Disruption of Cholinergic Circuits as an Area for Targeted Drug Treatment of Alzheimer’s Disease: In Vivo Assessment of Short-Term Plasticity in Rat Brain

Abstract
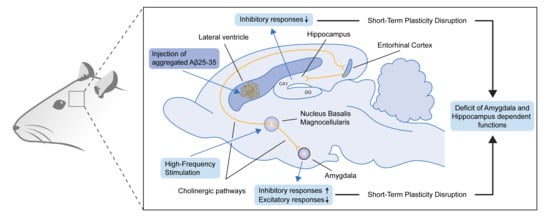
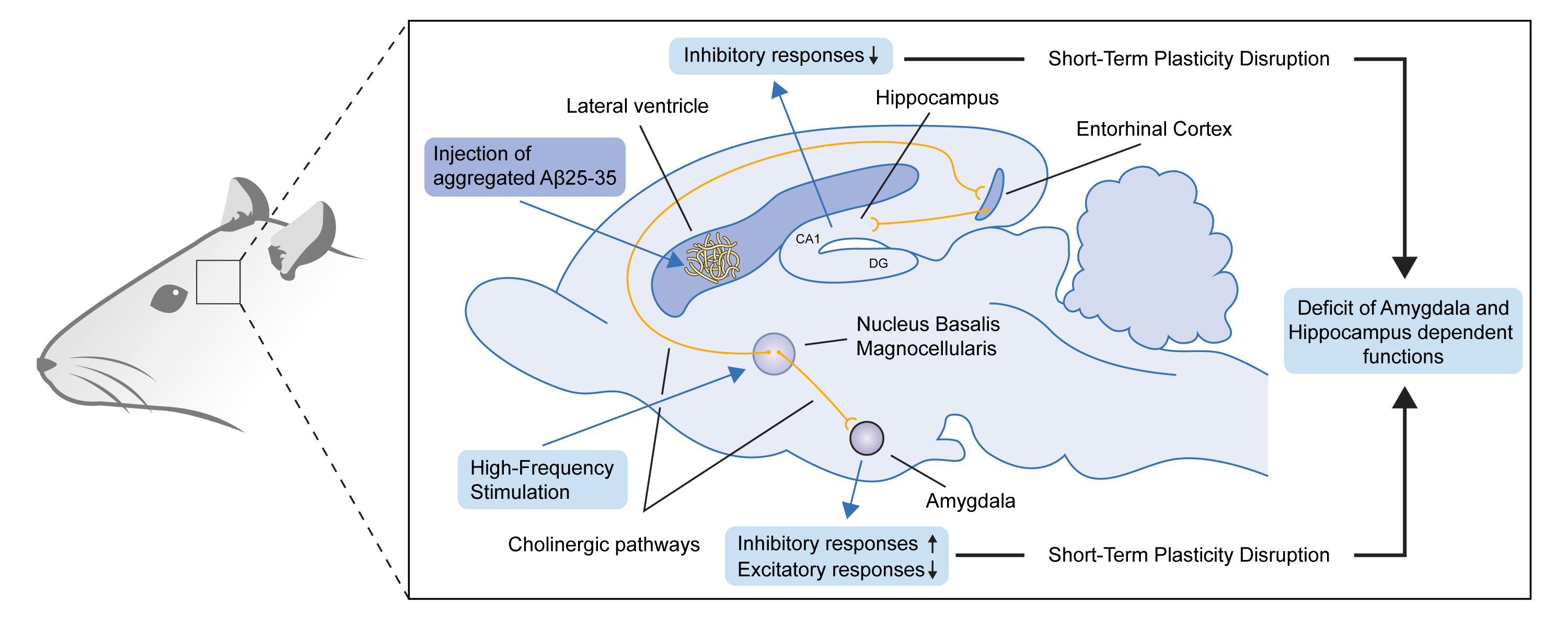
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
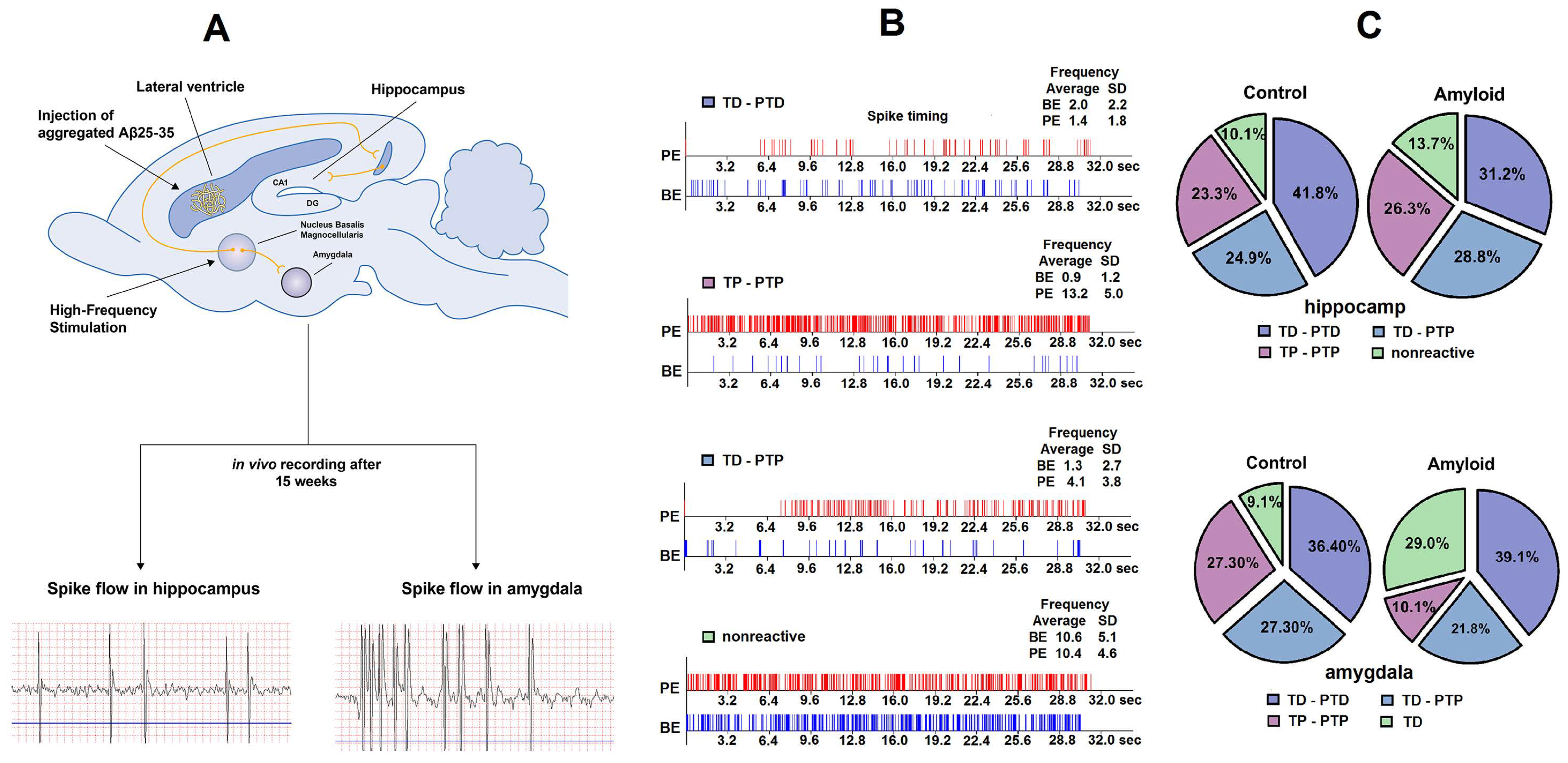
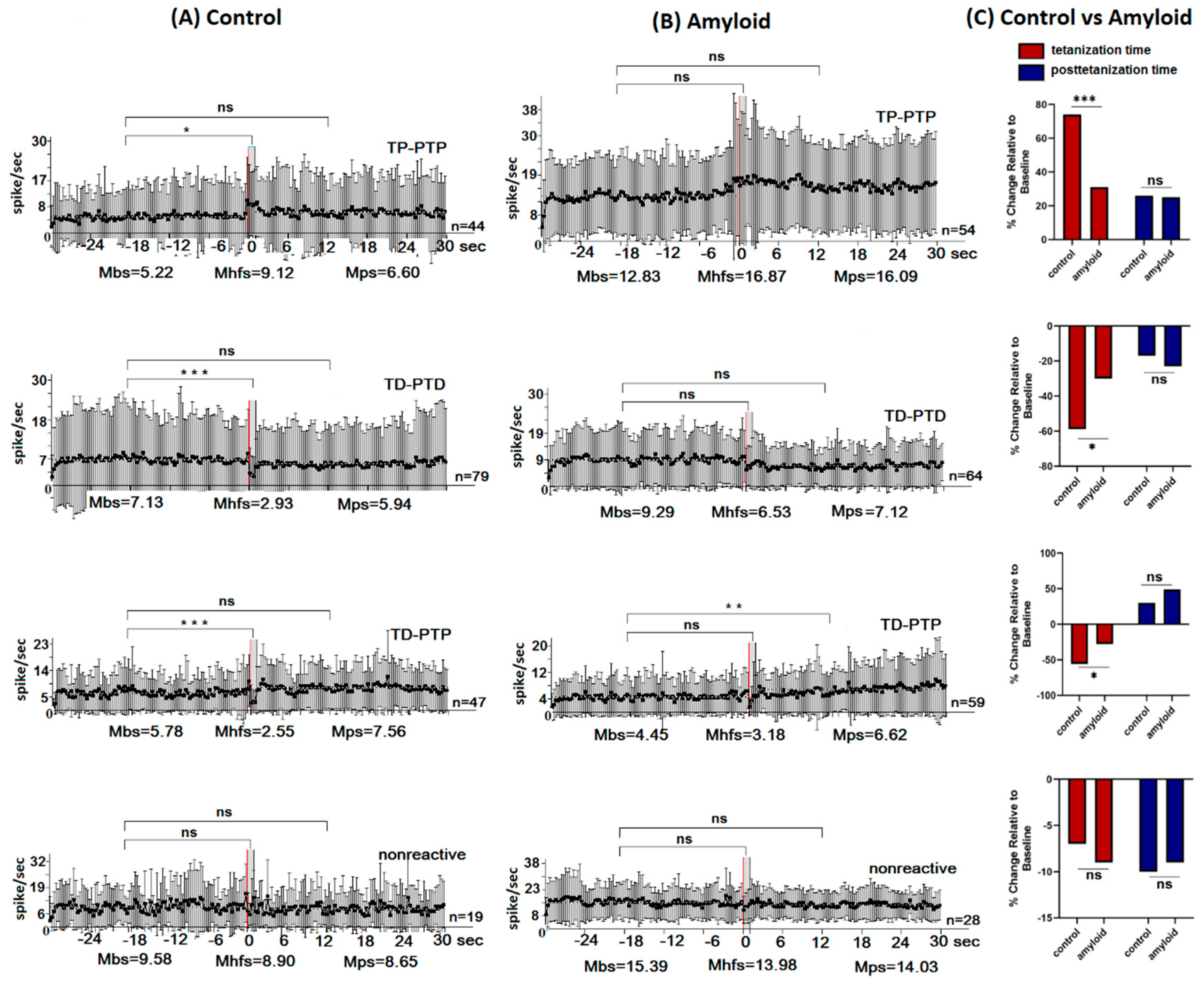
2.1. Excitatory and Inhibitory Responses of Hippocampal and Amygdala Neurons to HFS of NBM
2.2. Real Time Pre- and Post-stimulus Spike Activity and Intensity of Responses of Hippocampal and Amygdala Neurons to HFS of NBM
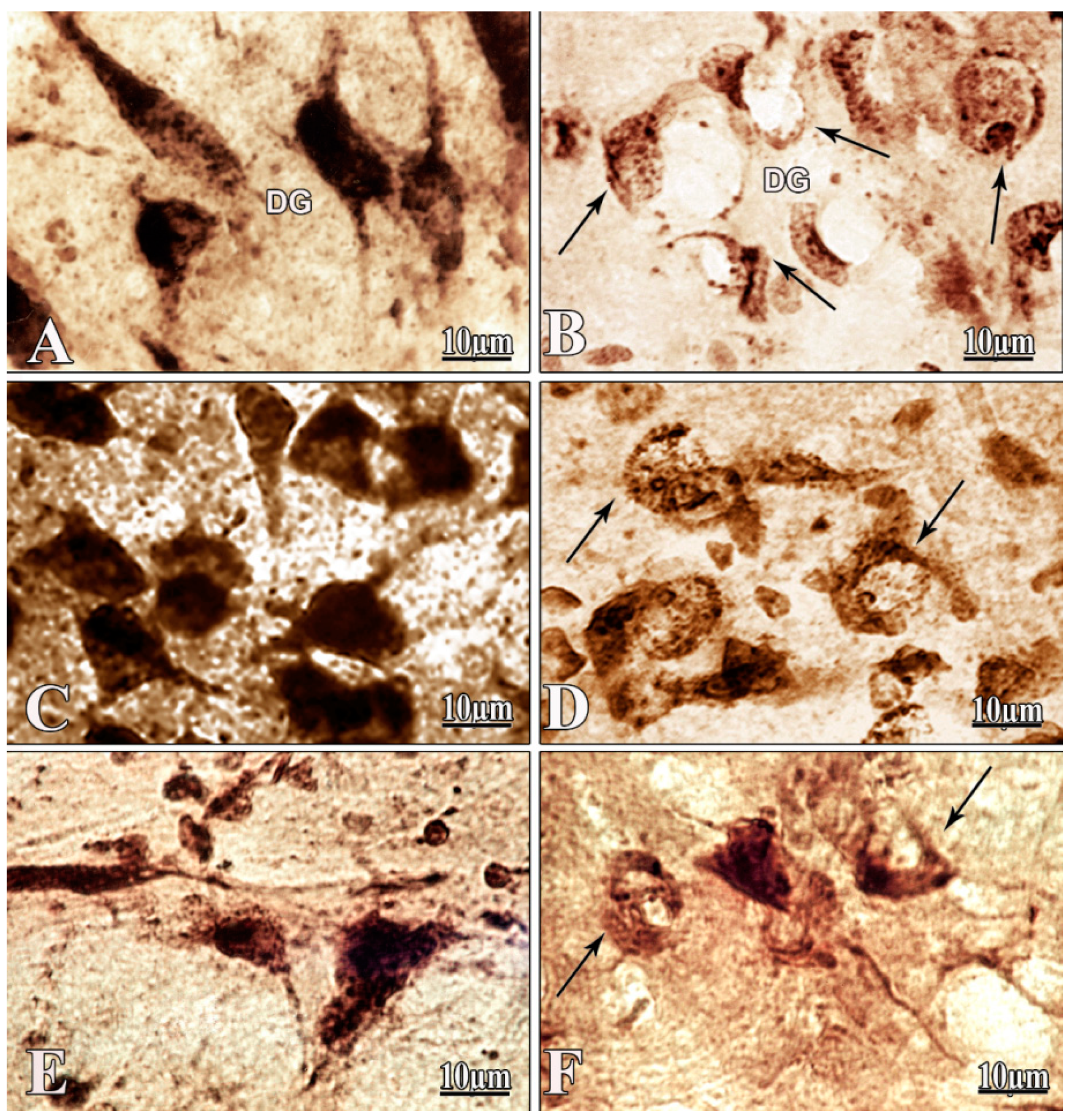
2.3. Morphological Study: Changes in Hippocampus, Amygdala and NBM
3. Discussion
4. Methods and Materials
4.1. Animals
4.2. Experimental Protocol
4.3. Aβ 25–35 Peptide Preparation
4.4. Surgical Procedure
4.5. In Vivo Electrophysiology and Statistical Analysis
4.6. Morphological Study
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wevers, A.; Burghaus, L.; Moser, N.; Witter, B.; Steinlein, O.K.; Schütz, U.; Achnitz, B.; Krempel, U.; Nowacki, S.; Pilz, K.; et al. Expression of nicotinic acetylcholine receptors in Alzheimer’s disease: Postmortem investigations and experimental approaches. Behav. Brain Res. 2000, 113, 207–215. [Google Scholar] [CrossRef]
- Schliebs, R.; Arendt, T. The significance of the cholinergic system in the brain during aging and in Alzheimer’s disease. J. Neural Transm. 2006, 113, 1625–1644. [Google Scholar] [CrossRef] [PubMed]
- Geula, C.; Nagykery, N.; Nicholas, A.; Wu, C.-K. Cholinergic Neuronal and Axonal Abnormalities Are Present Early in Aging and in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2008, 67, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Giovannelli, L.; Casamenti, F.; Scali, C.; Bartolini, L.; Pepeu, G. Differential effects of amyloid peptides β-(1–40) and β-(25–35) injections into the rat nucleus basalis. Neuroscience 1995, 66, 781–792. [Google Scholar] [CrossRef]
