CDK4/6 and MAPK—Crosstalk as Opportunity for Cancer Treatment


Abstract
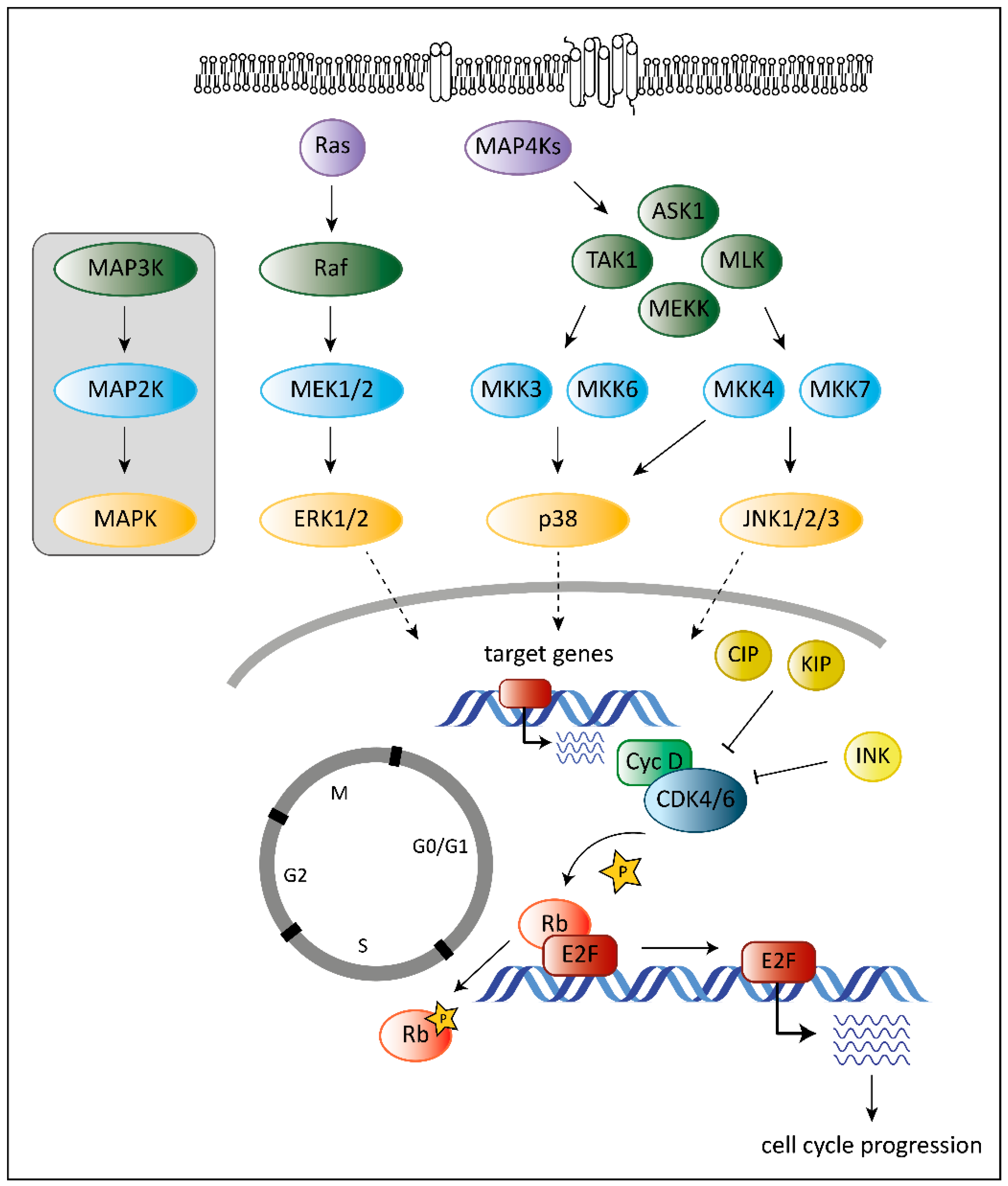
:1. Introduction
2. The Role of CDK6 in Cancer
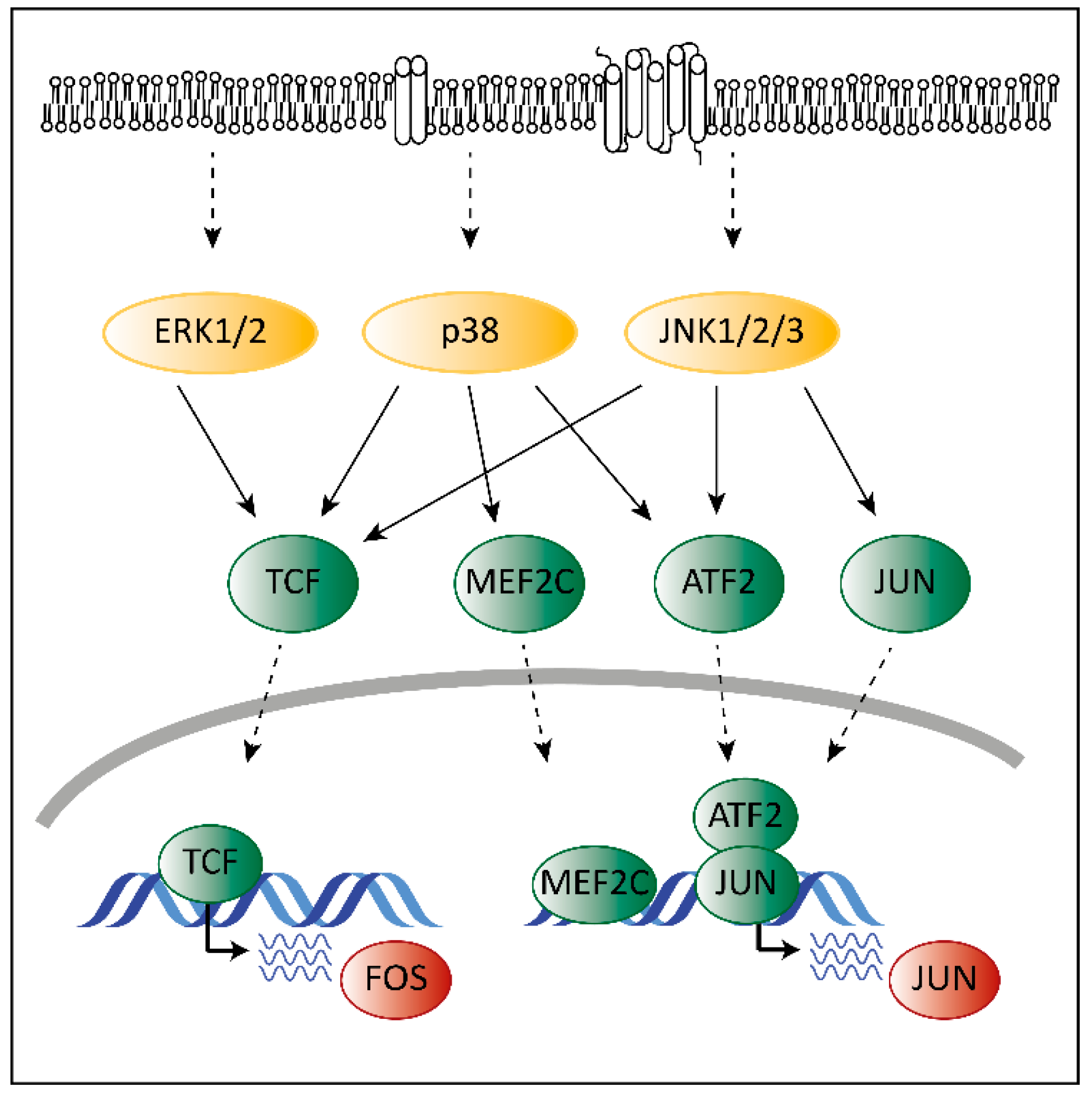
3. The Role of MAPKs in Cancer
4. The Unique Dual Role of p38
5. CDK6 as a Target in Cancer Therapy
6. Resistance to CDK4/6 Inhibitors
7. MAPK as Target in Cancer Therapy
8. Combinatorial Treatment with CDK4/6 and MAPK Inhibitors
9. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, J.N.; L’Allemain, G.; Brunei, A.; Müller, R.; Pouysségur, J. Cyclin D1 expression is regulated positively by the p42/p44(MAPK) and negatively by the p38/HOG(MAPK) pathway. J. Biol. Chem. 1996, 271, 20608–20616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyerson, M.; Harlow, E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol. Cell. Biol. 1994, 14, 2077–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tigan, A.S.; Bellutti, F.; Kollmann, K.; Tebb, G.; Sexl, V. CDK6-a review of the past and a glimpse into the future: From cell-cycle control to transcriptional regulation. Oncogene 2016, 35, 3083–3091. [Google Scholar] [CrossRef] [PubMed]
- Classon, M.; Harlow, E. The retinoblastoma tumour suppressor in development and cancer. Nat. Rev. Cancer 2002, 2, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Roovers, K.; Assoian, R.K. Integrating the MAP kinase signal into the G1 phase cell cycle machinery. BioEssays 2000, 22, 818–826. [Google Scholar] [CrossRef]
- Sewing, A.; Wiseman, B.; Lloyd, A.C.; Land, H. High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol. Cell. Biol. 1997, 17, 5588–5597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.Y.; Mercer, S.E.; Ewton, D.Z.; Yan, Z.; Jin, K.; Friedman, E. The stress-activated protein kinases p38 alpha and JNK1 stabilize p21(Cip1) by phosphorylation. J. Biol. Chem. 2002, 277, 29792–29802. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Kim, G.Y.; Deng, X.; Friedman, E. Transforming growth factor β1 induces proliferation in colon carcinoma cells by ras-dependent, smad-independent down-regulation of p21cip1. J. Biol. Chem. 2002, 277, 9870–9879. [Google Scholar] [CrossRef] [Green Version]
- Bulavin, D.V.; Higashimoto, Y.; Popoff, I.J.; Gaarde, W.A.; Basrur, V.; Potapova, O.; Appella, E.; Fornace, A.J. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 2001, 411, 102–107. [Google Scholar] [CrossRef]
- Munshi, A.; Ramesh, R. Mitogen-Activated Protein Kinases and Their Role in Radiation Response. Genes Cancer 2013, 4, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Manzano, J.L.; Layos, L.; Bugés, C.; De los Llanos Gil, M.; Vila, L.; Martínez-Balibrea, E.; Martínez-Cardús, A. Resistant mechanisms to BRAF inhibitors in melanoma. Ann. Transl. Med. 2016, 4, 237. [Google Scholar] [CrossRef] [Green Version]
- Khatib, Z.A.; Matsushime, H.; Valentine, M.; Shapiro, D.N.; Sherr, C.J.; Look, A.T. Coamplification of the CDK4 gene with MDM2 and GLI in human sarcomas. Cancer Res. 1993, 53, 5535–5541. [Google Scholar] [PubMed]
- Schmidt, E.E.; Ichimura, K.; Reifenberger, G.; Collins, V.P. CDKN2 (p16/MTS1) gene deletion or CDK4 amplification occurs in the majority of glioblastomas. Cancer Res. 1994, 54, 6321–6324. [Google Scholar]
- Wölfel, T.; Hauer, M.; Schneider, J.; Serrano, M.; Wölfel, C.; Klehmann-Hieb, E.; De Plaen, E.; Hankeln, T.; Meyer zum Büschenfelde, K.H.; Beach, D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995, 269, 1281–1284. [Google Scholar] [CrossRef]
- Chilosi, M.; Doglioni, C.; Yan, Z.; Lestani, M.; Menestrina, F.; Sorio, C.; Benedetti, A.; Vinante, F.; Pizzolo, G.; Inghirami, G. Differential expression of cyclin-dependent kinase 6 in cortical thymocytes and T-cell lymphoblastic lymphoma/leukemia. Am. J. Pathol. 1998, 152, 209–217. [Google Scholar] [PubMed]
- Nagel, S.; Leich, E.; Quentmeier, H.; Meyer, C.; Kaufmann, M.; Drexler, H.G.; Zettl, A.; Rosenwald, A.; MacLeod, R.A.F. Amplification at 7q22 targets cyclin-dependent kinase 6 in T-cell lymphoma. Leukemia 2008, 22, 387–392. [Google Scholar] [CrossRef] [Green Version]
- Kollmann, K.; Heller, G.; Schneckenleithner, C.; Warsch, W.; Scheicher, R.; Ott, R.G.; Schäfer, M.; Fajmann, S.; Schlederer, M.; Schiefer, A.-I.; et al. A Kinase-Independent Function of CDK6 Links the Cell Cycle to Tumor Angiogenesis. Cancer Cell 2013, 24, 167–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef]
- Kollmann, K.; Sexl, V. CDK6 and p16INK4A in lymphoid malignancies. Oncotarget 2013, 4, 1858–1859. [Google Scholar] [CrossRef]
