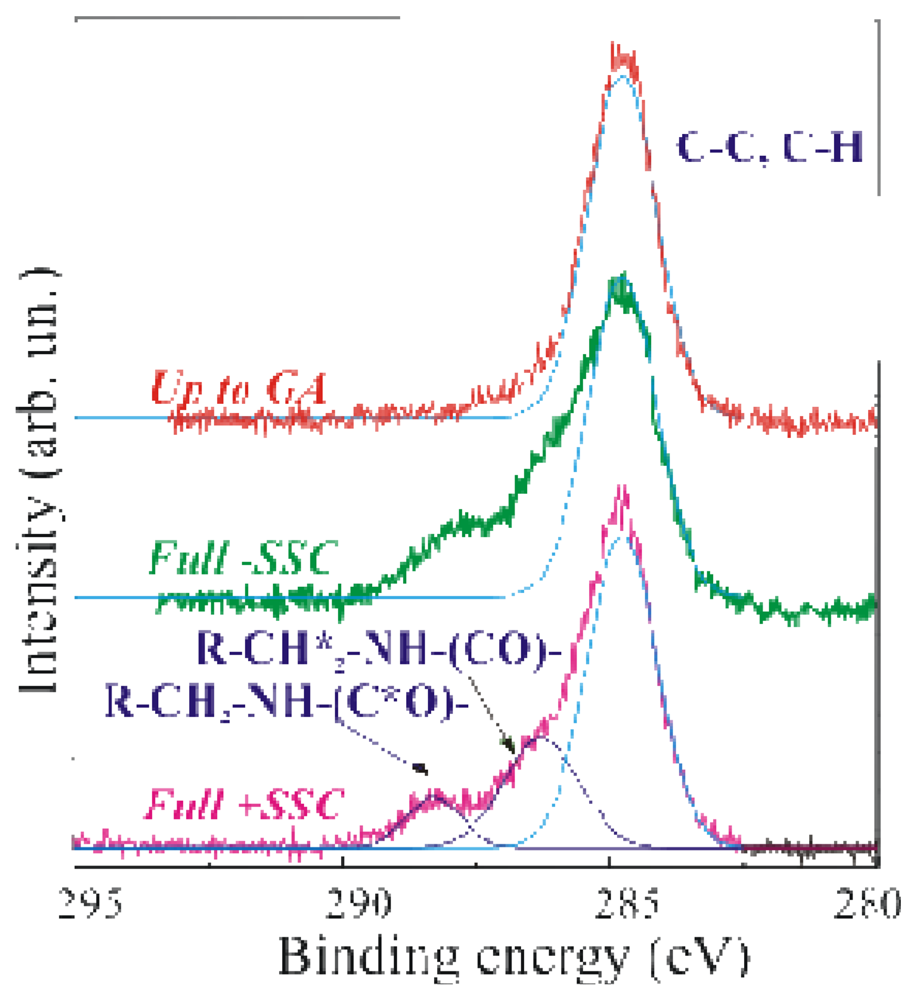
A further confirmation of the GOx presence was easily obtained by the inspection of the C 1
s peak reported in
Figure 2, where the XPS spectra of a sample stopped after the GA immobilization step (SSC+APTES+GA, labelled up-to-GA, red line), the Full-SSC (green line) and the Fully processed (Full+SSC, magenta line) samples are compared in the binding energy range 295–280eV.
The C 1
s peak, centred at 284.8–285eV, is due to C-C and C-H bonds. The light blue line superimposed to the experimental spectrum was a simulation of the C-C and C-H XPS peak. The up-to-GA sample showed only this peak, and an additional weak shoulder at about 287eV attributed to the R-CHO groups of GA. It should be mentioned that the GA is a linear molecule (CHO-(CH
2)
3-CHO) with one aldehydic group (CHO) at each end. The other two samples showed at least other two components at 286.3–286.5eV, assigned to R-CH
2*-NH-(CO)-, and at 288.3eV, assigned to R-CH
2-NH-(C*O)-chemical groups respectively. The dark blue lines in
Figure 2 are the simulated peaks superimposed to the experimental data to allow one an easier identification of the different peaks. Due to the sensitivity of the XPS technique, phosphorous and the other functions of the FAD groups were not detected.
The comparison of the two different preparation methods (Full-SSC and Full+SSC) allowed us to conclude that the C peak inspection does not allow to observe differences between our immobilization protocol and the former (Full-SSC). A more interesting data to compare the two immobilization methods is provided by the inspection of the Si related peaks.
