
Pathophysiological Significance of Dermatan Sulfate Proteoglycans Revealed by Human Genetic Disorders



Abstract
:1. Introduction
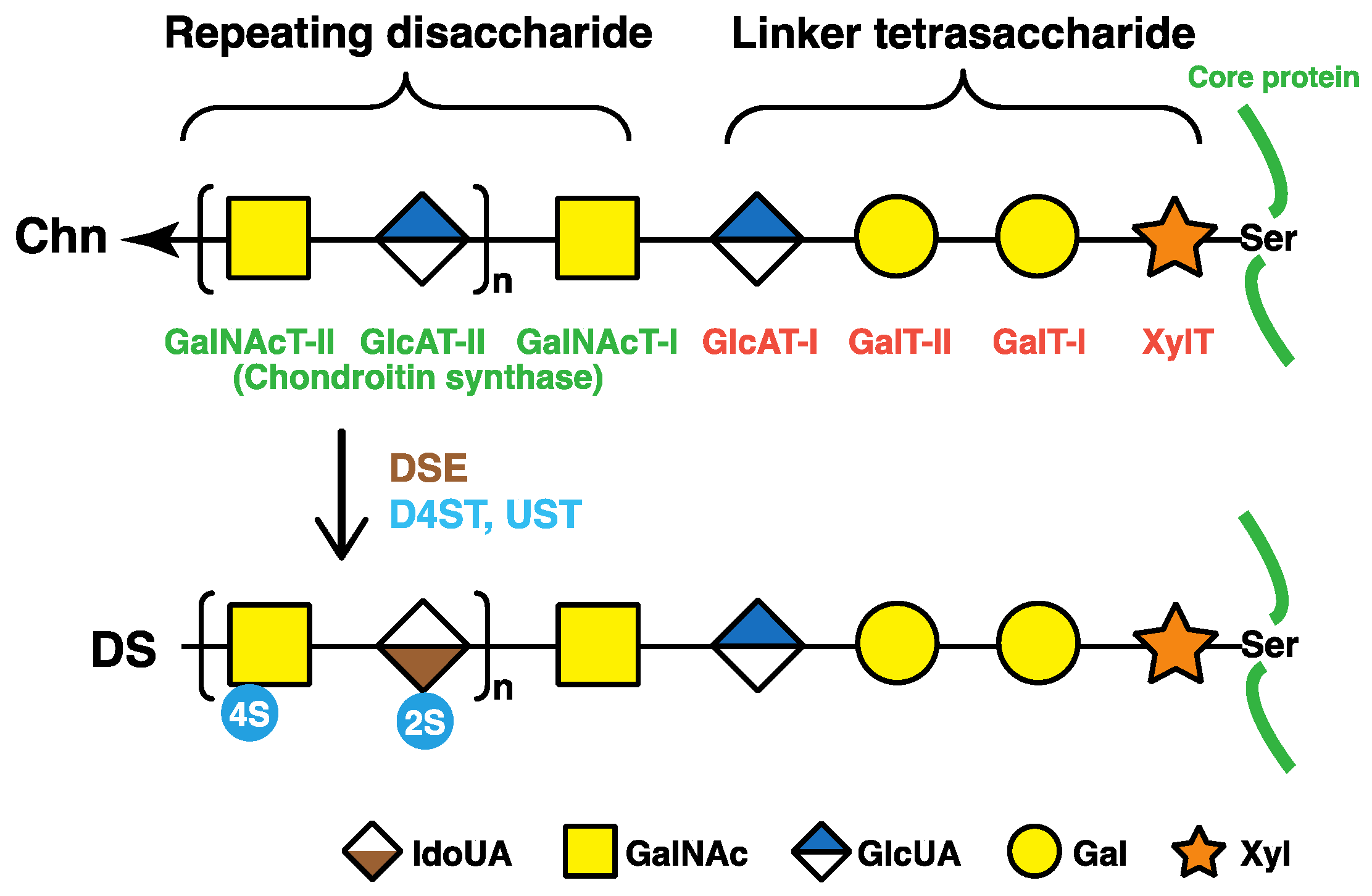
2. Biosynthesis of DS Chains
2.1. Glycosaminoglycan–Protein Linker Region
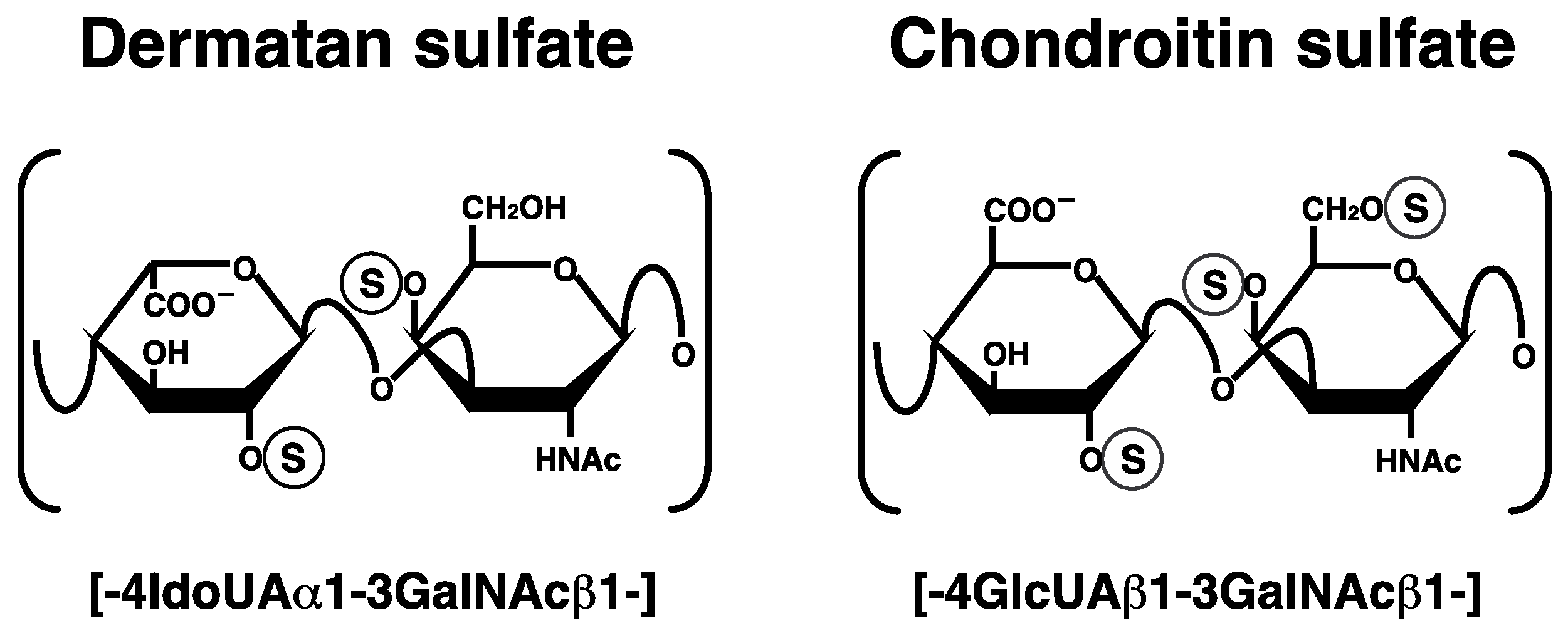
2.2. Repeating Disaccharide Region of DS
3. Human Disorders Affecting the Skeleton and Skin Caused by Disturbances in DS Biosynthetic Enzymes and DS-Proteoglycans
3.1. B4GALT7 (GalT-I) Deficiency
3.2. B3GALT6 (GalT-II) Deficiency
3.3. DSE Deficiency
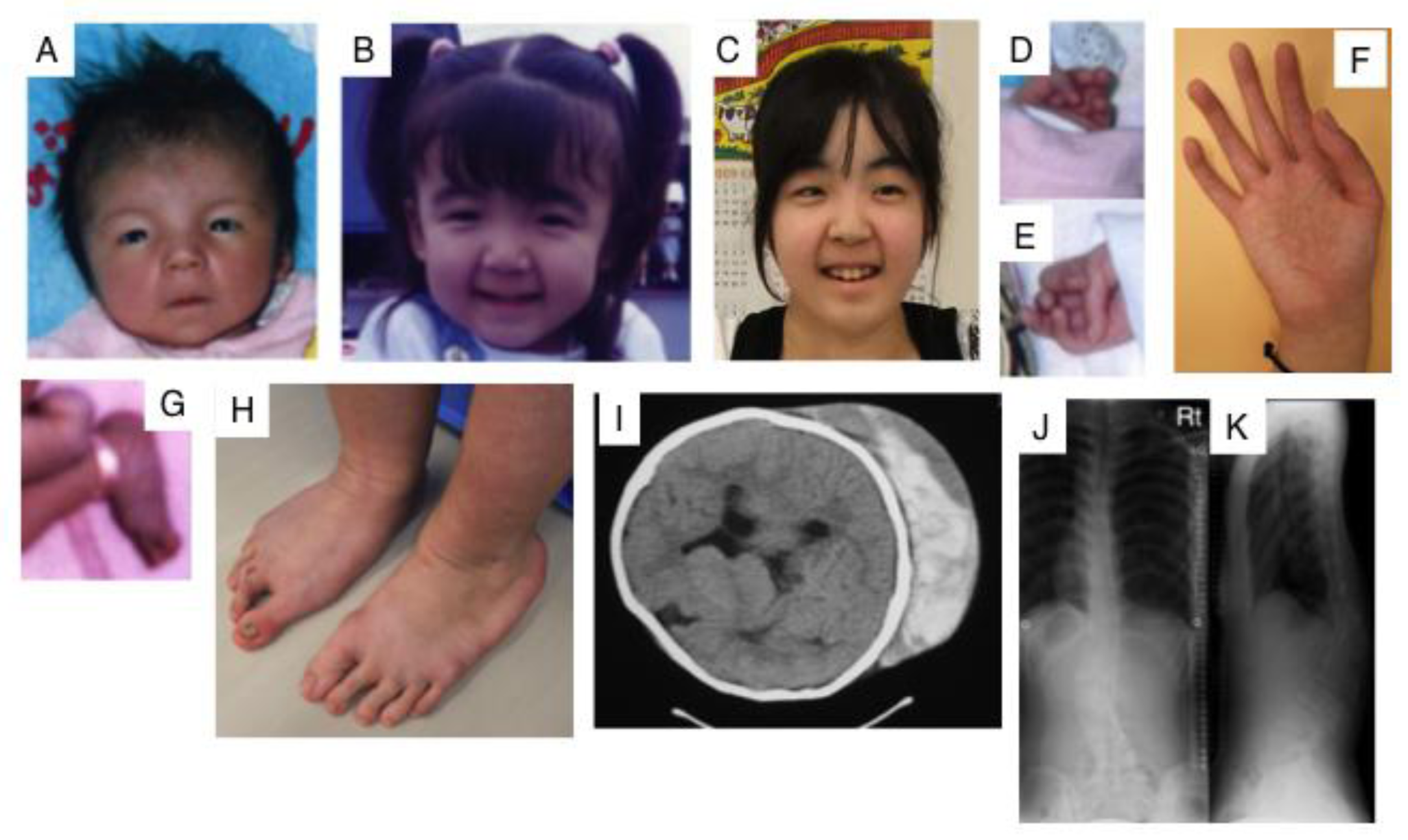
3.4. CHST14 (D4ST1) Deficiency
3.5. UST Deficiency
3.6. DCN Deficiecny
3.7. BGN Deficiency
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Iozzo, R.V. Matrix proteoglycans: From molecular design to cellular function. Annu. Rev. Biochem. 1998, 67, 609–652. [Google Scholar] [CrossRef] [PubMed]
- Neill, T.; Schaefer, L.; Iozzo, R.V. Decoding the matrix: Instructive roles of proteoglycan receptors. Biochemistry 2015, 54, 4583–4598. [Google Scholar] [CrossRef] [PubMed]
- Trowbridge, J.M.; Gallo, R.L. Dermatan sulfate: New functions from an old glycosaminoglycan. Glycobiology 2002, 12, 117R–125R. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, S.; Yamada, S.; Sugahara, K. Molecular interactions between chondroitin-dermatan sulfate and growth factors/receptors/matrix proteins. Curr. Opin. Struct. Biol. 2015, 34, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Danielson, K.G.; Baribault, H.; Holmes, D.F.; Graham, H.; Kadler, K.E.; Iozzo, R.V. Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J. Cell Biol. 1997, 136, 729–743. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Bianco, P.; Fisher, L.W.; Longenecker, G.; Smith, E.; Goldstein, S.; Bonadio, J.; Boskey, A.; Heegaard, A.M.; Sommer, B.; et al. Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nat. Genet. 1998, 20, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Svensson, L.; Aszódi, A.; Reinholt, F.P.; Fässler, R.; Heinegård, D.; Oldberg, A. Fibromodulin-null mice have abnormal collagen fibrils, tissue organization, and altered lumican deposition in tendon. J. Biol. Chem. 1999, 274, 9636–9647. [Google Scholar] [CrossRef] [PubMed]
- Ameye, L.; Young, M.F. Mice deficient in small leucine-rich proteoglycans: novel in vivo models for osteoporosis, osteoarthritis, Ehlers-Danlos syndrome, muscular dystrophy, and corneal diseases. Glycobiology 2002, 12, 107R–116R. [Google Scholar] [CrossRef] [PubMed]
- Corsi, A.; Xu, T.; Chen, X.D.; Boyde, A.; Liang, J.; Mankani, M.; Sommer, B.; Iozzo, R.V.; Eichstetter, I.; Robey, P.G.; et al. Phenotypic effects of biglycan deficiency are linked to collagen fibril abnormalities, are synergized by decorin deficiency, and mimic Ehlers-Danlos-like changes in bone and other connective tissues. J. Bone Miner. Res. 2002, 17, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.R.; Bristow, J. The Ehlers-Danlos syndrome: On beyond collagens. J. Clin. Investig. 2001, 107, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Kosho, T. Discovery and delineation of dermatan 4-O-sulfotransferase-1 (D4ST1)-deficient Ehlers-Danlos syndrome. In Current Genetics in Dermatology; Oiso, N., Kawada, A., Eds.; InTech: Rijeka, Croatia, 2013; pp. 73–86. [Google Scholar]