- Kollmann, K.; Briand, C.; Bellutti, F.; Schicher, N.; Blunder, S.; Zojer, M.; Hoeller, C.; Kollmann, K.; Briand, C.; Bellutti, F.; et al. The interplay of CDK4 and CDK6 in melanoma. Oncotarget 2019, 10, 1346–1359. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Sicinska, E.; Geng, Y.; Ahnström, M.; Zagozdzon, A.; Kong, Y.; Gardner, H.; Kiyokawa, H.; Harris, L.N.; Stål, O.; et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell 2006, 9, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Bartkova, J.; Lukas, J.; Müller, H.; Lützhøt, D.; Strauss, M.; Bartek, J. Cyclin D1 protein expression and function in human breast cancer. Int. J. Cancer 1994, 57, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Dickson, C.; Fantl, V.; Gillett, C.; Brookes, S.; Bartek, J.; Smith, R.; Fisher, C.; Barnes, D.; Peters, G. Amplification of chromosome band 11q13 and a role for cyclin D1 in human breast cancer. Cancer Lett. 1995, 90, 43–50. [Google Scholar] [CrossRef]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Foster, J.S.; Wimalasena, J. Estrogen regulates activity of cyclin-dependent kinases and retinoblastoma protein phosphorylation in breast cancer cells. Mol. Endocrinol. 1996, 10, 488–498. [Google Scholar] [PubMed] [Green Version]
- Lim, J.T.E.; Mansukhani, M.; Weinstein, I.B. Cyclin-dependent kinase 6 associates with the androgen receptor and enhances its transcriptional activity in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5156–5161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, T.; Anderson, K.; Jacobsen, S.E.W.; Nishikawa, S.; Nerlov, C. Cdk6 blocks myeloid differentiation by interfering with Runx1 DNA binding and Runx1-C/EBPα interaction. Embo J. 2007, 26, 2361–2370. [Google Scholar] [CrossRef] [Green Version]
- Placke, T.; Faber, K.; Nonami, A.; Putwain, S.L.; Salih, H.R.; Heidel, F.H.; Krämer, A.; Root, D.E.; Barbie, D.A.; Krivtsov, A.V.; et al. Requirement for CDK6 in MLL-rearranged acute myeloid leukemia. Blood 2014, 124, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Sebolt-Leopold, J.S.; Herrera, R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat. Rev. Cancer 2004, 4, 937–947. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Map kinase signaling pathways and hematologic malignancies. Blood 2003, 101, 4667–4679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Towatari, M.; Iida, H.; Tanimoto, M.; Iwata, H.; Hamaguchi, M.; Saito, H. Constitutive activation of mitogen-activated protein kinase pathway in acute leukemia cells. Leukemia 1997, 11, 479–484. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Krystal, G.; Varticovksi, L.; McKinstry, R.; Rahmani, M.; Dent, P.; Grant, S. Pharmacologic mitogen-activated protein/extracellular signal-regulated kinase kinase/mitogen-activated protein kinase inhibitors interact synergistically with STI571 to induce apoptosis in Bcr/Abl-expressing human leukemia cells. Cancer Res. 2002, 62, 188–199. [Google Scholar]
- Whitmarsh, A.J.; Davis, R.J. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. 1996, 74, 589–607. [Google Scholar] [CrossRef]
- Raingeaud, J.; Gupta, S.; Rogers, J.S.; Dickens, M.; Han, J.; Ulevitch, R.J.; Davis, R.J. Pro-inflammatory Cytokines and Environmental Stress Cause p38 Mitogen-Activated Protein Kinase Activation by Dual Phosphorylation on Tyrosine and Threonine. J. Biol. Chem. 1995, 270, 7420–7426. [Google Scholar] [CrossRef] [Green Version]
- Van Dam, H.; Wilhelm, D.; Herr, I.; Steffen, A.; Herrlich, P.; Angel, P. ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. Embo J. 1995, 14, 1798–1811. [Google Scholar] [CrossRef]
- Kollmann, K.; Heller, G.; Sexl, V. C-JUN prevents methylation of p16INK4a (and Cdk6): The villain turned bodyguard. Oncotarget 2011, 2, 422–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igea, A.; Nebreda, A.R. The stress kinase p38α as a target for cancer therapy. Cancer Res. 2015, 75, 3997–4002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Sun, P. The pathways to tumor suppression via route p38. Trends Biochem. Sci. 2007, 32, 364–371. [Google Scholar] [CrossRef]
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.-X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras Proteins Induce Senescence by Altering the Intracellular Levels of Reactive Oxygen Species. J. Biol. Chem. 1999, 274, 7936–7940. [Google Scholar] [CrossRef] [Green Version]
- Nicke, B.; Bastien, J.; Khanna, S.J.; Warne, P.H.; Cowling, V.; Cook, S.J.; Peters, G.; Delpuech, O.; Schulze, A.; Berns, K.; et al. Involvement of MINK, a ste20 family kinase, in ras oncogene-induced growth arrest in human ovarian surface epithelial cells. Mol. Cell 2005, 20, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Dolado, I.; Swat, A.; Ajenjo, N.; De Vita, G.; Cuadrado, A.; Nebreda, A.R. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell 2007, 11, 191–205. [Google Scholar] [CrossRef] [Green Version]
- Iwasa, H.; Han, J.; Ishikawa, F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells 2003, 8, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, J.X.; Liao, R.; Deng, Q.; Zhou, J.J.; Huang, S.; Sun, P. Sequential Activation of the MEK–Extracellular Signal-Regulated Kinase and MKK3/6–p38 Mitogen-Activated Protein Kinase Pathways Mediates Oncogenic ras-Induced Premature Senescence. Mol. Cell. Biol. 2002, 22, 3389–3403. [Google Scholar] [CrossRef] [Green Version]