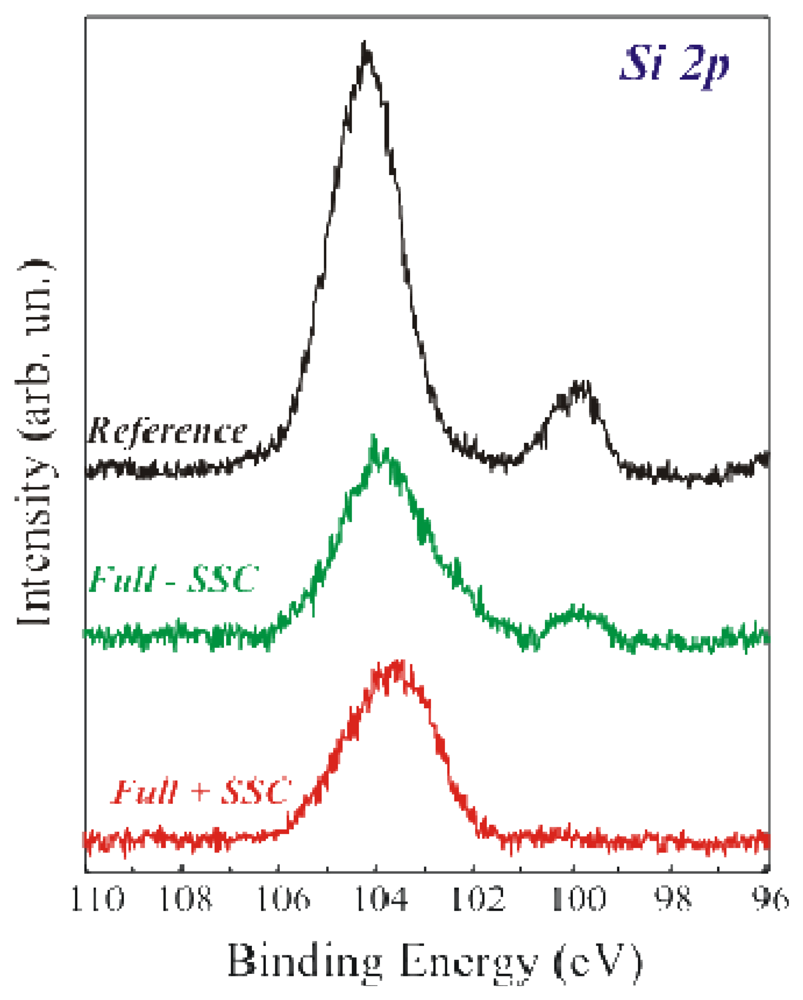
The Si 2
p spectra of the above mentioned samples are shown in
Figure 3. The reference sample (in black) exhibited two components having binding energies of 99.7 eV and 104 eV, assigned to Si° and SiO
2 respectively [
15,
16]. The SiO
2 component is centred at 104 eV instead of 103.4 eV, as expected, due to differential charging between the SiO
2 layer and the Si substrate [
17]. The Full+SSC sample (bottom spectrum) does not exhibit the Si° component, while the Full-SSC sample (centre spectrum) still exhibits the Si° component.
Recent literature data confirm that if the SiO
2 layer is thin (6.5 nm in our case), it is possible to infer information on the uniformity of the biological layer [
13,
14]. If the coverage was uniform, after GA immobilization, there should be a film fully shielding the Si° signal from the Si under the oxide. Two conditions must be fulfilled to have a complete substrate shield. The first one is a film thickness above the photoelectron penetration depth. The second is the uniformity of APTES+GA and, eventually, +GOx layer. The real shielding was confirmed by our measurements. In fact, the fully processed sample Si 2
p spectrum showed only SiO
2 component. The intensity of the Si substrate was attenuated by the two added layers (SiO
2+organic) [
14]. On the other hand, if the immobilization procedure did not produce a uniform film, the Si° signal was still visible after the full immobilization protocol, despite of the fact that the enzyme was correctly immobilized, as demonstrated by the Full-SSC spectrum. The results confirmed that the immobilization procedure without oxide activation was not the best achievable on our SiO
2 surfaces. This conclusion was possible only thanks to the direct comparison of the two immobilization methods on the same kind of sample (oxide thickness and SiO
2 preparation method).
The great improvement in film uniformity measured on the samples that underwent Full+SSC protocol can be explained considering the SiO
2 growing procedure. In fact, the oxide layer was grown in a dry environment (see experimental). Recent studies [
18] have demonstrated that silicon oxides grown in a dry environment have a density of Si–OH surface endings lower than wet grown oxides. As a result, they have a lower reactivity against silanization, leading, in our case, to a lower enzyme uniformity. The oxide activation we introduced as the first step allowed us to increase the number of available sites for enzyme bonding and to obtain a final uniform deposition. The enzymatic activity measurements confirmed these results (see after).