- Harkany, T.; Lengyel, Z.; Soós, K.; Penke, B.; Luiten, P.G.M.; Gulya, K. Cholinotoxic effects of β-amyloid(1–42) peptide on cortical projections of the rat nucleus basalis magnocellularis. Brain Res. 1995, 695, 71–75. [Google Scholar] [CrossRef] [Green Version]
- Itoh, A.; Nitta, A.; Nadai, M.; Nishimura, K.; Hirose, M.; Hasegawa, T.; Nabeshima, T. Dysfunction of Cholinergic and Dopaminergic Neuronal Systems in β-Amyloid Protein-Infused Rats. J. Neurochem. 2002, 66, 1113–1117. [Google Scholar] [CrossRef]
- Guela, C.; Wu, C.-K.; Saroff, D.; Lorenzo, A.; Yuan, M.; Yankner, B.A. Aging renders the brain vulnerable to amyloid β-protein neurotoxicity. Nat. Med. 1998, 4, 827–831. [Google Scholar] [CrossRef]
- Jhamandas, J.H.; Cho, C.; Jassar, B.; Harris, K.; MacTavish, D.; Easaw, J. Cellular Mechanisms for Amyloid β-Protein Activation of Rat Cholinergic Basal Forebrain Neurons. J. Neurophysiol. 2001, 86, 1312–1320. [Google Scholar] [CrossRef] [Green Version]
- Bronfman, F.C.; Moechars, D.; Van Leuven, F. Acetylcholinesterase-Positive Fiber Deafferentation and Cell Shrinkage in the Septohippocampal Pathway of Aged Amyloid Precursor Protein London Mutant Transgenic Mice. Neurobiol. Dis. 2000, 7, 152–168. [Google Scholar] [CrossRef] [Green Version]
- Wong, T.P.; Debeir, T.; Duff, K.; Cuello, A.C. Reorganization of cholinergic terminals in the cerebral cortex and hippocampus in transgenic mice carrying mutated presenilin-1 and amyloid precursor protein transgenes. J. Neurosci. 1999, 19, 2706–2716. [Google Scholar] [CrossRef] [Green Version]
