CDK11 Loss Induces Cell Cycle Dysfunction and Death of BRAF and NRAS Melanoma Cells

 , ,
, , 
Abstract
:1. Introduction
2. Results
2.1. CDK11A and CDK11B mRNA Expression in Non-Transformed Melanocytes and Melanoma Cell Lines
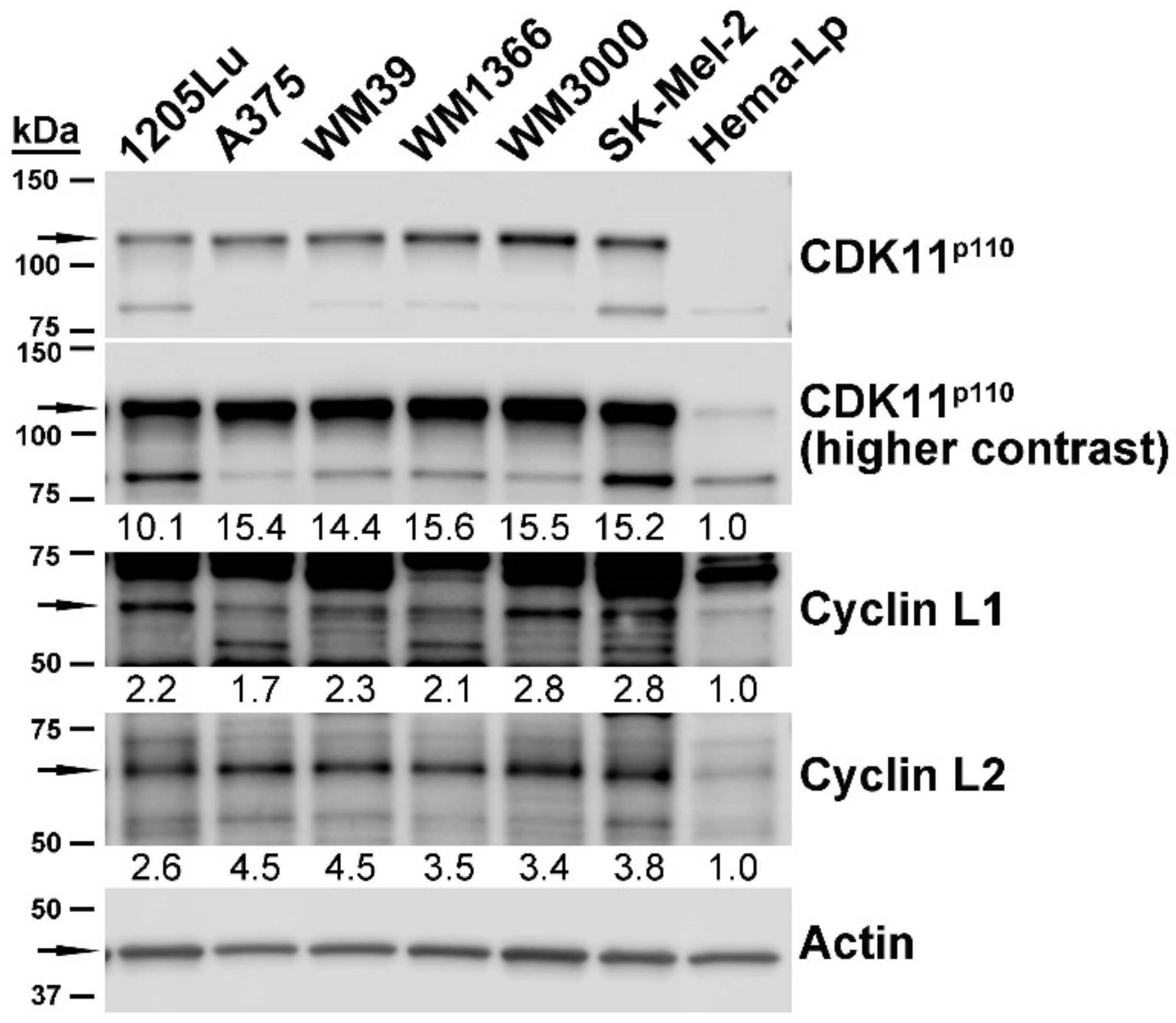
2.2. Expression of CDK11 Protein Complex Constituents in Primary Melanocytes and Melanoma Cell Lines
2.3. Down-Regulation of CDK11 Using siRNA Decreases BRAF- and NRAS-Mutant Melanoma Cell Viability
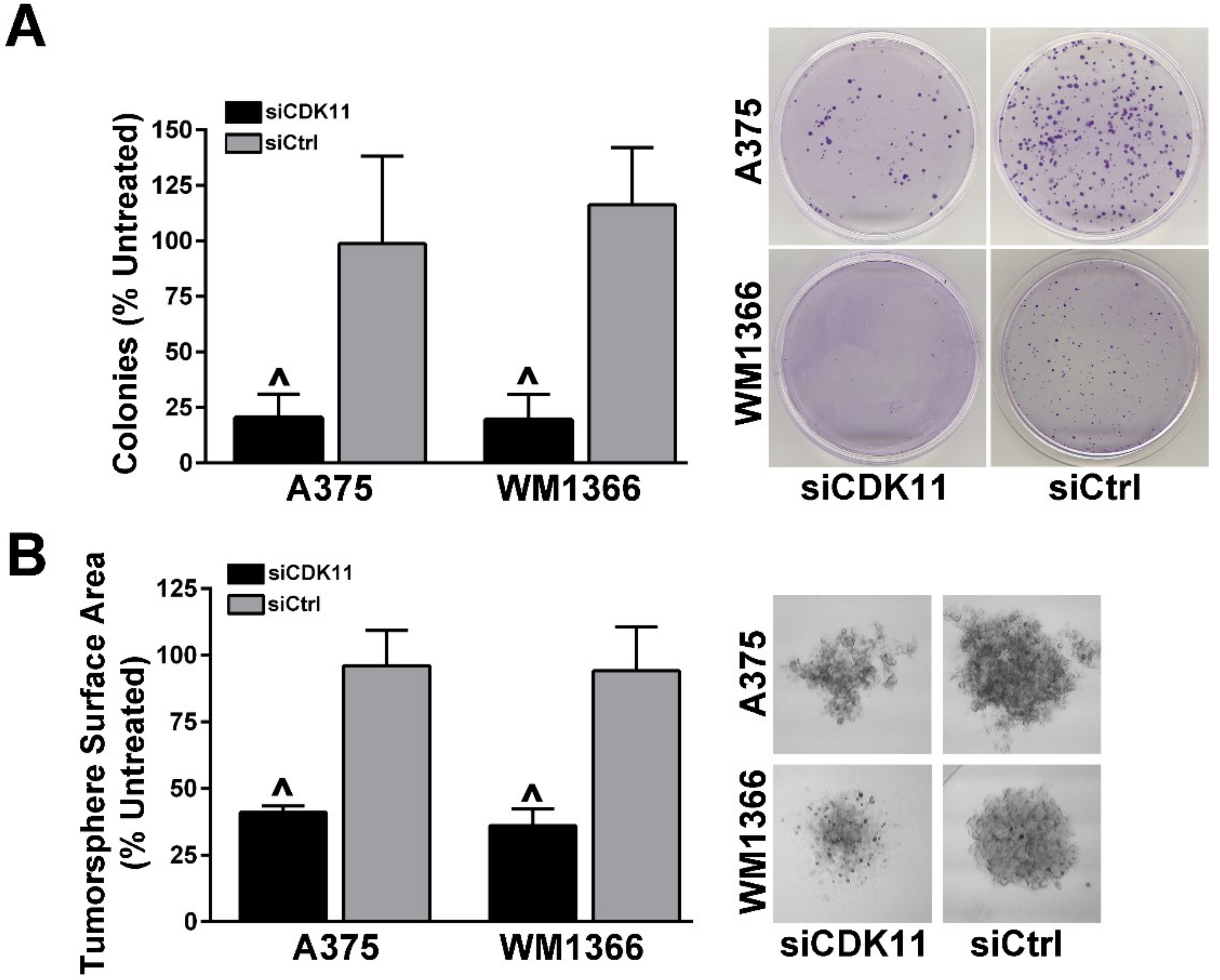
2.4. Loss of CDK11 Expression Has a Negative Impact on the Ability of Melanoma Cells to Form Colonies and Tumorspheres
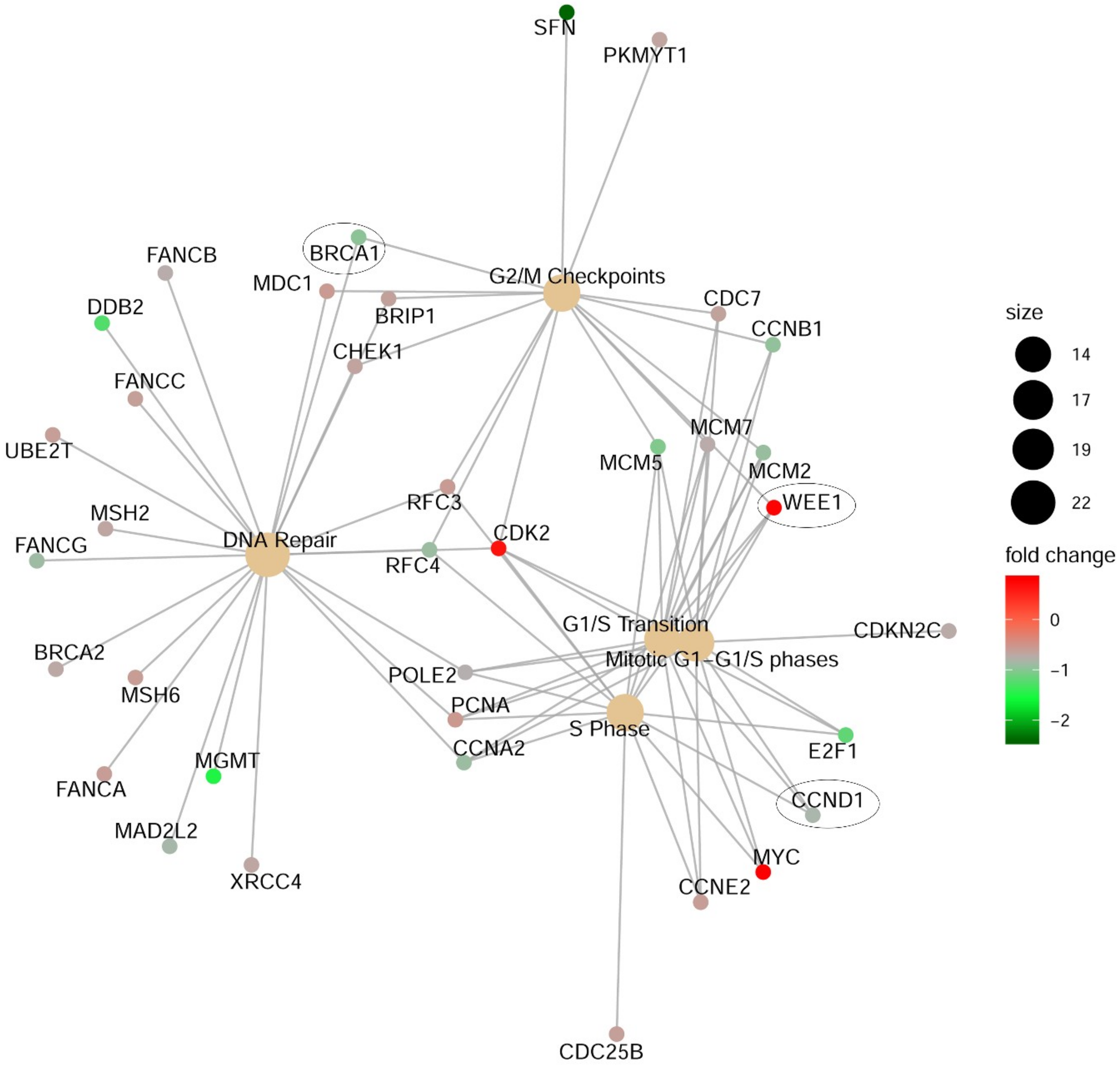
2.5. Effects of CDK11 Signaling Reduction on Melanoma Growth Pathways
2.6. CDK11 Impact on Pan-Cancer Gene Expression
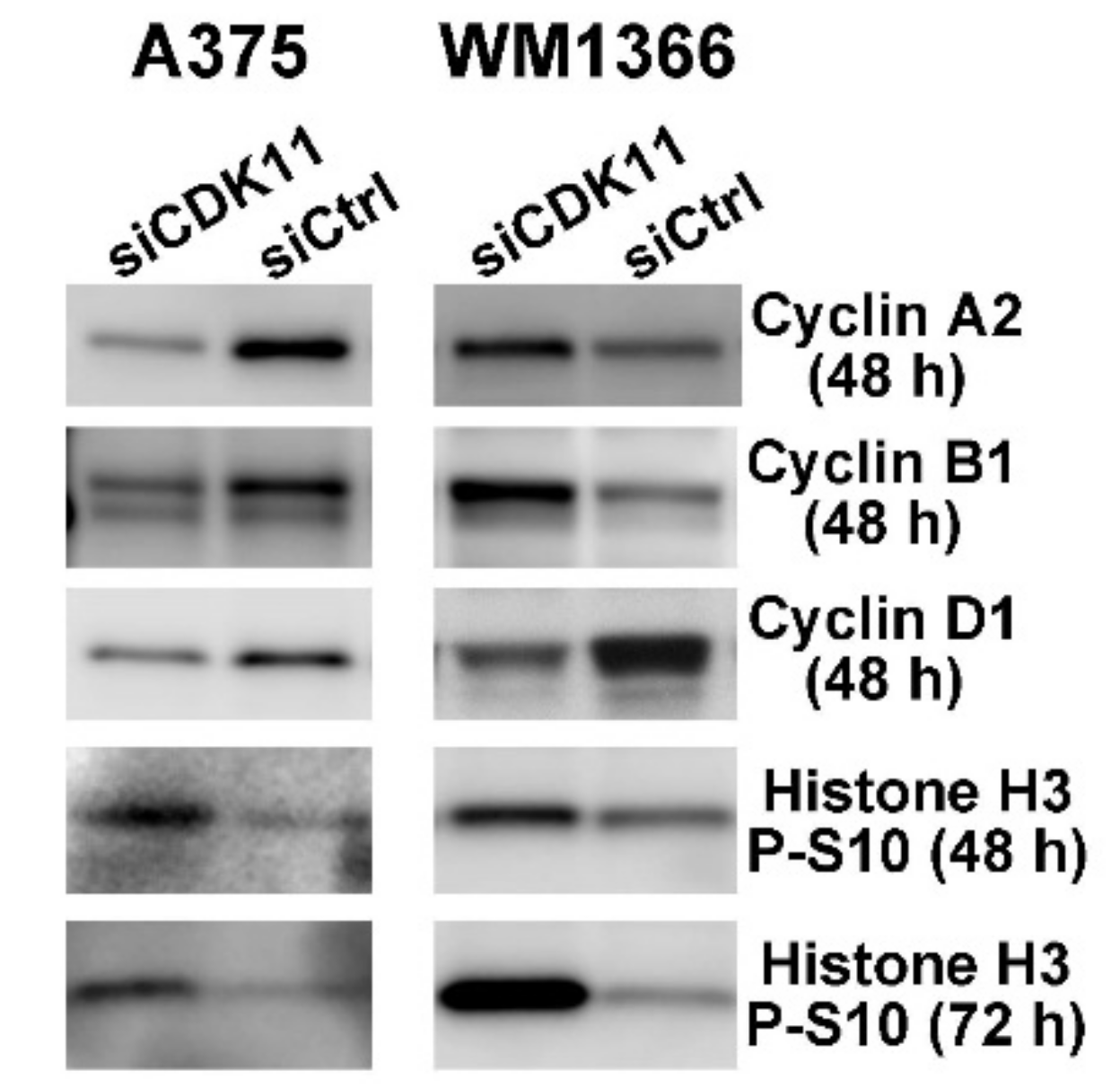
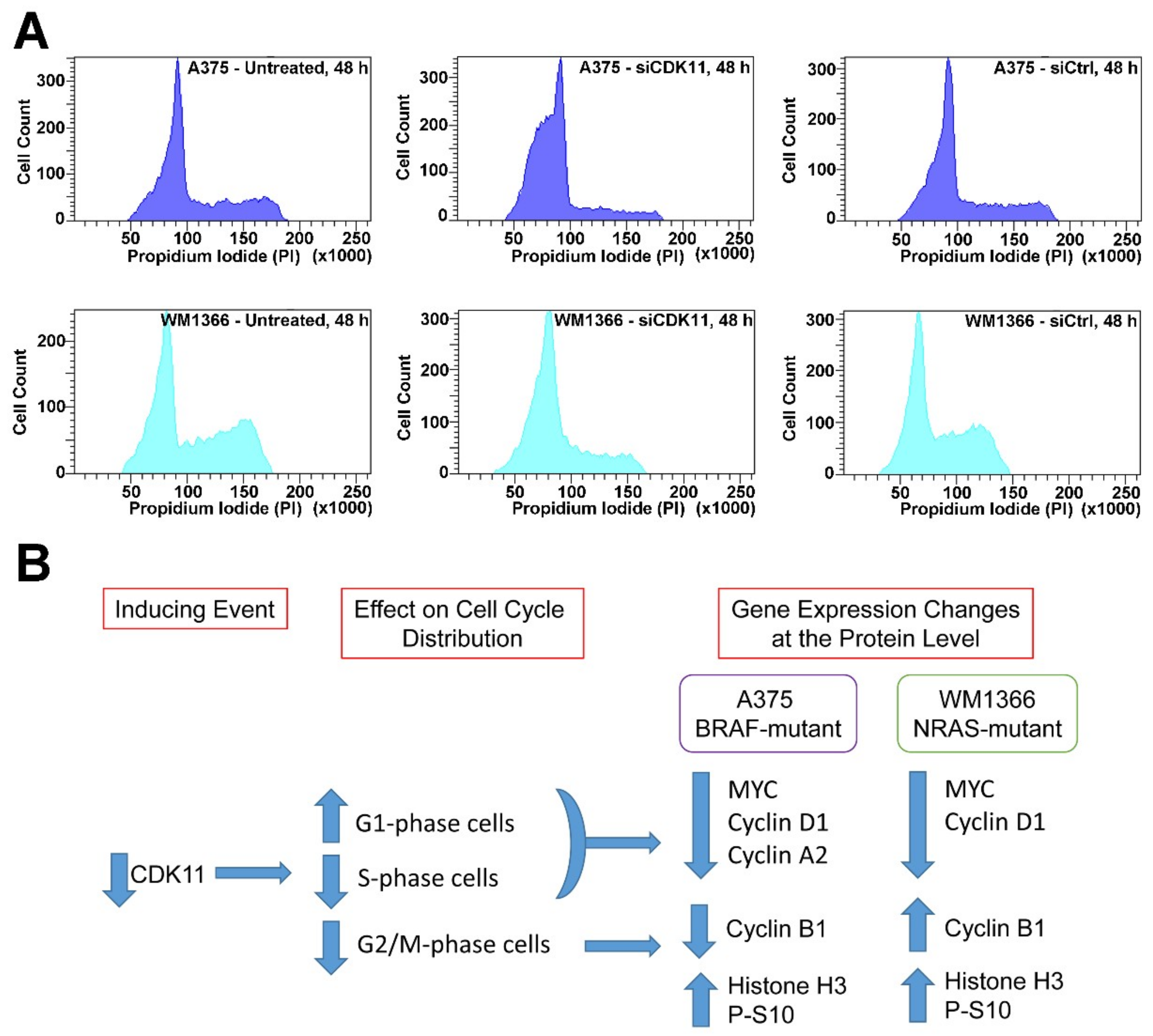
2.7. CDK11 Loss Rapidly Disrupts Cell Cycle Function in Melanoma
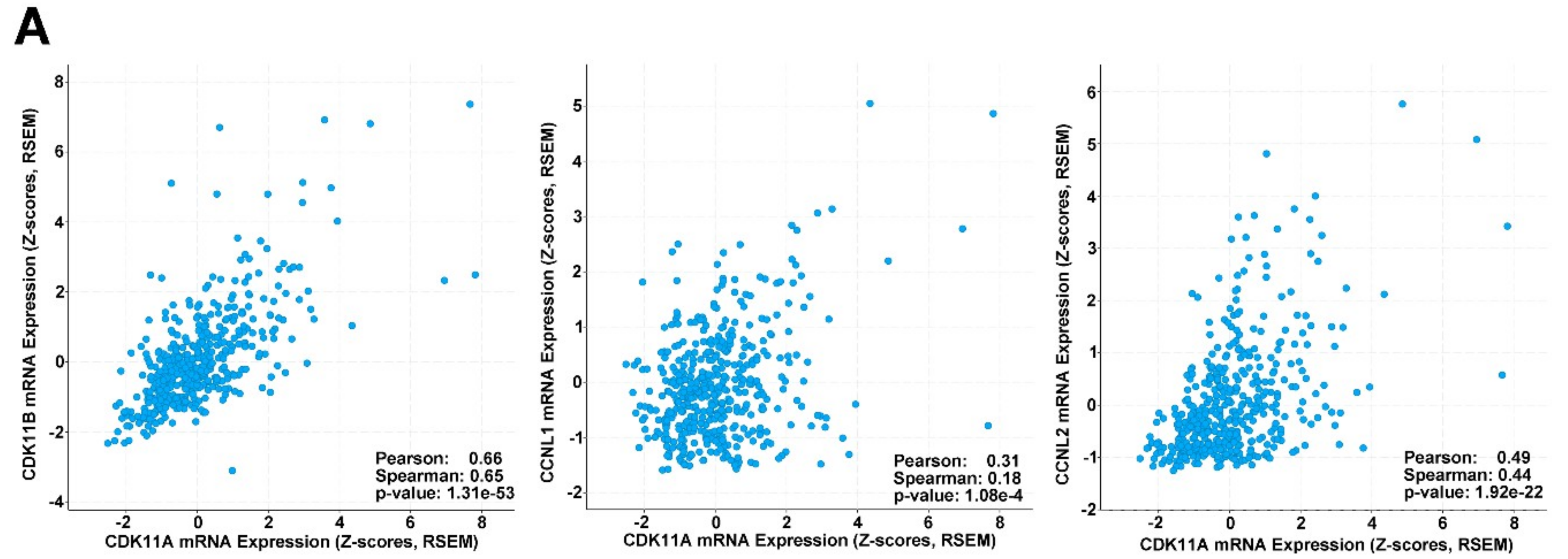
2.8. CDK11, CCNL1 and CCNL2 Are Co-Expressed in Human Melanoma Patient Samples
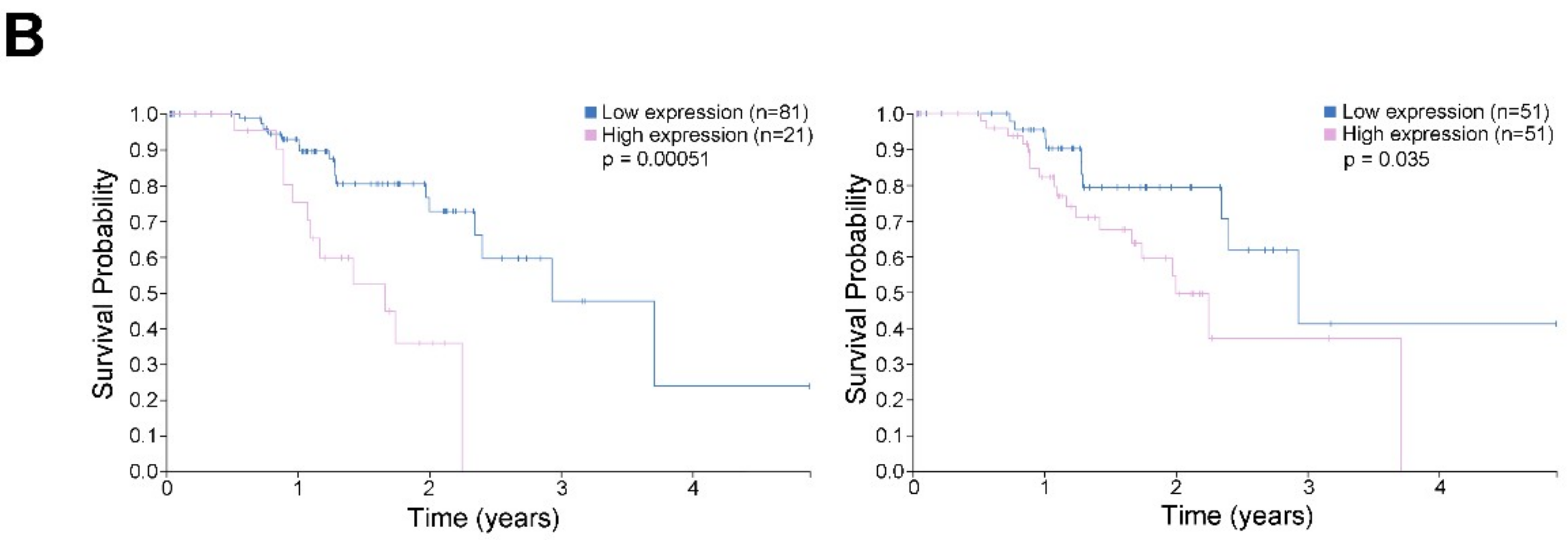
2.9. High CDK11A mRNA Expression is Associated with Decreased Patient Survival
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Oligonucleotides
4.3. Cell Viability Assays
4.4. Clonal Survival Assays
4.5. Tumorsphere Formation Assays
4.6. Immunoblot Analysis
4.7. Quantitative Real-Time RT-PCR Analysis
4.8. FACS Analysis of Cultured Cells
4.9. NanoString nCounter Expression and Bioinformatics Analysis
4.10. Human Data Survival Analysis
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Jemal, A.; Saraiya, M.; Patel, P.; Cherala, S.S.; Barnholtz-Sloan, J.; Kim, J.; Wiggins, C.L.; Wingo, P.A. Recent trends in cutaneous melanoma incidence and death rates in the United States, 1992–2006. J. Am. Acad. Dermatol. 2011, 65, S17.e1–S17.e11. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Eggermont, A.M.; Spatz, A.; Robert, C. Cutaneous melanoma. Lancet 2014, 383, 816–827. [Google Scholar] [CrossRef]
- Broussard, L.; Howland, A.; Ryu, S.; Song, K.; Norris, D.; Armstrong, C.A.; Song, P.I. Melanoma cell death mechanisms. Chonnam Med. J. 2018, 54, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Jenkins, R.W.; Sullivan, R.J. Mechanisms of resistance to immune checkpoint blockade. Am. J. Clin. Dermatol. 2019, 20, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Zhou, Y.; Shen, J.K.; Hornicek, F.J.; Kan, Q.; Duan, Z. The emerging roles and therapeutic potential of cyclin-dependent kinase 11 (cdk11) in human cancer. Oncotarget 2016, 7, 40846–40859. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Paparidis, N.F.; Canduri, F. The emerging picture of cdk11: Genetic, functional and medicinal aspects. Curr. Med. Chem. 2018, 25, 880–888. [Google Scholar] [CrossRef]