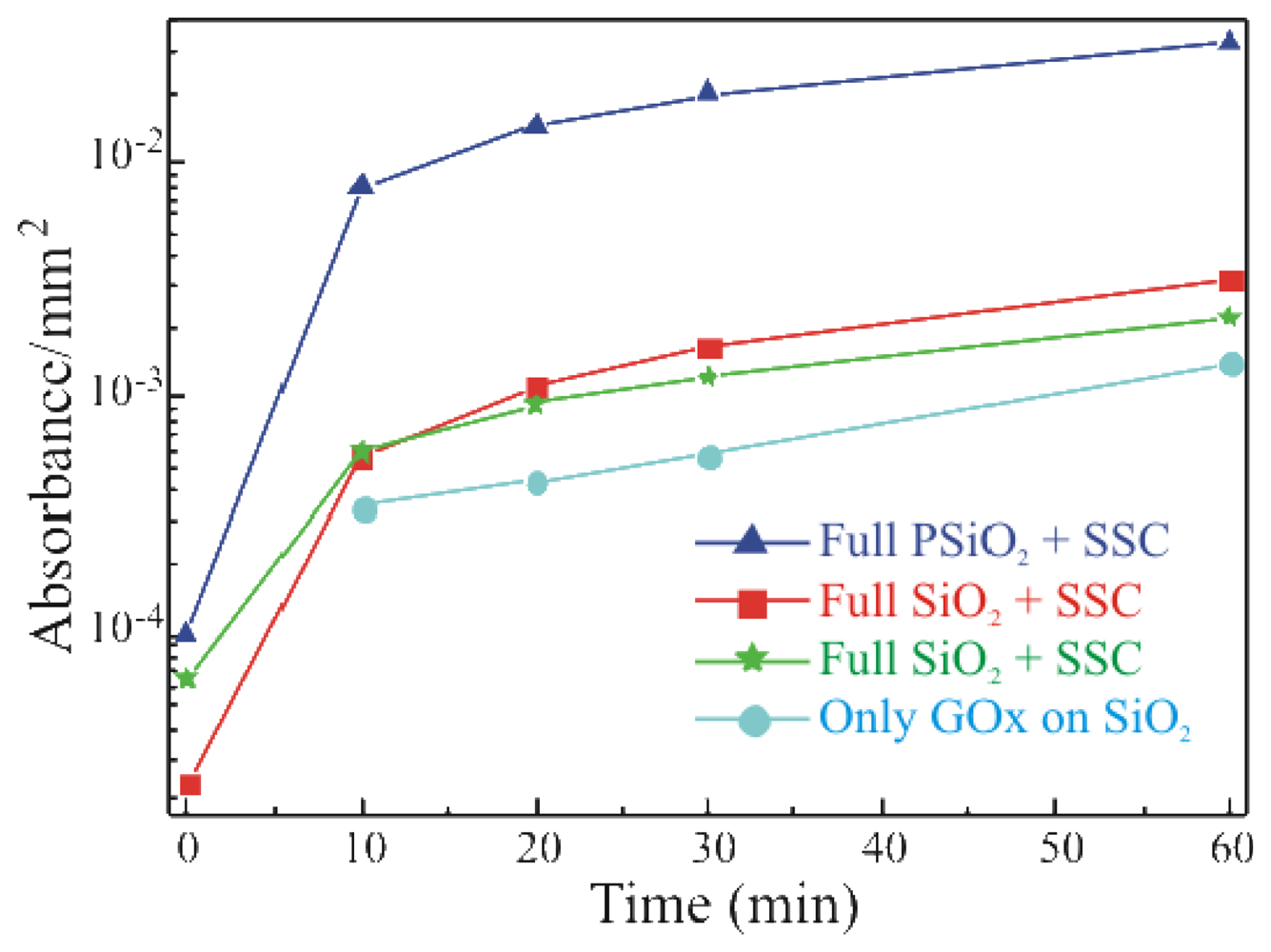
To study the activity of the immobilized enzyme, a simple, spectrophotometric assay was used. GOx activity was monitored on the Full+SSC sample, on the Full-SSC (former protocol) sample and on the sample that underwent only GOx deposition without previous surface functionalization (only GOx). The results are summarized in
Figure 4 and compared with the activity of a porous silicon dioxide (PSiO
2) fully processed with SSC sample. All data were normalized to the real area of the sample.