- Mesulam, M.-M.; Hersh, L.B.; Mash, D.C.; Geula, C. Differential cholinergic innervation within functional subdivisions of the human cerebral cortex: A choline acetyltransferase study. J. Comp. Neurol. 1992, 318, 316–328. [Google Scholar] [CrossRef]
- Botly, L.C.P.; Baxter, M.G.; de Rosa, E. Basal Forebrain and Memory. Encycl. Neurosci. 2009, 47–52. [Google Scholar] [CrossRef]
- Mesulam, M. The Cholinergic Lesion of Alzheimer’s Disease: Pivotal Factor or Side Show? Learn. Mem. 2004, 11, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geula, C.; Mesulam, M.M. Systematic regional variations in the loss of cortical cholinergic fibers in Alzheimer’s disease. Cereb. Cortex 1996, 6, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geula, C.; Musalum, M. Cholinergic systems in Alzheimer’s disease. In Alzheimer Disease, 2nd ed.; Terry, R.D., Katzman, R., Bick, K.L., Sisodia, S.S., Eds.; Lippinkott, Williams & Wilkins: Philadelphia, PA, USA, 1999; pp. 69–295. [Google Scholar]
- Woolf, N.J.; Butcher, L.L. Cholinergic projections to the basolateral amygdala: A combined Evans Blue and acetylcholinesterase analysis. Brain Res. Bull. 1982, 8, 751–763. [Google Scholar] [CrossRef]
- Hosseini, N.; Alaei, H.; Reisi, P.; Radahmadi, M. The effects of NBM- lesion on synaptic plasticity in rats. Brain Res. 2017, 1655, 122–127. [Google Scholar] [CrossRef]
- Goldman, M.S.; Maldonado, P.; Abbott, L.F.; Lewis, J.E. Redundancy reduction and sustained firing with stochastic depressing synapses. J. Neurosci. 2002, 22, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Mongillo, G.; Barak, O.; Tsodyks, M. Synaptic Theory of Working Memory. Science 2008, 319, 1543–1546. [Google Scholar] [CrossRef] [Green Version]
- Ghanbari, A.; Malyshev, A.; Volgushev, M.; Stevenson, I.H. Estimating short-term synaptic plasticity from pre- and postsynaptic spiking. PLoS Comput. Biol. 2017, 13, e1005738. [Google Scholar] [CrossRef] [Green Version]