- Bulavin, D.V.; Phillips, C.; Nannenga, B.; Timofeev, O.; Donehower, L.A.; Anderson, C.W.; Appella, E.; Fornace, A.J. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16Ink4a-p19 Arf pathway. Nat. Genet. 2004, 36, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Esteva, F.J.; Sahin, A.A.; Smith, T.L.; Yang, Y.; Pusztai, L.; Nahta, R.; Buchholz, T.A.; Buzdar, A.U.; Hortobagyi, G.N.; Bacus, S.S. Prognostic Significance of Phosphorylated P38 Mitogen-Activated Protein Kinase and HER-2 Expression in Lymph Node-Positive Breast Carcinoma. Cancer 2004, 100, 499–506. [Google Scholar] [CrossRef]
- Benhar, M.; Dalyot, I.; Engelberg, D.; Levitzki, A. Enhanced ROS Production in Oncogenically Transformed Cells Potentiates c-Jun N-Terminal Kinase and p38 Mitogen-Activated Protein Kinase Activation and Sensitization to Genotoxic Stress. Mol. Cell. Biol. 2001, 21, 6913–6926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelberg, D. Stress-activated protein kinases—Tumor suppressors or tumor initiators? Semin. Cancer Biol. 2004, 14, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Bragado, P.; Armesilla, A.; Silva, A.; Porras, A. Apoptosis by cisplatin requires p53 mediated p38α MAPK activation through ROS generation. Apoptosis 2007, 12, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Parmar, S.; Katsoulidis, E.; Verma, A.; Li, Y.; Sassano, A.; Lal, L.; Majchrzak, B.; Ravandi, F.; Tallman, M.S.; Fish, E.N.; et al. Role of the p38 mitogen-activated protein kinase pathway in the generation of the effects of imatinib mesylate (STI571) in BCR-ABL-expressing cells. J. Biol. Chem. 2004, 279, 25345–25352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giafis, N.; Katsoulidis, E.; Sassano, A.; Tallman, M.S.; Higgins, L.S.; Nebreda, A.R.; Davis, R.J.; Platanias, L.C. Role of the p38 mitogen-activated protein kinase pathway in the generation of arsenic trioxide-dependent cellular responses. Cancer Res. 2006, 66, 6763–6771. [Google Scholar] [CrossRef] [Green Version]
- Cánovas, B.; Igea, A.; Sartori, A.A.; Gomis, R.R.; Paull, T.T.; Isoda, M.; Pérez-Montoyo, H.; Serra, V.; González-Suárez, E.; Stracker, T.H.; et al. Targeting p38α Increases DNA Damage, Chromosome Instability, and the Anti-tumoral Response to Taxanes in Breast Cancer Cells. Cancer Cell 2018, 33, 1094–1110.e8. [Google Scholar]
- Paillas, S.; Boissière, F.; Bibeau, F.; Denouel, A.; Mollevi, C.; Causse, A.; Denis, V.; Vezzio-Vié, N.; Marzi, L.; Cortijo, C.; et al. Targeting the p38 MAPK pathway inhibits irinotecan resistance in colon adenocarcinoma. Cancer Res. 2011, 71, 1041–1049. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Martínez, C.; Gelbert, L.M.; Lallena, M.J.; De Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Bioorganic Med. Chem. Lett. 2015, 25, 3420–3435. [Google Scholar] [CrossRef]
- Malínková, V.; Vylíčil, J.; Kryštof, V. Cyclin-dependent kinase inhibitors for cancer therapy: A patent review (2009–2014). Expert Opin. Pat. 2015, 25, 953–970. [Google Scholar] [CrossRef]
- Toogood, P.L.; Harvey, P.J.; Repine, J.T.; Sheehan, D.J.; VanderWel, S.N.; Zhou, H.; Keller, P.R.; McNamara, D.J.; Sherry, D.; Zhu, T.; et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J. Med. Chem. 2005, 48, 2388–2406. [Google Scholar] [CrossRef]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: In-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investig. New Drugs 2014, 32, 825–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Loo, A.; Chopra, R.; Caponigro, G.; Huang, A.; Vora, S.; Parasuraman, S.; Howard, S.; Keen, N.; Sellers, W.; et al. LEE011: An orally bioavailable, selective small molecule inhibitor of CDK4/6– Reactivating Rb in cancer. Mol. Cancer Ther. 2013, 12, 2528. [Google Scholar]
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol. 2018, 28, 911–925. [Google Scholar] [CrossRef]
- Gong, X.; Litchfield, L.M.; Webster, Y.; Chio, L.C.; Wong, S.S.; Stewart, T.R.; Dowless, M.; Dempsey, J.; Zeng, Y.; Torres, R.; et al. Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer Cell 2017, 32, 761–776.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2014, 16, 25–35. [Google Scholar] [CrossRef]
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Huang Bartlett, C.; Zhang, K.; et al. Palbociclib in Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.-A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Yardley, D.A. MONALEESA clinical program: A review of ribociclib use in different clinical settings. Futur. Oncol. 2019, 15, 2673–2686. [Google Scholar] [CrossRef] [Green Version]