The data show an increase in the enzymatic activity when the oxide activation is carried out before silanization (Full+SSC; red squares) with respect to the Full-SSC sample (green stars). The only-GOx sample (light blue circles) exhibited an enzymatic activity lower than the one measured for both the samples that underwent chemical immobilization. The comparison of the absorbance values with those obtained from the free enzymes in solution, allowed us to estimate a concentration of active GOx on the SiO
2 samples of about 0.002 U ml
-1[
21]. The immobilization protocol carried out on porous SiO
2 surfaces (dark blue triangles) clearly indicated a higher enzymatic activity, more than one order of magnitude, for this sample with respect to the bulk SiO
2 samples. It is due to the greater surface available in such material. In fact, PSiO
2 has a porous structure, like a sponge, hence a greater surface to volume ratio. Values up to 1000 can be obtained. An SEM image of the PSiO
2 layer is provided in the inset of
Figure 6 Now, if the GOx permeates the PSiO
2 pores a higher enzyme amount can be deposited on the same sample area with respect to a bulk SiO
2 surface. Similar results are reported in literature [
19] and provide an indirect measure of the enzyme presence within the pores.
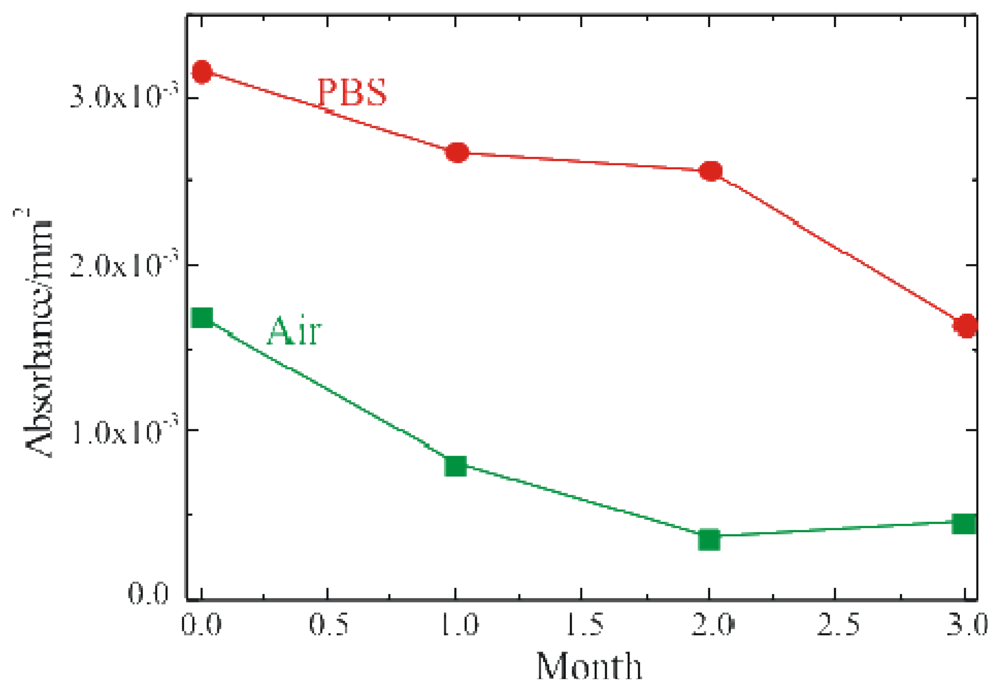
The enzymatic activity of the fully processed sample, stored either in buffer solution (PBS, 0.1M pH 6.5) at 4°C or in air at RT, was monitored. The results of these measurements are summarized in
Figure 5 where the red circles represent the sample stored in PBS, while the green squares represent the sample stored in air. Both samples were monitored immediately after immobilization and after 1, 2 and 3 months. The absorbance values reported in figure were detected 60 min after the reaction started. The samples stored in PBS retained activity over longer periods of time. On the other hand, the samples stored in air showed an immediate decrease in the enzymatic activity, already after the first month of aging.
The presence of carbon in the full porous thickness confirmed the result already observed from the enzymatic activity measurements: the enzyme permeated the pores. This last measurement provides a direct experimental evidence of the enzyme presence in pores, while its functionality is witnessed by the spectrophotometric measurements of
Figure 4.