- Markram, H.; Tsodyks, M. Redistribution of synaptic efficacy between neocortical pyramidal neurons. Nature 1996, 382, 807–810. [Google Scholar] [CrossRef]
- Hiratani, N.; Fukai, T. Interplay between Short- and Long-Term Plasticity in Cell-Assembly Formation. PLoS ONE 2014, 9, e101535. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Wei, H.; Li, X.; Xie, X.; Zhou, Y. Short-term synaptic plasticity in the rat geniculo-cortical pathway during development in vivo. Neurosci. Lett. 2006, 398, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Blackman, A.V.; Abrahamsson, T.; Costa, R.P.; Lalanne, T.; Sjöström, P.J. Target-cell-specific short-term plasticity in local circuits. Front. Synaptic Neurosci. 2013, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Briggs, C.A.; Chakroborty, S.; Stutzmann, G.E. Emerging pathways driving early synaptic pathology in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 988–997. [Google Scholar] [CrossRef] [Green Version]
- Chakroborty, S.; Hill, E.S.; Christian, D.T.; Helfrich, R.; Riley, S.; Schneider, C.; Kapecki, N.; Mustaly-Kalimi, S.; Seiler, F.A.; Peterson, D.A.; et al. Reduced presynaptic vesicle stores mediate cellular and network plasticity defects in an early-stage mouse model of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Aghajanov, M.I.; Yenkoyan, K.B.; Chavushyan, V.A.; Sarkissian, J.S. The proline-rich hypothalamic peptide is a modulator of functions of neurotrophins and neuronal activity in amyloid-induced neurodegeneration. Neurochem. J. 2014, 8, 47–57. [Google Scholar] [CrossRef]
- Yenkoyan, K.; Safaryan, K.; Chavushyan, V.; Meliksetyan, I.; Navasardyan, G.; Sarkissian, J.; Galoyan, A.; Aghajanov, M. Neuroprotective action of proline-rich polypeptide-1 in β-amyloid induced neurodegeneration in rats. Brain Res. Bull. 2011, 86, 262–271. [Google Scholar] [CrossRef]