- Hortobagyi, G.N. Ribociclib for the first-line treatment of advanced hormone receptor-positive breast cancer: A review of subgroup analyses from the MONALEESA-2 trial 11 Medical and Health Sciences 1112 Oncology and Carcinogenesis 11 Medical and Health Sciences 1103 Clinical Sciences. Breast Cancer Res. 2018, 20, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.-A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martín, M.; et al. Phase III Randomized Study of Ribociclib and Fulvestrant in Hormone Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer: MONALEESA-3. J. Clin. Oncol. 2018, 36, 2465–2472. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and safety of Abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non–small cell lung cancer, and other solid tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, J.W.; Mazieres, J.; Barlesi, F.; Dragnev, K.H.; Koczywas, M.; Göskel, T.; Cortot, A.B.; Girard, N.; Wesseler, C.; Bischoff, H.; et al. A Randomized Phase III Study of Abemaciclib Versus Erlotinib in Patients with Stage IV Non-small Cell Lung Cancer With a Detectable KRAS Mutation Who Failed Prior Platinum-Based Therapy: JUNIPER. Front. Oncol. 2020, 10, 2338. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.-C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef]
- Dickler, M.N.; Tolaney, S.M.; Rugo, H.S.; Cortes, J.; Dieras, V.; Patt, D.; Wildiers, H.; Hudis, C.A.; O’Shaughnessy, J.; Zamora, E.; et al. MONARCH 1, a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, n patients with refractory HR+/HER2- metastatic breast cancer. Clin. Cancer Res. 2017, 23, 5218–5224. [Google Scholar] [CrossRef] [Green Version]
- Sledge, G.W.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in combination with fulvestrant in women with HR+/HER2-advanced breast cancer who had progressed while receiving endocrine therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef]
- Hafner, M.; Mills, C.E.; Subramanian, K.; Chen, C.; Chung, M.; Boswell, S.A.; Everley, R.A.; Liu, C.; Walmsley, C.S.; Juric, D.; et al. Multiomics Profiling Establishes the Polypharmacology of FDA-Approved CDK4/6 Inhibitors and the Potential for Differential Clinical Activity. Cell Chem. Biol. 2019, 26, 1067–1080.e8. [Google Scholar] [CrossRef]
- Schoninger, S.F.; Blain, S.W. The ongoing search for biomarkers of CDK4/6 inhibitor responsiveness in breast cancer. Mol. Cancer Ther. 2020, 19, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; LoRusso, P.M.; DeMichele, A.; Abramson, V.G.; Courtney, R.; Randolph, S.S.; Shaik, M.N.; Wilner, K.D.; O’Dwyer, P.J.; Schwartz, G.K. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Bozhenko, V.K.; Kulinich, T.M.; Kudinova, E.A.; Boldyrev, A.; Solodkij, V.A. New targeted anti CDK4/6 peptide MM-D37K. J. Clin. Oncol. 2013, 31, e13545. [Google Scholar] [CrossRef]
- Bisi, J.E.; Sorrentino, J.A.; Roberts, P.J.; Tavares, F.X.; Strum, J.C. Preclinical Characterization of G1T28: A Novel CDK4/6 Inhibitor for Reduction of Chemotherapy-Induced Myelosuppression. Mol. Cancer Ther. 2016, 15, 783–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Roberts, P.J.; Sorrentino, J.A.; Bisi, J.E.; Storrie-White, H.; Tiessen, R.G.; Makhuli, K.M.; Wargin, W.A.; Tadema, H.; Van Hoogdalem, E.J.; et al. Transient CDK4/6 inhibition protects hematopoietic stem cells from chemotherapy-induced exhaustion. Sci. Transl. Med. 2017, 9, aal3986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, L.L.; Ferrarotto, R.; Andric, Z.G.; Beck, J.T.; Subramanian, J.; Radosavljevic, D.Z.; Zaric, B.; Hanna, W.T.; Aljumaily, R.; Owonikoko, T.K.; et al. Myelopreservation with Trilaciclib in Patients Receiving Topotecan for Small Cell Lung Cancer: Results from a Randomized, Double-Blind, Placebo-Controlled Phase II Study. Adv. Ther. Available online: https://link.springer.com/article/10.1007/s12325-020-01538-0 (accessed on 15 October 2020). [CrossRef]
- Weiss, J.M.; Csoszi, T.; Maglakelidze, M.; Hoyer, R.J.; Beck, J.T.; Domine Gomez, M.; Lowczak, A.; Aljumaily, R.; Rocha Lima, C.M.; Boccia, R.V.; et al. Myelopreservation with the CDK4/6 inhibitor trilaciclib in patients with small-cell lung cancer receiving first-line chemotherapy: A phase Ib/randomized phase II trial. Ann. Oncol. 2019, 30, 1613–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, F.; He, Y.; Fu, H.; Li, Y.; Bao, X.; Wang, Q.; Wang, Y.; Xie, C.; Lou, L. Preclinical characterization of SHR6390, a novel CDK 4/6 inhibitor, in vitro and in human tumor xenograft models. Cancer Sci. 2019, 110, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Q.; Yuan, J.; Wang, J.; Chen, Z.; Liu, Z.; Li, Z.; Lai, Y.; Gao, J.; Shen, L. CDK4/6 inhibitor-SHR6390 exerts potent antitumor activity in esophageal squamous cell carcinoma by inhibiting phosphorylated Rb and inducing G1 cell cycle arrest. J. Transl. Med. 2017, 15, 127. [Google Scholar] [CrossRef]
- Watts, C.K.W.; Sweeney, K.J.E.; Warlters, A.; Musgrove, E.A.; Sutherland, R.L. Antiestrogen regulation of cell cycle progression and cyclin D1 gene expression in MCF-7 human breast cancer cells. Breast Cancer Res. Treat. 1994, 31, 95–105. [Google Scholar] [CrossRef]