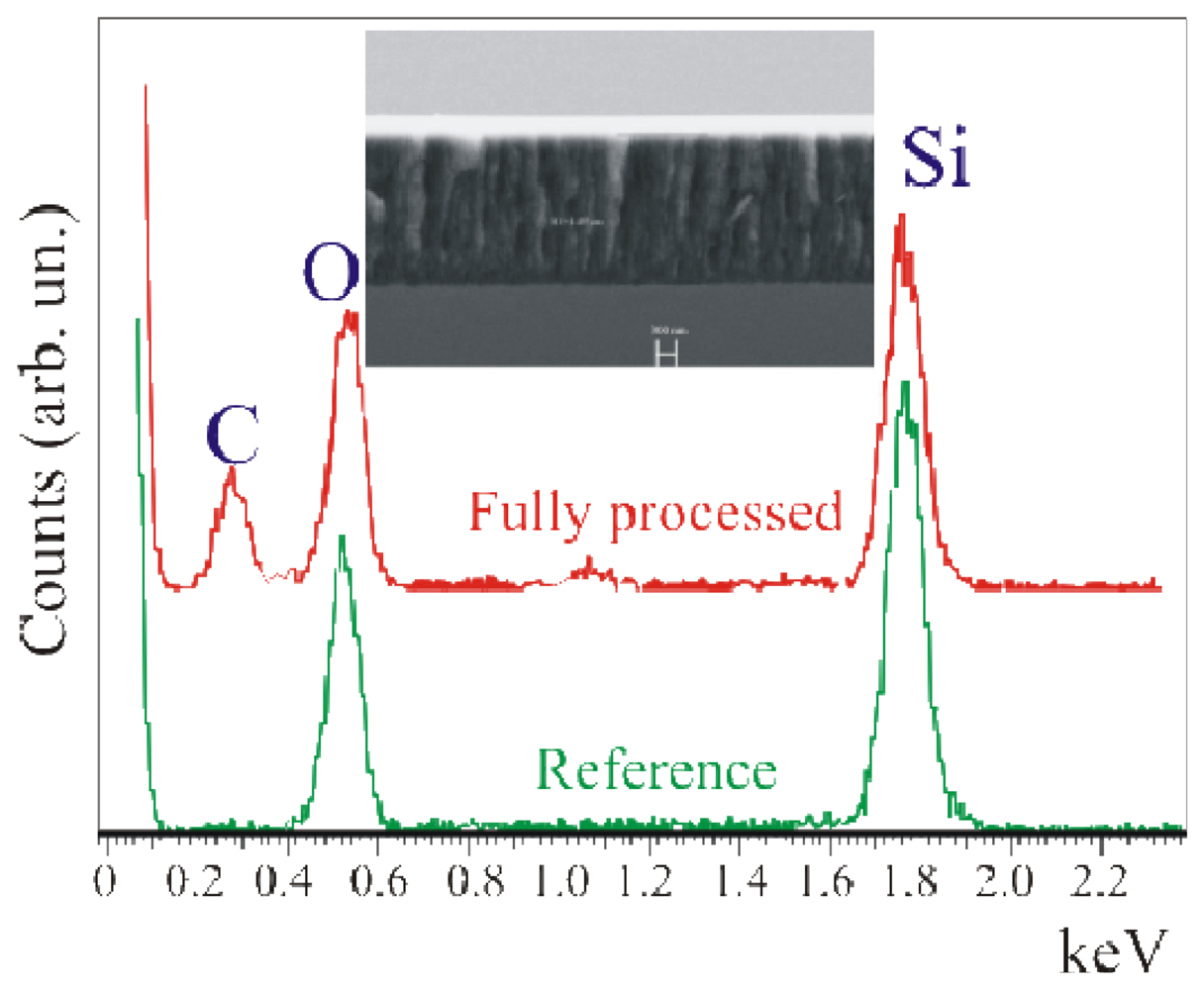
Using SEM-EDX measurements [
22] some interesting observations were made (
Figure 6). Both the thickness and the porous nature of the layer are clearly visible in the insert of
Figure 6, where the SEM image of the device cross-section shows a layer thickness of 2.26 μm. EDX measurements were carried out on the fully processed sample (Full + SSC) and on a reference sample. This comparison is needed to exclude any carbon contamination of the porous layer. In fact, since the PSi is obtained through electrochemical reaction in a cell containing a C-rich solution (see Experimental), the sample could absorb C (or have C bonded to the surface) after processing. Such C could diffuse in the sample during the subsequent oxidation process. As a result a C peak not necessarily related to the biological molecules could be detected. In our case, the reference did not show any C contamination (within the detection limits) and exhibited only the presence of O and Si, as observed from the K
α emission of the two elements at 0.51 keV and 1.73 keV, respectively. On the other hand, the sample that underwent the full immobilization protocol exhibited, in addition to the Si and O signals, a clear emission at 0.34 keV due to C.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