- Chavushyan, V.; Matinyan, S.; Danielyan, M.; Aghajanov, M.; Yenkoyan, K. Embryonic proteoglycans regulate monoamines in the rat frontal cortex and hippocampus in Alzheimer’s disease-like pathology. Neurochem. Int. 2020, 140. [Google Scholar] [CrossRef]
- Yenkoyan, K.; Safaryan, K.; Navasardyan, G.; Mkrtchyan, L.; Aghajanov, M. Effects of beta-amyloid on behavioral and amino acids spectrum in rats’ brain and their modulation by embryonic proteins. Neurochem. Int. 2009, 54, 292–298. [Google Scholar] [CrossRef]
- Galoyan, A.A.; Sarkissian, J.S.; Chavushyan, V.A. Neuroprotection by hypothalamic peptide proline-rich peptide-1 in Aβ 25–35 model of Alzheimer’ s disease. Alzheimer’s Dement. 2008, 4, 332–344. [Google Scholar] [CrossRef]
- Busche, M.A.; Eichhoff, G.; Adelsberger, H.; Abramowski, D.; Wiederhold, K.H.; Haass, C.; Staufenbiel, M.; Konnerth, A.; Garaschuk, O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 2008, 321, 1686–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busche, M.A.; Konnerth, A. Impairments of neural circuit function in Alzheimer’s disease. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150429. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Amyloid-β Induced Neuronal Dysfunction in Alzheimer’s Disease: From Synapses toward Neural Networks. Nat. Neurosci. 2010, 13, 812. [Google Scholar] [CrossRef] [PubMed]
- Goel, P.; Nishimura, S.; Chetlapalli, K.; Li, X.; Chen, C.; Dickman, D. Distinct Target-Specific Mechanisms Homeostatically Stabilize Transmission at Pre- and Post-synaptic Compartments. Front. Cell. Neurosci. 2020, 14. [Google Scholar] [CrossRef]