- Kosho, T. CHST14/D4ST1 deficiency: new form of Ehlers-Danlos syndrome. Pediatr. Int. 2016, 58, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Götting, C.; Kuhn, J.; Zahn, R.; Brinkmann, T.; Kleesiek, K. Molecular cloning and expression of Human UDP-d-xylose: Proteoglycan core protein β-d-xylosyltransferase and its first isoform XT-II. J. Mol. Biol. 2000, 304, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Pönighaus, C.; Ambrosius, M.; Casanova, J.C.; Prante, C.; Kuhn, J.; Esko, J.D.; Kleesiek, K.; Götting, C. Human xylosyltransferase II is involved in the biosynthesis of the uniform tetrasaccharide linkage region in chondroitin sulfate and heparan sulfate proteoglycans. J. Biol. Chem. 2007, 282, 5201–5206. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.; Levery, S.B.; Mandel, U.; Kresse, H.; Schwientek, T.; Bennett, E.P.; Clausen, H. Cloning and expression of a proteoglycan UDP-galactose: β-xylose β1,4-galactosyltransferase I: A seventh member of the human β4-galactosyltransferase gene family. J. Biol. Chem. 1999, 274, 26165–26171. [Google Scholar] [CrossRef] [PubMed]
- Okajima, T.; Yoshida, K.; Kondo, T.; Furukawa, K. Human homolog of Caenorhabditis elegans sqv-3 gene is galactosyltransferase I involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 1999, 274, 22915–22918. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Zhou, D.; Brown, J.R.; Crawford, B.E.; Hennet, T.; Esko, J.D. Biosynthesis of the linkage region of glycosaminoglycans: Cloning and activity of galactosyltransferase II, the sixth member of the β1,3-galactosyltransferase family (β3GalT6). J. Biol. Chem. 2001, 276, 48189–48195. [Google Scholar] [PubMed]
- Kitagawa, H.; Tone, Y.; Tamura, J.; Neumann, K.W.; Ogawa, T.; Oka, S.; Kawasaki, T.; Sugahara, K. Molecular cloning and expression of glucuronyltransferase I involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 1998, 273, 6615–6618. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, H.; Uyama, T.; Sugahara, K. Molecular cloning and expression of a human chondroitin synthase. J. Biol. Chem. 2001, 276, 38721–38726. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, H.; Izumikawa, T.; Uyama, T.; Sugahara, K. Molecular cloning of a chondroitin polymerizing factor that cooperates with chondroitin synthase for chondroitin polymerization. J. Biol. Chem. 2003, 278, 23666–23671. [Google Scholar] [CrossRef] [PubMed]
- Izumikawa, T.; Uyama, T.; Okuura, Y.; Sugahara, K.; Kitagawa, H. Involvement of chondroitin sulfate synthase-3 (chondroitin synthase-2) in chondroitin polymerization through its interaction with chondroitin synthase-1 or chondroitin polymerizing factor. Biochem. J. 2007, 403, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Izumikawa, T.; Koike, T.; Shiozawa, S.; Sugahara, K.; Tamura, J.; Kitagawa, H. Identification of chondroitin sulfate glucuronyltransferase as chondroitin synthase-3 involved in chondroitin polymerization: Chondroitin polymerization is achieved by multiple enzyme complexes consisting of chondroitin synthase family members. J. Biol. Chem. 2008, 283, 11396–11406. [Google Scholar] [CrossRef] [PubMed]
- Uyama, T.; Kitagawa, H.; Tamura, J.; Sugahara, K. Molecular cloning and expression of human chondroitin N-acetylgalactosaminyltransferase: The key enzyme for chain initiation and elongation of chondroitin/dermatan sulfate on the protein linkage region tetrasaccharide shared by heparin/heparan sulfate. J. Biol. Chem. 2002, 277, 8841–8846. [Google Scholar] [CrossRef] [PubMed]
- Uyama, T.; Kitagawa, H.; Tanaka, J.; Tamura, J.; Ogawa, T.; Sugahara, K. Molecular cloning and expression of a second chondroitin N-acetylgalactosaminyltransferase involved in the initiation and elongation of chondroitin/dermatan sulfate. J. Biol. Chem. 2003, 278, 3072–3078. [Google Scholar] [CrossRef] [PubMed]