- Prall, O.W.J.; Sarcevic, B.; Musgrove, E.A.; Watts, C.K.W.; Sutherland, R.L. Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J. Biol. Chem. 1997, 272, 10882–10894. [Google Scholar] [CrossRef] [Green Version]
- Miller, T.W.; Balko, J.M.; Fox, E.M.; Ghazoui, Z.; Dunbier, A.; Anderson, H.; Dowsett, M.; Jiang, A.; Adam Smith, R.; Maira, S.M.; et al. ERα-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov. 2011, 1, 338–351. [Google Scholar] [CrossRef] [Green Version]
- Geoerger, B.; Bourdeaut, F.; DuBois, S.G.; Fischer, M.; Geller, J.I.; Gottardo, N.G.; Marabelle, A.; Pearson, A.D.J.; Modak, S.; Cash, T.; et al. A phase I study of the CDK4/6 inhibitor ribociclib (LEE011) in pediatric patients with malignant rhabdoid tumors, neuroblastoma, and other solid tumors. Clin. Cancer Res. 2017, 23, 2433–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, M.A.; Schwartz, G.K.; Louise Keohan, M.; D’Angelo, S.P.; Gounder, M.M.; Chi, P.; Antonescu, C.R.; Landa, J.; Qin, L.X.; Crago, A.M.; et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with cdk4 inhibitor palbociclib a phase 2 clinical trial. JAMA Oncol. 2016, 2, 937–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, J.P.; LaCasce, A.S.; Smith, M.R.; Noy, A.; Chirieac, L.R.; Rodig, S.J.; Yu, J.Q.; Vallabhajosula, S.; Schoder, H.; English, P.; et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood 2012, 119, 4597–4607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uras, I.Z.; Sexl, V.; Kollmann, K. CDK6 inhibition: A novel approach in AML management. Int. J. Mol. Sci. 2020, 21, 2528. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, J.; Blaser, B.W.; Duchemin, A.M.; Kusewitt, D.F.; Liu, T.; Caligiuri, M.A.; Briesewitz, R. Pharmacologic inhibition of CDK4/6: Mechanistic evidence for selective activity or acquired resistance in acute myeloid leukemia. Blood 2007, 110, 2075–2083. [Google Scholar] [CrossRef]
- Uras, I.Z.; Walter, G.J.; Scheicher, R.; Bellutti, F.; Prchal-Murphy, M.; Tigan, A.S.; Valent, P.; Heidel, F.H.; Kubicek, S.; Scholl, C.; et al. Palbociclib treatment of FLT3-ITD+ AML cells uncovers a kinase-dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood 2016, 127, 2890–2902. [Google Scholar] [CrossRef] [Green Version]
- Keegan, K.; Li, C.; Li, Z.; Ma, J.; Ragains, M.; Coberly, S.; Hollenback, D.; Eksterowicz, J.; Liang, L.; Weidner, M.; et al. Preclinical evaluation of AMG 925, a FLT3/CDK4 dual kinase inhibitor for treating acute myeloid leukemia. Mol. Cancer Ther. 2014, 13, 880–889. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, X.; Eksterowicz, J.; Gribble, M.W.; Alba, G.Q.; Ayres, M.; Carlson, T.J.; Chen, A.; Chen, X.; Cho, R.; et al. Discovery of AMG 925, a FLT3 and CDK4 dual kinase inhibitor with preferential affinity for the activated state of FLT3. J. Med. Chem. 2014, 57, 3430–3449. [Google Scholar] [CrossRef]
- Uras, I.Z.; Maurer, B.; Nivarthi, H.; Jodl, P.; Kollmann, K.; Prchal-Murphy, M.; Milosevic Feenstra, J.D.; Zojer, M.; Lagger, S.; Grausenburger, R.; et al. CDK6 coordinates JAK2V617F mutant MPN via NF-kB and apoptotic networks. Blood 2019, 133, 1677–1690. [Google Scholar] [CrossRef] [Green Version]
- De Dominici, M.; Porazzi, P.; Xiao, Y.; Chao, A.; Tang, H.-Y.; Kumar, G.; Fortina, P.; Spinelli, O.; Rambaldi, A.; Peterson, L.F.; et al. Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and independent effects by CDK6-specific PROTACs. Blood 2020, 135, 1560–1573. [Google Scholar] [CrossRef]
- Bellutti, F.; Tigan, A.-S.; Nebenfuehr, S.; Dolezal, M.; Zojer, M.; Grausenburger, R.; Hartenberger, S.; Kollmann, S.; Doma, E.; Prchal-Murphy, M.; et al. CDK6 Antagonizes p53-Induced Responses during Tumorigenesis. Cancer Discov. 2018, 8, 884–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, J.L.; McClendon, A.K.; Knudsen, E.S. Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J. Biol. Chem. 2012, 287, 29075–29087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvador-Barbero, B.; Álvarez-Fernández, M.; Zapatero-Solana, E.; El Bakkali, A.; del Menéndez, M.C.; López-Casas, P.P.; Di Domenico, T.; Xie, T.; VanArsdale, T.; Shields, D.J.; et al. CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer Cell 2020, 37, 340–353.e6. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.; Dustin, D.; Fuqua, S.A. Targeted therapy for breast cancer and molecular mechanisms of resistance to treatment. Curr. Opin. Pharm. 2016, 31, 97–103. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Goel, S. CDK4/6 Inhibition in Breast Cancer: Mechanisms of Response and Treatment Failure. Curr. Breast Cancer Rep. 2017, 9, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Comstock, C.E.S.; Augello, M.A.; Goodwin, J.F.; De Leeuw, R.; Schiewer, M.J.; Ostrander, W.F.; Burkhart, R.A.; McClendon, A.K.; McCue, P.A.; Trabulsi, E.J.; et al. Targeting cell cycle and hormone receptor pathways in cancer. Oncogene 2013, 32, 5481–5491. [Google Scholar] [CrossRef] [Green Version]