- Scott Gratz, X.J.; Pragya Goel, X.; Joseph Bruckner, X.J.; Roberto Hernandez, X.X.; Karam Khateeb, X.; Gregory Macleod, X.T.; Dion Dickman, X.; Kate, O.X.M. Development/Plasticity/Repair Endogenous Tagging Reveals Differential Regulation of Ca 2 Channels at Single Active Zones during Presynaptic Homeostatic Potentiation and Depression. J. Neurosci. 2019. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.; Wu, L.-G. Post-tetanic potentiation is caused by two signalling mechanisms affecting quantal size and quantal content. J. Physiol. 2010, 588, 4987–4994. [Google Scholar] [CrossRef]
- Zucker, R.S.; Regehr, W.G. Short-Term Synaptic Plasticity. Annu. Rev. Physiol. 2002, 64, 355–405. [Google Scholar] [CrossRef] [Green Version]
- Korogod, N.; Lou, X.; Schneggenburger, R. Presynaptic Ca2+ Requirements and Developmental Regulation of Posttetanic Potentiation at the Calyx of Held. J. Neurosci. 2005, 25, 5127–5137. [Google Scholar] [CrossRef]
- Lee, J.S.; Ho, W.-K.; Lee, S.-H. Post-tetanic increase in the fast-releasing synaptic vesicle pool at the expense of the slowly releasing pool. J. Gen. Physiol. 2010, 136, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Rovira, C.; Arbez, N.; Mariani, J. Aβ (25–35) and Aβ (1–40) act on different calcium channels in CA1 hippocampal neurons. Biochem. Biophys. Res. Commun. 2002, 296, 1317–1321. [Google Scholar] [CrossRef]
- Nanou, E.; Lee, A.; Catterall, W.A. Control of Excitation/Inhibition Balance in a Hippocampal Circuit by Calcium Sensor Protein Regulation of Presynaptic Calcium Channels. J. Neurosci. 2018, 38, 4430–4440. [Google Scholar] [CrossRef] [PubMed]
- Ni, R.; Marutle, A.; Nordberg, A. Modulation of α7 Nicotinic Acetylcholine Receptor and Fibrillar Amyloid-β Interactions in Alzheimer’s Disease Brain. J. Alzheimer’s Dis. 2013, 33, 841–851. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Wu, J. Nicotinic cholinergic mechanisms in Alzheimer’s disease. In International Review of Neurobiology; Academic Press: Cambridge, MA, USA, 2015; Volume 124, pp. 275–292. [Google Scholar]