- Maccarana, M.; Olander, B.; Malmström, J.; Tiedemann, K.; Aebersold, R.; Lindahl, U.; Li, J.P.; Malmström, A. Biosynthesis of dermatan sulfate: Chondroitin-glucuronate C5-epimerase is identical to SART2. J. Biol. Chem. 2006, 281, 11560–11568. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, B.; Malmström, A.; Maccarana, M. Two dermatan sulfate epimerases form iduronic acid domains in dermatan sulfate. J. Biol. Chem. 2009, 284, 9788–9795. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.R.; Xia, G.; Kang, H.G.; Schachner, M.; Baenziger, J.U. Molecular cloning and characterization of a dermatan-specific N-acetylgalactosamine 4-O-sulfotransferase. J. Biol. Chem. 2001, 276, 36344–36353. [Google Scholar] [CrossRef] [PubMed]
- Mikami, T.; Mizumoto, S.; Kago, N.; Kitagawa, H.; Sugahara, K. Specificities of three distinct human chondroitin/dermatan N-acetylgalactosamine 4-O-sulfotransferases demonstrated using partially desulfated dermatan sulfate as an acceptor: Implication of differential roles in dermatan sulfate biosynthesis. J. Biol. Chem. 2003, 278, 36115–36127. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Sugumaran, G.; Liu, J.; Shworak, N.W.; Silbert, J.E.; Rosenberg, R.D. Molecular cloning and characterization of a human uronyl 2-sulfotransferase that sulfates iduronyl and glucuronyl residues in dermatan/chondroitin sulfate. J. Biol. Chem. 1999, 274, 10474–10480. [Google Scholar] [CrossRef] [PubMed]
- Quentin, E.; Gladen, A.; Rodén, L.; Kresse, H. A genetic defect in the biosynthesis of dermatan sulfate proteoglycan: Galactosyltransferase I deficiency in fibroblasts from a patient with a progeroid syndrome. Proc. Natl. Acad. Sci. USA 1990, 87, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Okajima, T.; Fukumoto, S.; Furukawa, K.; Urano, T.; Furukawa, K. Molecular basis for the progeroid variant of Ehlers-Danlos syndrome: Identification and characterization of two mutations in galactosyltransferase I gene. J. Biol. Chem. 1999, 274, 28841–28844. [Google Scholar] [CrossRef] [PubMed]
- Faiyaz-Ul-Haque, M.; Zaidi, S.H.E.; Al-Ali, M.; Al-Mureikhi, M.S.; Kennedy, S.; Al-Thani, G.; Tsui, L.C.; Teebi, A.S. A novel missense mutation in the galactosyltransferase-I (B4GALT7) gene in a family exhibiting facioskeletal anomalies and Ehlers-Danlos syndrome resembling the progeroid type. Am. J. Med. Genet. Part A 2004, 128, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Seidler, D.G.; Faiyaz-Ul-Haque, M.; Hansen, U.; Yip, G.W.; Zaidi, S.H.; Teebi, A.S.; Kiesel, L.; Götte, M. Defective glycosylation of decorin and biglycan, altered collagen structure, and abnormal phenotype of the skin fibroblasts of an Ehlers-Danlos syndrome patient carrying the novel Arg270Cys substitution in galactosyltransferase I (β4GalT-7). J. Mol. Med. (Berl.) 2006, 84, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Götte, M.; Kresse, H. Defective glycosaminoglycan substitution of decorin in a patient with progeroid syndrome is a direct consequence of two point mutations in the galactosyltransferase I (β4GalT-7) gene. Biochem. Genet. 2005, 43, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Götte, M.; Spillmann, D.; Yip, G.W.; Versteeg, E.; Echtermeyer, F.G.; van Kuppevelt, T.H.; Kiesel, L. Changes in heparan sulfate are associated with delayed wound repair, altered cell migration, adhesion and contractility in the galactosyltransferase I (β4GalT-7) deficient form of Ehlers-Danlos syndrome. Hum. Mol. Genet. 2008, 17, 996–1009. [Google Scholar] [CrossRef] [PubMed]