- Sharifi, M.N.; Anandan, A.; Grogan, P.; O’Regan, R.M. Therapy after cyclin-dependent kinase inhibition in metastatic hormone receptor-positive breast cancer: Resistance mechanisms and novel treatment strategies. Cancer 2020, 126, 3400–3416. [Google Scholar] [CrossRef]
- DeMichele, A.; Clark, A.S.; Tan, K.S.; Heitjan, D.F.; Gramlich, K.; Gallagher, M.; Lal, P.; Feldman, M.; Zhang, P.; Colameco, C.; et al. CDK 4/6 Inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: Phase II activity, safety, and predictive biomarker assessment. Clin. Cancer Res. 2015, 21, 995–1001. [Google Scholar] [CrossRef] [Green Version]
- Raspé, E.; Coulonval, K.; Pita, J.M.; Paternot, S.; Rothé, F.; Twyffels, L.; Brohée, S.; Craciun, L.; Larsimont, D.; Kruys, V.; et al. CDK 4 phosphorylation status and a linked gene expression profile predict sensitivity to palbociclib. Embo Mol. Med. 2017, 9, 1052–1066. [Google Scholar] [CrossRef]
- Finn, R.S.; Liu, Y.; Zhu, Z.; Martin, M.; Rugo, H.S.; Dieras, V.; Im, S.A.; Gelmon, K.A.; Harbeck, N.; Lu, D.R.; et al. Biomarker analyses of response to cyclin-dependent kinase 4/6 inhibition and endocrine therapy in women with treatment-naïve metastatic breast cancer. Clin. Cancer Res. 2020, 26, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017, 36, 2255–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, J.L.; Thangavel, C.; McClendon, A.K.; Reed, C.A.; Knudsen, E.S. Therapeutic CDK4/6 inhibition in breast cancer: Key mechanisms of response and failure. Oncogene 2010, 29, 4018–4032. [Google Scholar] [CrossRef] [Green Version]
- Malorni, L.; Piazza, S.; Ciani, Y.; Guarducci, C.; Bonechi, M.; Biagioni, C.; Hart, C.D.; Verardo, R.; Di Leo, A.; Migliaccio, I. A gene expression signature of retinoblastoma loss-of-function is a predictive biomarker of resistance to palbociclib in breast cancer cell lines and is prognostic in patients with ER positive early breast cancer. Oncotarget 2016, 7, 68012–68022. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Razavi, P.; Li, Q.; Toy, W.; Liu, B.; Ping, C.; Hsieh, W.; Sanchez-Vega, F.; Brown, D.N.; Da Cruz Paula, A.F.; et al. Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 2018, 34, 893–905.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alevizopoulos, K.; Vlach, J.; Hennecke, S.; Amati, B. Cyclin E and c-Myc promote cell proliferation in the presence of p16(INK4a) of hypophosphorylated retinoblastoma family proteins. Embo J. 1997, 16, 5322–5333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, P.; Tsiperson, V.; Gottesman, S.R.S.; Somma, J.; Blain, S.W. Dual inhibition of CDK4 and CDK2 via targeting p27 tyrosine phosphorylation induces a potent and durable response in breast cancer cells. Mol. Cancer Res. 2018, 16, 361–377. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef] [Green Version]
- Guarducci, C.; Bonechi, M.; Benelli, M.; Biagioni, C.; Boccalini, G.; Romagnoli, D.; Verardo, R.; Schiff, R.; Osborne, C.K.; De Angelis, C.; et al. Cyclin E1 and Rb modulation as common events at time of resistance to palbociclib in hormone receptor-positive breast cancer. Npj Breast Cancer 2018, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.C.; Liu, Y.; Zhu, Z.; Loi, S.; Colleoni, M.; Loibl, S.; DeMichele, A.; Harbeck, N.; André, F.; Bayar, M.A.M.; et al. Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor-positive metastatic breast cancer. J. Clin. Oncol. 2019, 37, 1169–1178. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, J.B.; Oliff, A.; Kohl, N.E. Farnesyltransferase inhibitors: Ras research yields a potential cancer therapeutic. Cell 1994, 77, 175–178. [Google Scholar] [CrossRef]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.K. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakih, M.; O’Neil, B.; Price, T.J.; Falchook, G.S.; Desai, J.; Kuo, J.; Govindan, R.; Rasmussen, E.; Morrow, P.K.H.; Ngang, J.; et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRAS G12C inhibitor, in advanced solid tumors. J. Clin. Oncol. 2019, 37, 3003. [Google Scholar] [CrossRef]
- Lyons, J.F.; Wilhelm, S.; Hibner, B.; Bollag, G. Discovery of a novel Raf kinase inhibitor. In Endocrine-Related Cancer; Bioscientifica: Bristol, UK, 2001; Volume 8, pp. 219–225. [Google Scholar]
- Lee, J.T.; McCubrey, J.A. BAY-43-9006 Bayer/Onyx. Curr. Opin. Investig. Drugs 2003, 4, 757–763. [Google Scholar] [PubMed]
- Wilhelm, S.M.; Carter, C.; Tang, L.Y.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [Green Version]
- Escudier, B.; Szczylik, C.; Eisen, T.; Stadler, W.M.; Schwartz, B.; Shan, M.; Bukowski, R.M. Randomized phase III trial of the Raf kinase and VEGFR inhibitor sorafenib (BAY 43–9006) in patients with advanced renal cell carcinoma (RCC). J. Clin. Oncol. 2005, 23, LBA4510. [Google Scholar] [CrossRef]
- Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; Testori, A.; Lorigan, P.C.; et al. Vemurafenib in patients with BRAFV600 mutation-positive metastatic melanoma: Final overall survival results of the randomized BRIM-3 study. Ann. Oncol. 2017, 28, 2581–2587. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, K.; Zhu, X.; Lin, G.; Song, F.; Zhao, Y.; Piao, Y.; Liu, J.; Cheng, W.; Bi, X.; et al. Encorafenib (LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAFV600E melanoma cells. Cancer Lett. 2016, 370, 332–344. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Cuyle, P.-J.A.R.; Huijberts, S.; Schellens, J.H.M.; Elez, E.; Fakih, M.; Montagut Viladot, C.; Peeters, M.; et al. Updated results of the BEACON CRC safety lead-in: Encorafenib (ENCO) + binimetinib (BINI) + cetuximab (CETUX) for BRAFV600E-mutant metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2019, 37, 688. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandal, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbiah, V.; Cabanillas, M.E.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Keam, B.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600–mutant anaplastic thyroid cancer. J. Clin. Oncol. 2018, 36, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planchard, D.; Kim, T.M.; Mazieres, J.; Quoix, E.; Riely, G.; Barlesi, F.; Souquet, P.J.; Smit, E.F.; Groen, H.J.M.; Kelly, R.J.; et al. Dabrafenib in patients with BRAFV600E-positive advanced non-small-cell lung cancer: A single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 642–650. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef]
- Robert, C.; Flaherty, K.T.; Hersey, P.; Nathan, P.D.; Garbe, C.; Milhem, M.M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. METRIC phase III study: Efficacy of trametinib (T), a potent and selective MEK inhibitor (MEKi), in progression-free survival (PFS) and overall survival (OS), compared with chemotherapy (C) in patients (pts) with BRAF V600E/K mutant advanced or metastatic melanoma (MM). J. Clin. Oncol. 2012, 30, LBA8509. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Girotti, M.R.; Pedersen, M.; Sanchez-Laorden, B.; Viros, A.; Turajlic, S.; Niculescu-Duvaz, D.; Zambon, A.; Sinclair, J.; Hayes, A.; Gore, M.; et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013, 3, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Moriceau, G.; Kong, X.; Lee, M.K.; Lee, H.; Koya, R.C.; Ng, C.; Chodon, T.; Scolyer, R.A.; Dahlman, K.B.; et al. Melanoma whole-exome sequencing identifies V600E B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 2012, 3, 724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trunzer, K.; Pavlick, A.C.; Schuchter, L.; Gonzalez, R.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; Kim, K.B.; Weber, J.S.; et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J. Clin. Oncol. 2013, 31, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Paraiso, K.H.T.; Fedorenko, I.V.; Cantini, L.P.; Munko, A.C.; Hall, M.; Sondak, V.K.; Messina, J.L.; Flaherty, K.T.; Smalley, K.S.M. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br. J. Cancer 2010, 102, 1724–1730. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Moffat, J.; Schaefer, G.; Chan, J.; Wang, X.; Orr, C.; Cheng, J.; Hunsaker, T.; Shao, L.; Wang, S.J.; et al. Combined MEK and ERK inhibition overcomes therapy-mediated pathway reactivation in RAS mutant tumors. PLoS ONE 2017, 12, e0185862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germann, U.A.; Furey, B.F.; Markland, W.; Hoover, R.R.; Aronov, A.M.; Roix, J.J.; Hale, M.; Boucher, D.M.; Sorrell, D.A.; Martinez-Botella, G.; et al. Targeting the MAPK signaling pathway in cancer: Promising preclinical activity with the novel selective ERK1/2 inhibitor BVD-523 (ulixertinib). Mol. Cancer Ther. 2017, 16, 2351–2363. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Jean Lee Wong, D.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Chin, H.; Lai, D.; Falchook, G. Extracellular signal-regulated kinase (ERK) inhibitors in oncology clinical trials. J. Immunother. Precis. Oncol. 2019, 2, 10. [Google Scholar] [CrossRef]
- Sunaga, N.; Miura, Y.; Tsukagoshi, Y.; Kasahara, N.; Masuda, T.; Sakurai, R.; Kaira, K.; Hisada, T. Dual inhibition of MEK and p38 impairs tumor growth in KRAS-mutated non-small cell lung cancer. Oncol. Lett. 2019, 17, 3569. [Google Scholar] [CrossRef] [Green Version]
- Vitos-Faleato, J.; Real, S.M.; Gutierrez-Prat, N.; Villanueva, A.; Llonch, E.; Drosten, M.; Barbacid, M.; Nebreda, A.R. Requirement for epithelial p38α in KRAS-driven lung tumor progression. Proc. Natl. Acad. Sci. USA 2020, 117, 2588–2596. [Google Scholar] [CrossRef]
- Pranteda, A.; Piastra, V.; Stramucci, L.; Fratantonio, D.; Bossi, G. The p38 mapk signaling activation in colorectal cancer upon therapeutic treatments. Int. J. Mol. Sci. 2020, 21, 2773. [Google Scholar] [CrossRef]