- Kren, B.T.; Unger, G.M.; Abedin, M.J.; Vogel, R.I.; Henzler, C.M.; Ahmed, K.; Trembley, J.H. Preclinical evaluation of cyclin dependent kinase 11 and casein kinase 2 survival kinases as RNA interference target for triple negative breast cancer therapy. Breast Cancer Res. 2015, 17, 19. [Google Scholar] [CrossRef]
- Duan, Z.; Zhang, J.; Choy, E.; Harmon, D.; Liu, X.; Nielsen, P.; Mankin, H.; Gray, N.S.; Hornicek, F.J. Systematic kinome shrna screening identifies cdk11 (pitslre) kinase expression is critical for osteosarcoma cell growth and proliferation. Clin. Cancer Res. 2012, 18, 4580–4588. [Google Scholar] [CrossRef]
- Jia, B.; Choy, E.; Cote, G.; Harmon, D.; Ye, S.; Kan, Q.; Mankin, H.; Hornicek, F.; Duan, Z. Cyclin-dependent kinase 11 (cdk11) is crucial in the growth of liposarcoma cells. Cancer Lett. 2014, 342, 104–112. [Google Scholar] [CrossRef]
- Zhou, Y.; Han, C.; Li, D.; Yu, Z.; Li, F.; Li, F.; An, Q.; Bai, H.; Zhang, X.; Duan, Z.; et al. Cyclin-dependent kinase 11(p110) (cdk11(p110)) is crucial for human breast cancer cell proliferation and growth. Sci. Rep. 2015, 5, 10433. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gao, Y.; Shen, J.; Yang, W.; Choy, E.; Mankin, H.; Hornicek, F.J.; Duan, Z. Cyclin-dependent kinase 11 (cdk11) is required for ovarian cancer cell growth in vitro and in vivo, and its inhibition causes apoptosis and sensitizes cells to paclitaxel. Mol. Cancer Ther. 2016, 15, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Inoue, A.; Lahti, J.M.; Kidd, V.J. Failure to proliferate and mitotic arrest of cdk11p110/p58-null mutant mice at the blastocyst stage of embryonic cell development. Mol. Cell. Biol. 2004, 24, 3188–3197. [Google Scholar] [CrossRef] [PubMed]
- MacKeigan, J.P.; Murphy, L.O.; Blenis, J. Sensitized rnai screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat. Cell Biol. 2005, 7, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Loyer, P.; Hu, D.; Li, T.; Grenet, J.; Lahti, J.M.; Kidd, V.J. Cyclin dependent kinase 11 in RNA transcription and splicing. In Progress in Nucleic acid Research and Molecular Biology; Moldave, K., Ed.; Elsevier Academic Press: San Diego, CA, USA, 2004; Volume 77, pp. 263–288. [Google Scholar]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Loyer, P.; Trembley, J.H.; Grenet, J.A.; Busson, A.; Corlu, A.; Zhao, W.; Kocak, M.; Kidd, V.J.; Lahti, J.M. Characterization of cyclin L1 and L2 interactions with cdk11 and splicing factors: Influence of cyclin l isoforms on splice site selection. J. Biol. Chem. 2008, 283, 7721–7732. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, L.A.; Edgar, A.J.; Ehley, J.; Gottesfeld, J.M. Cyclin L is an RS domain protein involved in pre-mrna splicing. J. Biol. Chem. 2002, 277, 25465–25473. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, K.; Hekerman, P.; Spelten, O.; Herrmann, A.; Packman, L.C.; Büssow, K.; Müller-Newen, G.; Becker, W. Characterization of cyclin L2, a novel cyclin with an arginine/serine-rich domain: Phosphorylation by dyrk1a and colocalization with splicing factors. J. Biol. Chem. 2004, 279, 4612–4624. [Google Scholar] [CrossRef] [PubMed]
- Berke, J.D.; Sgambato, V.; Zhu, P.-P.; Lavoie, B.; Vincent, M.; Krause, M.; Hyman, S.E. Dopamine and glutamate induce distinct striatal splice forms of ania-6, an RNA polymerase ii-associated cyclin. Neuron 2001, 32, 277–287. [Google Scholar] [CrossRef]
- Yang, L.; Li, N.; Wang, C.; Yu, Y.; Yuan, L.; Zhang, M.; Cao, X. Cyclin l2, a novel RNA polymerase ii-associated cyclin, is involved in pre-mrna splicing and induces apoptosis of human hepatocellular carcinoma cells. J. Biol. Chem. 2004, 279, 11639–11648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cai, M.; Xu, S.; Chen, S.; Chen, X.; Chen, C.; Gu, J. Interaction of p58(pitslre), a g2/m-specific protein kinase, with cyclin d3. J. Biol. Chem. 2002, 277, 35314–35322. [Google Scholar] [CrossRef]
- Xiang, J.; Lahti, J.M.; Grenet, J.; Easton, J.; Kidd, V.J. Molecular cloning and expression of alternatively spliced pitslre protein kinase isoforms. J. Biol. Chem. 1994, 269, 15786–15794. [Google Scholar]
- Hu, D.; Mayeda, A.; Trembley, J.H.; Lahti, J.M.; Kidd, V.J. Cdk11 complexes promote pre-mrna splicing. J. Biol. Chem. 2003, 278, 8623–8629. [Google Scholar] [CrossRef]
- Loyer, P.; Busson, A.; Trembley, J.H.; Hyle, J.; Grenet, J.; Zhao, W.; Ribault, C.; Montier, T.; Kidd, V.J.; Lahti, J.M. The RNA binding motif protein 15b (rbm15b/ott3) is a functional competitor of serine-arginine (sr) proteins and antagonizes the positive effect of the cdk11p110-cyclin l2α complex on splicing. J. Biol. Chem. 2011, 286, 147–159. [Google Scholar] [CrossRef]
- Loyer, P.; Trembley, J.H.; Lahti, J.M.; Kidd, V.J. The rnp protein, rnps1, associates with specific isoforms of the p34cdc2-related pitslre protein kinase in vivo. J. Cell Sci. 1998, 111 Pt 11, 1495–1506. [Google Scholar]