- Cartault, F.; Munier, P.; Jacquemont, M.L.; Vellayoudom, J.; Doray, B.; Payet, C.; Randrianaivo, H.; Laville, J.M.; Munnich, A.; Cormier-Daire, V. Expanding the clinical spectrum of B4GALT7 deficiency: Homozygous p.R270C mutation with founder effect causes Larsen of Reunion Island syndrome. Eur. J. Hum. Genet. 2015, 23, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Larsen, L.J.; Schottstaedt, E.R.; Bost, F.C. Multiple congenital dislocations associated with characteristic facial abnormality. J. Pediatr. 1950, 37, 574–581. [Google Scholar] [CrossRef]
- Nakajima, M.; Mizumoto, S.; Miyake, N.; Kogawa, R.; Iida, A.; Ito, H.; Kitoh, H.; Hirayama, A.; Mitsubuchi, H.; Miyazaki, O.; et al. Mutations in B3GALT6, which encodes a glycosaminoglycan linker region enzyme, cause a spectrum of skeletal and connective tissue disorders. Am. J. Hum. Genet. 2013, 92, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Kariminejad, A.; Van Damme, T.; Gauche, C.; Syx, D.; Merhi-Soussi, F.; Gulberti, S.; Symoens, S.; Vanhauwaert, S.; Willaert, A.; et al. A. Defective initiation of glycosaminoglycan synthesis due to B3GALT6 mutations causes a pleiotropic Ehlers-Danlos-syndrome-like connective tissue disorder. Am. J. Hum. Genet. 2013, 92, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Vorster, A.A.; Beighton, P.; Ramesar, R.S. Spondyloepimetaphyseal dysplasia with joint laxity (Beighton type): Mutation analysis in 8 affected South African families. Clin. Genet. 2015, 87, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Alazami, A.M.; Al-Qattan, S.M.; Faqeih, E.; Alhashem, A.; Alshammari, M.; Alzahrani, F.; Al-Dosari, M.S.; Patel, N.; Alsagheir, A.; Binabbas, B.; et al. Expanding the clinical and genetic heterogeneity of hereditary disorders of connective tissue. Hum. Genet. 2016, 135, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Mizumoto, S.; Suresh, I.; Komatsu, Y.; Vodopiutz, J.; Dundar, M.; Straub, V.; Lingenhel, A.; Melmer, A.; Lechner, S.; et al. Loss of dermatan sulfate epimerase (DSE) function results in musculocontractural Ehlers-Danlos syndrome. Hum. Mol. Genet. 2013, 22, 3761–3772. [Google Scholar] [CrossRef] [PubMed]
- Syx, D.; Van Damme, T.; Symoens, S.; Maiburg, M.C.; van de Laar, I.; Morton, J.; Suri, M.; Del Campo, M.; Hausser, I.; Hermanns-Lê, T.; et al. Genetic heterogeneity and clinical variability in musculocontractural Ehlers-Danlos syndrome caused by impaired dermatan sulfate biosynthesis. Hum. Mutat. 2015, 36, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Kosho, T.; Takahashi, J.; Ohashi, H.; Nishimura, G.; Kato, H.; Fukushima, Y. Ehlers-Danlos syndrome type VIB with characteristic facies, decreased curvatures of the spinal column, and joint contractures in two unrelated girls. Am. J. Med. Genet. Part A 2005, 138, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Kosho, T.; Miyake, N.; Hatamochi, A.; Takahashi, J.; Kato, H.; Miyahara, T.; Igawa, Y.; Yasui, H.; Ishida, T.; Ono, K.; et al. A new Ehlers-Danlos syndrome with craniofacial characteristics, multiple congenital contractures, progressive joint and skin laxity, and multisystem fragility-related manifestations. Am. J. Med. Genet. Part A 2010, 152, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Miyake, N.; Kosho, T.; Mizumoto, S.; Furuichi, T.; Hatamochi, A.; Nagashima, Y.; Arai, E.; Takahashi, K.; Kawamura, R.; Wakui, K.; et al. Loss-of-function mutations of CHST14 in a new type of Ehlers-Danlos syndrome. Hum. Mutat. 2010, 31, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Dündar, M.; Müller, T.; Zhang, Q.; Pan, J.; Steinmann, B.; Vodopiutz, J.; Gruber, R.; Sonoda, T.; Krabichler, B.; Utermann, G.; et al. Loss of dermatan-4-sulfotransferase 1 function results in adducted thumb-clubfoot syndrome. Am. J. Hum. Genet. 