- Ji, D.; Lape, R.; Dani, J.A. Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron 2001, 31, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Tong, R.; Emptage, N.J.; Padamsey, Z. A two-compartment model of synaptic computation and plasticity. Mol. Brain 2020, 13, 79. [Google Scholar] [CrossRef] [PubMed]
- Regehr, W.G. Short-term presynaptic plasticity. Cold Spring Harb. Perspect. Biol. 2012, 4, a005702. [Google Scholar] [CrossRef] [PubMed]
- Griffith, W.H. Voltage-clamp analysis of posttetanic potentiation of the mossy fiber to CA3 synapse in hippocampus. J. Neurophysiol. 1990, 63, 491–501. [Google Scholar] [CrossRef]
- Foster, K.A.; Kreitzer, A.C.; Regehr, W.G. Interaction of postsynaptic receptor saturation with presynaptic mechanisms produces a reliable synapse. Neuron 2002, 36, 1115–1126. [Google Scholar] [CrossRef] [Green Version]
- Xu-Friedman, M.A.; Regehr, W.G. Structural Contributions to Short-Term Synaptic Plasticity. Physiol. Rev. 2004, 84, 69–85. [Google Scholar] [CrossRef] [Green Version]
- McDonnell, M.D.; Graham, B.P. Phase changes in neuronal postsynaptic spiking due to short term plasticity. PLoS Comput. Biol. 2017, 13, e1005634. [Google Scholar] [CrossRef] [Green Version]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef]
- Dong, X.; Wang, Y.; Qin, Z. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugam, G.; Polavarapu, P.L. Structure of Aβ(25–35) peptide in different environments. Biophys. J. 2004, 87, 622–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurice, T.; Su, T.P.; Privat, A. Sigma1 (sigma 1) receptor agonists and neurosteroids attenuate B25–35-amyloid peptide-induced amnesia in mice through a common mechanism. Neuroscience 1998, 83, 413–428. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates: Compact, 6th ed.; Academic Press: Cambridge, MA, USA, 2005. [Google Scholar]