- Trembley, J.H.; Hu, D.; Hsu, L.-C.; Yeung, C.-Y.; Slaughter, C.; Lahti, J.M.; Kidd, V.J. Pitslre p110 protein kinases associate with transcription complexes and affect their activity. J. Biol. Chem. 2002, 277, 2589–2596. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.H.; Choi, H.K.; Jung, S.Y.; Hyle, J.; Kim, B.J.; Yoon, K.; Cho, E.J.; Youn, H.D.; Lahti, J.M.; Qin, J.; et al. Chk2 kinase promotes pre-mrna splicing via phosphorylating cdk11p110. Oncogene 2014, 33, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Drogat, J.; Migeot, V.; Mommaerts, E.; Mullier, C.; Dieu, M.; Van Bakel, H.; Hermand, D. Cdk11-cyclinl controls the assembly of the RNA polymerase ii mediator complex. Cell Rep. 2012, 2, 1068–1076. [Google Scholar] [CrossRef]
- Cornelis, S.; Bruynooghe, Y.; Denecker, G.; Van Huffel, S.; Tinton, S.; Beyaert, R. Identification and characterization of a novel cell cycle-regulated internal ribosome entry site. Mol. Cell 2000, 5, 597–605. [Google Scholar] [CrossRef]
- Petretti, C.; Savoian, M.; Montembault, E.; Glover, D.M.; Prigent, C.; Giet, R. The pitslre/cdk11p58 protein kinase promotes centrosome maturation and bipolar spindle formation. EMBO Rep. 2006, 7, 418–424. [Google Scholar] [CrossRef]
- Hu, D.; Valentine, M.; Kidd, V.J.; Lahti, J.M. Cdk11p58 is required for the maintenance of sister chromatid cohesion. J. Cell Sci. 2007, 120, 2424–2434. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, H.; Gruss, O.J.; Rybina, S.; Caudron, M.; Schelder, M.; Wilm, M.; Mattaj, I.W.; Karsenti, E. Cdk11 is a rangtp-dependent microtubule stabilization factor that regulates spindle assembly rate. J. Cell Biol. 2008, 180, 867–875. [Google Scholar] [CrossRef]
- Kong, X.; Gan, H.; Hao, Y.; Cheng, C.; Jiang, J.; Hong, Y.; Yang, J.; Zhu, H.; Chi, Y.; Yun, X.; et al. Cdk11p58 phosphorylation of pak1 ser174 promotes dlc2 binding and roles on cell cycle progression. J. Biochem. 2009, 146, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Franck, N.; Montembault, E.; Romé, P.; Pascal, A.; Cremet, J.-Y.; Giet, R. Cdk11p58 is required for centriole duplication and plk4 recruitment to mitotic centrosomes. PLoS ONE 2011, 6, e14600. [Google Scholar] [CrossRef] [PubMed]
- Rakkaa, T.; Escude, C.; Giet, R.; Magnaghi-Jaulin, L.; Jaulin, C. Cdk11(p58) kinase activity is required to protect sister chromatid cohesion at centromeres in mitosis. Chromosome Res. 2014, 22, 267–276. [Google Scholar] [CrossRef]