2009, 85, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Dundar, M.; Demiryilmaz, F.; Demiryilmaz, I.; Kumandas, S.; Erkilic, K.; Kendirci, M.; Tuncel, M.; Ozyazgan, I.; Tolmie, J.L. An autosomal recessive adducted thumb-club foot syndrome observed in Turkish cousins. Clin. Genet. 1997, 51, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, T.; Kouno, K. Two brothers with distal arthrogryposis, peculiar facial appearance, cleft palate, short stature, hydronephrosis, retentio testis, and normal intelligence: A new type of distal arthrogryposis? Am. J. Med. Genet. 2000, 91, 280–285. [Google Scholar] [CrossRef]
- Malfait, F.; Syx, D.; Vlummens, P.; Symoens, S.; Nampoothiri, S.; Hermanns-Lê, T.; Van Laer, L.; De Paepe, A. Musculocontractural Ehlers-Danlos Syndrome (former EDS type VIB) and adducted thumb clubfoot syndrome (ATCS) represent a single clinical entity caused by mutations in the dermatan-4-sulfotransferase 1 encoding CHST14 gene. Hum. Mutat. 2010, 31, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Kosho, T.; Miyake, N.; Mizumoto, S.; Hatamochi, A.; Fukushima, Y.; Yamada, S.; Sugahara, K.; Matsumoto, N. A response to: loss of dermatan-4-sulfotransferase 1 (D4ST1/CHST14) function represents the first dermatan sulfate biosynthesis defect, “dermatan sulfate-deficient Adducted Thumb-Clubfoot Syndrome”. Which name is appropriate, “Adducted Thumb-Clubfoot Syndrome” or “Ehlers-Danlos syndrome?”. Hum. Mutat. 2011, 32, 1507–1509. [Google Scholar] [PubMed]
- Shimizu, K.; Okamoto, N.; Miyake, N.; Taira, K.; Sato, Y.; Matsuda, K.; Akimaru, N.; Ohashi, H.; Wakui, K.; Fukushima, Y.; et al. Delineation of dermatan 4-O-sulfotransferase 1 deficient Ehlers-Danlos syndrome: observation of two additional patients and comprehensive review of 20 reported patients. Am. J. Med. Genet. Part A 2011, 155, 1949–1958. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Londono, R.; Chitayat, D.; Kahr, W.H.; Hinek, A.; Blaser, S.; Dupuis, L.; Goh, E.; Badilla-Porras, R.; Howard, A.; Mittaz, L.; et al. Extracellular matrix and platelet function in patients with musculocontractural Ehlers-Danlos syndrome caused by mutations in the CHST14 gene. Am. J. Med. Genet. Part A 2012, 158, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Voermans, N.C.; Kempers, M.; Lammens, M.; van Alfen, N.; Janssen, M.C.; Bönnemann, C.; van Engelen, B.G.; Hamel, B.C. Myopathy in a 20-year-old female patient with D4ST-1 deficient Ehlers-Danlos syndrome due to a homozygous CHST14 mutation. Am. J. Med. Genet. Part A 2012, 158, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Winters, K.A.; Jiang, Z.; Xu, W.; Li, S.; Ammous, Z.; Jayakar, P.; Wierenga, K.J. Re-assigned diagnosis of D4ST1-deficient Ehlers-Danlos syndrome (adducted thumb-clubfoot syndrome) after initial diagnosis of Marden-Walker syndrome. Am. J. Med. Genet. Part A 2012, 158, 2935–2940. [Google Scholar] [CrossRef] [PubMed]
- Janecke, A.R.; Li, B.; Boehm, M.; Krabichler, B.; Rohrbach, M.; Müller, T.; Fuchs, I.; Golas, G.; Katagiri, Y.; Ziegler, S.G.; et al. The phenotype of the musculocontractural type of Ehlers-Danlos syndrome due to CHST14 mutations. Am. J. Med. Genet. Part A 2016, 170, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Hasegawa-Murakami, Y.; Sugiura, K.; Ono, M.; Toriyama, K.; Miyake, N.; Hatamochi, A.; Kamei, Y.; Kosho, T.; Akiyama, M. A 45-year-old woman with Ehlers-Danlos syndrome caused by dermatan 4-O-sulfotransferase-1 deficiency: Implications for early ageing. Acta Derm. Venereol. 2016, 96, 830–831. [Google Scholar] [CrossRef] [PubMed]