- Meliksetyan, I.B.; Nazaryan, O.A.; Sahakyan, I.K.; Abrahamyan, D.O.; Abrahamyan, S.S. Application of a histochemical method for detection of Ca2+ dependent acid phosphatase activity for studies of morpho-functional state of the cell structures in the rat brain. Neurochem. J. 2008, 2, 315. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chavushyan, V.; Soghomonyan, A.; Karapetyan, G.; Simonyan, K.; Yenkoyan, K. Disruption of Cholinergic Circuits as an Area for Targeted Drug Treatment of Alzheimer’s Disease: In Vivo Assessment of Short-Term Plasticity in Rat Brain. Pharmaceuticals 2020, 13, 297. https://doi.org/10.3390/ph13100297
Chavushyan V, Soghomonyan A, Karapetyan G, Simonyan K, Yenkoyan K. Disruption of Cholinergic Circuits as an Area for Targeted Drug Treatment of Alzheimer’s Disease: In Vivo Assessment of Short-Term Plasticity in Rat Brain. Pharmaceuticals. 2020; 13(10):297. https://doi.org/10.3390/ph13100297
Chicago/Turabian StyleChavushyan, Vergine, Ani Soghomonyan, Gohar Karapetyan, Karen Simonyan, and Konstantin Yenkoyan. 2020. "Disruption of Cholinergic Circuits as an Area for Targeted Drug Treatment of Alzheimer’s Disease: In Vivo Assessment of Short-Term Plasticity in Rat Brain" Pharmaceuticals 13, no. 10: 297. https://doi.org/10.3390/ph13100297
APA StyleChavushyan, V., Soghomonyan, A., Karapetyan, G., Simonyan, K., & Yenkoyan, K. (2020). Disruption of Cholinergic Circuits as an Area for Targeted Drug Treatment of Alzheimer’s Disease: In Vivo Assessment of Short-Term Plasticity in Rat Brain. Pharmaceuticals, 13(10), 297. https://doi.org/10.3390/ph13100297