- Lahti, J.M.; Xiang, J.; Heath, L.S.; Campana, D.; Kidd, V.J. Pitslre protein kinase activity is associated with apoptosis. Mol. Cell. Biol. 1995, 15, 1–11. [Google Scholar] [CrossRef]
- Tang, D.; Gururajan, R.; Kidd, V.J. Phosphorylation of pitslre p110 isoforms accompanies their processing by caspases during fas-mediated cell death. J. Biol. Chem. 1998, 273, 16601–16607. [Google Scholar] [CrossRef] [PubMed]
- Ariza, M.E.; Broome-Powell, M.; Lahti, J.M.; Kidd, V.J.; Nelson, M.A. Fas-induced apoptosis in human malignant melanoma cell lines is associated with the activation of the p34(cdc2)-related pitslre protein kinases. J. Biol. Chem. 1999, 274, 28505–28513. [Google Scholar] [CrossRef] [PubMed]
- Beyaert, R.; Kidd, V.J.; Cornelis, S.; Van de Craen, M.; Denecker, G.; Lahti, J.M.; Gururajan, R.; Vandenabeele, P.; Fiers, W. Cleavage of pitslre kinases by ice/casp-1 and cpp32/casp-3 during apoptosis induced by tumor necrosis factor. J. Biol. Chem. 1997, 272, 11694–11697. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Hershey, J.W.; Nelson, M.A. Phosphorylation of the eukaryotic initiation factor 3f by cyclin-dependent kinase 11 during apoptosis. FEBS Lett. 2009, 583, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Ariza, M.E.; Goulet, A.C.; Shi, J.; Nelson, M.A. Death-signal-induced relocalization of cyclin-dependent kinase 11 to mitochondria. Biochem. J. 2005, 392, 65–73. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Rusan, M.; Li, K.; Li, Y.; Christensen, C.L.; Abraham, B.J.; Kwiatkowski, N.; Buczkowski, K.A.; Bockorny, B.; Chen, T.; Li, S.; et al. Suppression of adaptive responses to targeted cancer therapy by transcriptional repression. Cancer Discov. 2018, 8, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. Cdk7-dependent transcriptional addiction in triple-negative breast cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef]
- Nelson, M.A.; Ariza, M.E.; Yang, J.M.; Thompson, F.H.; Taetle, R.; Trent, J.M.; Wymer, J.; Massey-Brown, K.; Broome-Powell, M.; Easton, J.; et al. Abnormalities in the p34cdc2-related pitslre protein kinase gene complex (cdc2l) on chromosome band 1p36 in melanoma. Cancer Genet. Cytogenet. 1999, 108, 91–99. [Google Scholar] [CrossRef]
- Chandramouli, A.; Shi, J.; Feng, Y.; Holubec, H.; Shanas, M.R.; Bhattacharyya, A.K.; Zheng, W.; Nelson, M.A. Haploinsufficiency of the cdc2l gene contributes to skin cancer development in mice. Carcinogenesis 2007, 28, 2028–2035. [Google Scholar] [CrossRef]