- Mochida, K.; Amano, M.; Miyake, N.; Matsumoto, N.; Hatamochi, A.; Kosho, T. Dermatan 4-O-sulfotransferase 1-deficient Ehlers-Danlos syndrome complicated by a large subcutaneous hematoma on the back. J. Dermatol. 2016, 43, 832–833. [Google Scholar] [CrossRef] [PubMed]
- Salpietro, V.; Ruggieri, M.; Mankad, K.; Di Rosa, G.; Granata, F.; Loddo, I.; Moschella, E.; Calabro, M.P.; Capalbo, A.; Bernardini, L.; et al. A de novo 0.63 Mb 6q25.1 deletion associated with growth failure, congenital heart defect, underdeveloped cerebellar vermis, abnormal cutaneous elasticity and joint laxity. Am. J. Med. Genet. Part A 2015, 167, 2042–2051. [Google Scholar] [CrossRef] [PubMed]
- Bredrup, C.; Knappskog, P.M.; Majewski, J.; Rodahl, E.; Boman, H. Congenital stromal dystrophy of the cornea caused by a mutation in the decorin gene. Investig. Ophthalmol. Vis. Sci. 2005, 46, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Rodahl, E.; Van Ginderdeuren, R.; Knappskog, P.M.; Bredrup, C.; Boman, H. A second decorin frame shift mutation in a family with congenital stromal corneal dystrophy. Am. J. Ophthalmol. 2006, 142, 520–521. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Ko, J.M.; Lee, I.; Kim, J.Y.; Kim, M.J.; Tchah, H. A novel mutation of the decorin gene identified in a Korean family with congenital hereditary stromal dystrophy. Cornea 2011, 30, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Kumar, P.R.; Zhu, L.; Edward, D.P.; Tao, S.; Wang, L.; Chuck, R.; Zhang, C. Novel decorin mutation in a Chinese family with congenital stromal corneal dystrophy. Cornea 2014, 33, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Van Ginderdeuren, R.; De Vos, R.; Casteels, I.; Foets, B. Report of a new family with dominant congenital heredity stromal dystrophy of the cornea. Cornea 2002, 21, 118–120. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Bae, J.S.; Kim, N.K.; Forzano, F.; Girisha, K.M.; Baldo, C.; Faravelli, F.; Cho, T.J.; Kim, D.; Lee, K.Y.; et al. BGN mutations in X-linked spondyloepimetaphyseal dysplasia. Am. J. Hum. Genet. 2016, 98, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Meester, J.A.; Vandeweyer, G.; Pintelon, I.; Lammens, M.; Van Hoorick, L.; De Belder, S.; Waitzman, K.; Young, L.; Markham, L.W.; Vogt, J.; et al. Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Maccarana, M.; Kalamajski, S.; Kongsgaard, M.; Magnusson, S.P.; Oldberg, A.; Malmström, A. Dermatan sulfate epimerase 1-deficient mice have reduced content and changed distribution of iduronic acids in dermatan sulfate and an altered collagen structure in skin. Mol. Cell. Biol. 2009, 29, 5517–5528. [Google Scholar] [CrossRef] [PubMed]
- Malmström, A. Biosynthesis of dermatan sulfate. II. Substrate specificity of the C-5 uronosyl epimerase. J. Biol. Chem. 1984, 259, 161–165. [Google Scholar] [PubMed]
- Akyüz, N.; Rost, S.; Mehanna, A.; Bian, S.; Loers, G.; Oezen, I.; Mishra, B.; Hoffmann, K.; Guseva, D.; Laczynska, E.; et al. Dermatan 4-O-sulfotransferase1 ablation accelerates peripheral nerve regeneration. Exp. Neurol. 2013, 247, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Bian, S.; Akyüz, N.; Bernreuther, C.; Loers, G.; Laczynska, E.; Jakovcevski, I.; Schachner, M. Dermatan sulfotransferase Chst14/d4st1, but not chondroitin sulfotransferase Chst11/C4st1, regulates proliferation and neurogenesis of neural progenitor cells. J. Cell Sci. 2011, 124, 4051–4063. [Google Scholar] [CrossRef] [PubMed]
- Thienpont, B.; Zhang, L.; Postma, A.V.; Breckpot, J.; Tranchevent, L.C.; Van Loo, P.; Møllgård, K.; Tommerup, N.; Bache, I.; Tümer, Z.; et al. Haploinsufficiency of TAB2 causes congenital heart defects in humans. Am. J. Hum. Genet. 2010, 86, 839–849. [Google Scholar] [CrossRef] [PubMed]
- St John, M.A.; Tao, W.; Fei, X.; Fukumoto, R.; Carcangiu, M.L.; Brownstein, D.G.; Parlow, A.F.; McGrath, J.; Xu, T. Mice deficient of Lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat. Genet. 1999, 21, 182–186. [Google Scholar] [PubMed]
- Gubbiotti, M.A.; Vallet, S.D.; Ricard-Blum, S.; Iozzo, R.V. Decorin interacting network: A comprehensive analysis of decorin-binding partners and their versatile functions. Matrix Biol. 2016, 55, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sun, M.; Meng, X.; Iozzo, R.V.; Kao, W.W.; Birk, D.E. Pathophysiological mechanisms of autosomal dominant congenital stromal corneal dystrophy: C-terminal-truncated decorin results in abnormal matrix assembly and altered expression of small leucine-rich proteoglycans. Am. J. Pathol. 2011, 179, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Mellgren, A.E.; Bruland, O.; Vedeler, A.; Saraste, J.; Schönheit, J.; Bredrup, C.; Knappskog, P.M.; Rødahl, E. Development of congenital stromal corneal dystrophy is dependent on export and extracellular deposition of truncated decorin. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2909–2915. [Google Scholar] [CrossRef] [PubMed]
- Massoudi, D.; Malecaze, F.; Galiacy, S.D. Collagens and proteoglycans of the cornea: importance in transparency and visual disorders. Cell Tissue Res. 2016, 363, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Häkkinen, L.; Strassburger, S.; Kähäri, V.M.; Scott, P.G.; Eichstetter, I.; Lozzo, R.V.; Larjava, H. A role for decorin in the structural organization of periodontal ligament. Lab. Investig. 2000, 80, 1869–1880. [Google Scholar] [CrossRef] [PubMed]
- Schönherr, E.; Witsch-Prehm, P.; Harrach, B.; Robenek, H.; Rauterberg, J.; Kresse, H. Interaction of biglycan with type I collagen. J. Biol. Chem. 1995, 270, 2776–2783. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, A.; Romarís, M.; Rasmussen, L.M.; Heinegård, D.; Twardzik, D.R.; Border, W.A.; Ruoslahti, E. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem. J. 1994, 302, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.D.; Fisher, L.W.; Robey, P.G.; Young, M.F. The small leucine-rich proteoglycan biglycan modulates BMP-4-induced osteoblast differentiation. FASEB J. 2004, 18, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, A.D.; Fisher, L.W.; Kilts, T.M.; Owens, R.T.; Robey, P.G.; Gutkind, J.S.; Young, M.F. Modulation of canonical Wnt signaling by the extracellular matrix component biglycan. Proc. Natl. Acad. Sci. USA 2011, 108, 17022–17027. [Google Scholar] [CrossRef] [PubMed]
- Van Laer, L.; Dietz, H.; Loeys, B. Loeys-Dietz syndrome. Adv. Exp. Med. Biol. 2014, 802, 95–105. [Google Scholar] [PubMed]
- Melchior-Becker, A.; Dai, G.; Ding, Z.; Schäfer, L.; Schrader, J.; Young, M.F.; Fischer, J.W. Deficiency of biglycan causes cardiac fibroblasts to differentiate into a myofibroblast phenotype. J. Biol. Chem. 2011, 286, 17365–17375. [Google Scholar] [CrossRef] [PubMed]
- Heegaard, A.M.; Corsi, A.; Danielsen, C.C.; Nielsen, K.L.; Jorgensen, H.L.; Riminucci, M.; Young, M.F.; Bianco, P. Biglycan deficiency causes spontaneous aortic dissection and rupture in mice. Circulation 2007, 115, 2731–2738. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Enzymes and DS-PG Core Proteins | Coding Genes | MIM Number | Human Genetic Disorders | Clinical Features | Refs. |
|---|---|---|---|---|---|
| β4Galactosyltransferase-I (GalT-I) | B4GALT7 | 130070 604327 | Ehlers-Danlos syndrome progeroid type 1 | Developmental delays, aged appearance, a short stature, craniofacial dysmorphism, and generalized osteopenia. | [30,31,32,33,34,35,36] |