- Trembley, J.H.; Hu, D.; Slaughter, C.A.; Lahti, J.M.; Kidd, V.J. Casein kinase 2 interacts with cyclin-dependent kinase 11 (cdk11) in vivo and phosphorylates both the RNA polymerase ii carboxyl-terminal domain and cdk11 in vitro. J. Biol. Chem. 2003, 278, 2265–2270. [Google Scholar] [CrossRef]
- Chi, Y.; Zhang, C.; Zong, H.; Hong, Y.; Kong, X.; Liu, H.; Zou, W.; Wang, Y.; Yun, X.; Gu, J. Thr-370 is responsible for cdk11p58 autophosphorylation, dimerization, and kinase activity. J. Biol. Chem. 2011, 286, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Perez-Cadahia, B.; Drobic, B.; Davie, J.R. H3 phosphorylation: Dual role in mitosis and interphase. Biochem. Cell Biol. 2009, 87, 695–709. [Google Scholar]
- Sawicka, A.; Seiser, C. Histone h3 phosphorylation—A versatile chromatin modification for different occasions. Biochimie 2012, 94, 2193–2201. [Google Scholar] [CrossRef]
- Du, Y.; Yan, D.; Yuan, Y.; Xu, J.; Wang, S.; Yang, Z.; Cheng, W.; Tian, X.; Kan, Q. Cdk11(p110) plays a critical role in the tumorigenicity of esophageal squamous cell carcinoma cells and is a potential drug target. Cell Cycle 2019, 18, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.H.; Wu, Y.F.; Dong, X.L.; Pan, C.X.; Du, G.Y.; Yang, J.G.; Wang, W.; Bao, X.Y.; Chen, P.; Pan, M.H.; et al. Identification and characterization of the bmcyclin l1-bmcdk11a/b complex in relation to cell cycle regulation. Cell Cycle 2017, 16, 861–868. [Google Scholar] [CrossRef]
- Freeman, A.K.; Morrison, D.K. 14-3-3 proteins: Diverse functions in cell proliferation and cancer progression. Semin. Cell Dev. Biol. 2011, 22, 681–687. [Google Scholar] [CrossRef]
- Feng, Y.; Qi, W.; Martinez, J.; Nelson, M.A. The cyclin-dependent kinase 11 interacts with 14-3-3 proteins. Biochem. Biophys. Res. Commun. 2005, 331, 1503–1509. [Google Scholar] [CrossRef]
- Wilker, E.W.; Van Vugt, M.A.T.M.; Artim, S.C.; Huang, P.H.; Petersen, C.P.; Reinhardt, H.C.; Feng, Y.; Sharp, P.A.; Sonenberg, N.; White, F.M.; et al. 14-3-3[sgr] controls mitotic translation to facilitate cytokinesis. Nature 2007, 446, 329–332. [Google Scholar] [CrossRef]
- Whittaker, S.R.; Mallinger, A.; Workman, P.; Clarke, P.A. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol. Ther. 2017, 173, 83–105. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Unger, G.M.; Tobolt, D.K.; Korman, V.L.; Wang, G.; Ahmad, K.A.; Slaton, J.W.; Kren, B.T.; Ahmed, K. Systemic administration of antisense oligonucleotides simultaneously targeting ck2alpha and alpha′ subunits reduces orthotopic xenograft prostate tumors in mice. Mol. Cell. Biochem. 