| Larsen of Reunion Island syndrome | Multiple dislocations, hyperlaxity, dwarfism, and distinctive facial features. | ||||
| β3Galactosyltransferase-II (GalT-II) | B3GALT6 | 615349 615291 | Ehlers-Danlos syndrome progeroid type 2 | Sparse hair, wrinkled skin, defective wound healing with atrophic scars, osteopenia, and radial head dislocation. | [38,39,40,41] |
| 271640 | Spondyloepimetaphyseal dysplasia with joint laxity type 1 | Spatulate fingers with short nails, hip dislocation, elbow contracture, clubfeet, and mild craniofacial dysmorphism including prominent eyes, blue sclera, a long upper lip, and small mandible with a cleft palate. | |||
| Dermatan sulfate epimerase | DSE | 615539 605942 | Ehlers-Danlos syndrome musculocontractural type 2 | Characteristic facial features, congenital contracture of the thumbs and feet, hypermobility of the finger, elbow, and knee joints, atrophic scarring of the skin, and myopathy. | [42,43] |
| Dermatan 4-O-sulfotransferase | CHST14 | 601776 608429 | Ehlers-Danlos syndrome musculocontractural type 1; EDS Kosho type | Craniofacial dysmorphism, multiple congenital contractures including adduction-flexion contracture of the thumbs and clubfeet, malformations of the heart, kidney, intestine, and eye; skin hyperextensibility, bruisability, and fragility with atrophic scars; recurrent joint dislocations, progressive foot or spinal deformities, pneumothorax, large subcutaneous hematomas, and diverticular perforation. | [41,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58] |
| Adducted thumb-clubfoot syndrome | |||||
| Uronosyl 2-O-sulfotransferase | UST | 610752 | Multiple congenital anomalies of the heart and central nervous system | Growth failure, congenital heart defect, underdeveloped cerebellar vermis, abnormal cutaneous elasticity, and joint laxity. | [59] |
| Decorin | DCN | 610048 125255 | Congenital stromal corneal dystrophy | Diffuse bilateral corneal clouding, corneal opacities, strabismus, nystagmus, photophobia, and esotropia. | [60,61,62,63,64] |
| Biglycan | BGN | 300106 301870 | Spondyloepimetaphyseal dysplasia, X-linked | A short stature and osteoarthritic changes in joints; anomalies of the spine, and epiphyses and metaphyses of the long bones. | [65,66] |
| 300989 | Meester-Loeys syndrome | Aortic aneurysm and dissection, hypertelorism, proptosis, downslanting palpebral fissures, frontal bossing, malar hypoplasia, pectus deformities, joint hypermobility or contracture, skin striae, a bifid uvula, cervical spine instability, ventricular dilation, hip dislocation, platyspondyly, phalangeal dysplasia, and dysplastic epiphyses of the long bones. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizumoto, S.; Kosho, T.; Yamada, S.; Sugahara, K. Pathophysiological Significance of Dermatan Sulfate Proteoglycans Revealed by Human Genetic Disorders. Pharmaceuticals 2017, 10, 34. https://doi.org/10.3390/ph10020034
Mizumoto S, Kosho T, Yamada S, Sugahara K. Pathophysiological Significance of Dermatan Sulfate Proteoglycans Revealed by Human Genetic Disorders. Pharmaceuticals. 2017; 10(2):34. https://doi.org/10.3390/ph10020034
Chicago/Turabian StyleMizumoto, Shuji, Tomoki Kosho, Shuhei Yamada, and Kazuyuki Sugahara. 2017. "Pathophysiological Significance of Dermatan Sulfate Proteoglycans Revealed by Human Genetic Disorders" Pharmaceuticals 10, no. 2: 34. https://doi.org/10.3390/ph10020034
APA StyleMizumoto, S., Kosho, T., Yamada, S., & Sugahara, K. (2017). Pathophysiological Significance of Dermatan Sulfate Proteoglycans Revealed by Human Genetic Disorders. Pharmaceuticals, 10(2), 34. https://doi.org/10.3390/ph10020034