2011, 356, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Nickles, D.; Sandmann, T.; Ziman, R.; Bourgon, R. Nanostringqcpro: Quality Metrics and Data Processing Methods for Nanostring mRNA Gene Expression Data; R Package Version 1.14.0. 2018. Available online: https://doi.org/doi:10.18129/B9.bioc.NanoStringQCPro. [CrossRef]
- Wang, H.; Horbinski, C.; Wu, H.; Liu, Y.; Sheng, S.; Liu, J.; Weiss, H.; Stromberg, A.J.; Wang, C. Nanostringdiff: A novel statistical method for differential expression analysis based on nanostring ncounter data. Nucleic Acids Res. 2016, 44, e151. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Yu, G.; He, Q.Y. Reactomepa: An r/bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | Transformed | BRAF Mutation | NRAS Mutation |
|---|---|---|---|
| Hema-Lp 1 | No | None | None |
| A375 | Yes | V600E | None |
| 1205Lu | Yes | V600E | None |
| WM39 | Yes | V600E | None |
| SK-Mel-2 | Yes | None | Q61R |
| WM1366 | Yes | None | Q61L |
| WM3000 | Yes | None | Q61R |
| Cell Line | CDK11A | CDK11B | MYC |
|---|---|---|---|
| Hema-Lp | 1 | 1 | 1 |
| A375 | 0.06 | 0.97 | 3.24 |
| 1205Lu | 0.28 | 0.18 | 1.01 |
| WM39 | 1.51 | 0.94 | 1.68 |
| WM1366 | 0.10 | 0.16 | 1.51 |
| WM3000 | 0.07 | 0.40 | 0.86 |
| Cell Line | Time Point (h) | CDK11 | rpS6 P-S235/236 | rpS6 | Erk 1/2 P-TY | Erk 1/2 | β-Catenin Active | β-Catenin | cMYC | MCL-1 | Survivin |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A375 | 48 | 0.22 | 0.89 | 1.11 | 1.94 | 0.87 | 1.24 | 1.37 | 0.90 | ND | ND |
| (0.09, 0.34) | (0.83, 0.96) | (0.81, 1.40) | (1.17, 2.71) | (0.72, 1.01) | (0.68, 1.81) | (0.78, 1.95) | (0.83, 0.96) | ||||
| 72 | 0.39 | 0.92 | 1.27 | 1.63 | ND | ND | 1.62 | 0.18 | 0.98 | 0.69 | |
| (0.35, 0.43) | (0.67, 1.17) | 0.80, 1.74) | (1.25, 2.02) | (1.21, 2.02) | (0.09, 0.26) | (0.74, 1.22) | (0.55, 0.84) | ||||
| WM-1366 | 48 | 0.27 | 1.28 | 0.88 | 0.90 | 0.92 | 1.13 | 0.89 | 0.36 | ND | ND |
| (0.15, 0.39) | (0.72, 1.84) | (0.77, 0.99) | (0.72, 1.08) | (0.77, 1.08) | (0.70, 1.56) | (0.65, 1.14) | (0.13, 0.60) | ||||
| 72 | 0.19 | 1.17 | 0.77 | 1.53 | 0.93 | 0.68 | 0.81 | 0.33 | 0.67 | 2.08 | |
| (−0.01, 0.39) | (−0.68, 4.82) | (0.35, 1.19) | (0.84, 2.22) | (0.76, 1.11) | (0.31, 1.06) | (0.27, 1.35) | (0.21, 0.44) | (0.44, 0.91) | (1.79, 2.37) |
| NanoString: A375 | q-RT-PCR: A375 | q-RT-PCR: WM1366 | ||
|---|---|---|---|---|
| Gene | Fold Change 1 | q-Value | Fold Change (95% CI) 2 | Fold Change (95% CI) 2 |
| CCNA2 | 0.53 | 1.35 × 10−9 | 0.55 (0.48, 0.62) | 1.08 (0.42, 1.75) |
| CCNB1 | 0.52 | 2.72 × 10−6 | 0.38 (0.34, 0.41) | 0.91 (0.53, 1.28) |
| CCND1 | 0.56 | 3.65 × 10−5 | 0.49 (0.41, 0.58) | 0.37 (0.37, 0.38) |
| CCNE2 | 0.66 | 1.15 × 10−3 | 0.72 (0.66, 0.77) | 1.13 (0.20, 1.22) |
| WEE1 | 1.70 | 5.39 × 10−3 | 1.97 (1.44, 2.51) | 1.56 (0.72, 2.41) |
| SFN | 0.19 | 1.95 × 10−2 | 0.25 (0.24, 0.26) | 1.10 (0.95, 1.25) |
| BRCA1 | 0.51 | 5.19 × 10−10 | 0.40 (0.31, 0.49) | 0.72 (0.51, 0.93) |
| MYC | 1.72 | 1.45 × 10−3 | 1.40 (1.34, 1.46) | 0.71 (0.20, 1.22) |
| Cell Line | Time (h) | Cyclin A2 | Cyclin B1 | Cyclin D1 | Histone H3 P-S10 |
|---|---|---|---|---|---|
| A375 | 48 | 0.27 | 0.45 | 0.72 | 1.57 |
| (0.15, 0.39) | (0.27, 0.64) | (0.66, 0.79) | (1.50, 1.65) | ||
| 72 | ND | ND | ND | 2.75 (1.72, 3.79) | |
| WM1366 | 48 | 1.14 | 1.35 | 0.32 | 1.99 |
| (0.48, 1.81) | (0.43, 2.28) | (0.12, 0.52) | (0.59, 3.39) | ||
| 72 | 3.48 | 2.02 | 0.51 | ND | |
| (2.80, 4.16) | (1.03, 3.01) | (0.11, 0.91) |
| Cell Line | Treatment | G1 | S | G2/M |
|---|---|---|---|---|
| A375 | Untreated | 62.6 ± 1.4 | 24.7 ± 1.0 | 12.7 ± 0.6 |
| siCDK11 | 84.0 ± 0.7 ** | 11.4 ± 0.5 *** | 4.5 ± 0.5 ** | |
| siControl | 68.4 ± 0.3 | 21.1 ± 0.3 | 10.5 ± 0.3 | |
| WM1366 | Untreated | 56.2 ± 4.3 | 23.6 ± 2.9 | 20.3 ± 1.5 |
| siCDK11 | 76.6 ± 0.3 * | 16.1 ± 0.1 * | 7.2 ± 0.5 ** | |
| siControl | 56.4 ± 2.7 | 23.1 ± 2.8 | 20.5 ± 0.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, R.L.; Shaughnessy, D.P.; Knutson, T.P.; Vogel, R.I.; Ahmed, K.; Kren, B.T.; Trembley, J.H. CDK11 Loss Induces Cell Cycle Dysfunction and Death of BRAF and NRAS Melanoma Cells. Pharmaceuticals 2019, 12, 50. https://doi.org/10.3390/ph12020050
Ahmed RL, Shaughnessy DP, Knutson TP, Vogel RI, Ahmed K, Kren BT, Trembley JH. CDK11 Loss Induces Cell Cycle Dysfunction and Death of BRAF and NRAS Melanoma Cells. Pharmaceuticals. 2019; 12(2):50. https://doi.org/10.3390/ph12020050
Chicago/Turabian StyleAhmed, Rehana L., Daniel P. Shaughnessy, Todd P. Knutson, Rachel I. Vogel, Khalil Ahmed, Betsy T. Kren, and Janeen H. Trembley. 2019. "CDK11 Loss Induces Cell Cycle Dysfunction and Death of BRAF and NRAS Melanoma Cells" Pharmaceuticals 12, no. 2: 50. https://doi.org/10.3390/ph12020050
APA StyleAhmed, R. L., Shaughnessy, D. P., Knutson, T. P., Vogel, R. I., Ahmed, K., Kren, B. T., & Trembley, J. H. (2019). CDK11 Loss Induces Cell Cycle Dysfunction and Death of BRAF and NRAS Melanoma Cells. Pharmaceuticals, 12(2), 50. https://doi.org/10.3390/ph12020050