- Pedersen, I.M.; Buhl, A.M.; Klausen, P.; Geisler, C.H.; Jurlander, J. The chimeric anti-CD20 antibody rituximab induces apoptosis in B-cell chronic lymphocytic leukemia cells through a p38 mitogen activated protein-kinase-dependent mechanism. Blood 2002, 99, 1314–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, L.; Igea, A.; Canovas, B.; Dolado, I.; Nebreda, A.R. Inhibition of p38 MAPK sensitizes tumour cells to cisplatin-induced apoptosis mediated by reactive oxygen species and JNK. Embo Mol. Med. 2013, 5, 1759–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Ebisuya, M.; Ashida, F.; Okamoto, K.; Yonehara, S.; Nishida, E. Continuous ERK Activation Downregulates Antiproliferative Genes throughout G1 Phase to Allow Cell-Cycle Progression. Curr. Biol. 2006, 16, 1171–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.-H.; Gong, X.; Zhang, Y.; Van Horn, R.D.; Yin, T.; Huber, L.; Burke, T.F.; Manro, J.; Iversen, P.W.; Wu, W.; et al. RAF inhibitor LY3009120 sensitizes RAS or BRAF mutant cancer to CDK4/6 inhibition by abemaciclib via superior inhibition of phospho-RB and suppression of cyclin D1. Nat. Publ. Gr. 2018, 37, 821–832. [Google Scholar] [CrossRef]
- Yadav, V.; Burke, T.F.; Huber, L.; Van Horn, R.D.; Zhang, Y.; Buchanan, S.G.; Chan, E.M.; Starling, J.J.; Beckmann, R.P.; Peng, S. Bin The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Mol. Cancer 2014, 13, 2253–2263. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, B.; Gu, S.; Jiang, P.; Sahu, A.; Chen, C.H.; Han, T.; Shi, S.; Wang, X.; Traugh, N.; et al. CRISPR Screens Identify Essential Cell Growth Mediators in BRAF Inhibitor-resistant Melanoma. Genom. Proteom. Bioinform. 2020, 18, 26–40. [Google Scholar] [CrossRef]
- Ramsdale, R.; Jorissen, R.N.; Li, F.Z.; Al-Obaidi, S.; Ward, T.; Sheppard, K.E.; Bukczynska, P.E.; Young, R.J.; Boyle, S.E.; Shackleton, M.; et al. The transcription cofactor c-JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci. Signal. 2015, 8, ra82. [Google Scholar] [CrossRef]
- Kim, H.S.; Jung, M.; Kang, H.N.; Kim, H.; Park, C.W.; Kim, S.M.; Shin, S.J.; Kim, S.H.; Kim, S.G.; Kim, E.K.; et al. Oncogenic BRAF fusions in mucosal melanomas activate the MAPK pathway and are sensitive to MEK/PI3K inhibition or MEK/CDK4/6 inhibition. Oncogene 2017, 36, 3334–3345. [Google Scholar] [CrossRef]
- Kwong, L.N.; Costello, J.C.; Liu, H.; Jiang, S.; Helms, T.L.; Langsdorf, A.E.; Jakubosky, D.; Genovese, G.; Muller, F.L.; Jeong, J.H.; et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat. Med. 2012, 18, 1503–1510. [Google Scholar] [CrossRef] [Green Version]
- Sosman, J.A.; Kittaneh, M.; Lolkema, M.P.J.K.; Postow, M.A.; Schwartz, G.; Franklin, C.; Matano, A.; Bhansali, S.; Parasuraman, S.; Kim, K. A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS -mutant melanoma: Early encouraging clinical activity. J. Clin. Oncol. 2014, 32, 9009. [Google Scholar] [CrossRef]
- Lee, M.S.; Helms, T.L.; Feng, N.; Gay, J.; Chang, Q.E.; Tian, F.; Wu, J.Y.; Toniatti, C.; Heffernan, T.P.; Powis, G.; et al. Efficacy of the combination of MEK and CDK4/6 inhibitors in vitro and in vivo in KRAS mutant colorectal cancer models. Oncotarget 2016, 7, 39595–39608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Zhang, S.; Chen, X.; Zheng, X.; Yao, Y.; Lu, G.; Zhou, J. Palbociclib, a selective CDK4/6 inhibitor, enhances the effect of selumetinib in RAS-driven non-small cell lung cancer. Cancer Lett. 2017, 408, 130–137. [Google Scholar] [CrossRef]
- Guiley, K.Z.; Stevenson, J.W.; Lou, K.; Barkovich, K.J.; Kumarasamy, V.; Wijeratne, T.U.; Bunch, K.L.; Tripathi, S.; Knudsen, E.S.; Witkiewicz, A.K.; et al. P27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 2019, 366, eaaw2106. [Google Scholar] [CrossRef]
- Nebenfuehr, S.; Kollmann, K.; Sexl, V. The role of CDK6 in cancer. Int. J. Cancer 2020, ijc.33054. [Google Scholar] [CrossRef]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Daage, L.C.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, L.S.; Rader, J.A.; Raman, P.; Batra, V.; Russell, M.R.; Tsang, M.; Gagliardi, M.; Chen, L.; Martinez, D.; Li, Y.; et al. Preclinical therapeutic synergy of MEK1/2 and CDK4/6 inhibition in neuroblastoma. Clin. Cancer Res. 2017, 23, 1785–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yang, C.; Wan, H.; Zhang, G.; Feng, J.; Zhang, L.; Chen, X.; Zhong, D.; Lou, L.; Tao, W.; et al. Discovery and development of pyrotinib: A novel irreversible EGFR/HER2 dual tyrosine kinase inhibitor with favorable safety profiles for the treatment of breast cancer. Eur. J. Pharm. Sci. 2017, 110, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, Q.; Chen, S.; Zhu, W.; Fan, Y.; Wang, J.; Luo, Y.; Xing, P.; Lan, B.; Li, M.; et al. Phase I Study and Biomarker Analysis of Pyrotinib, a Novel Irreversible Pan-ErbB Receptor Tyrosine Kinase Inhibitor, in Patients With Human Epidermal Growth Factor Receptor 2–Positive Metastatic Breast Cancer. J. Clin. Oncol. 2017, 35, 3105–3112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Hong, R.; Kaping, L.; Xu, F.; Xia, W.; Qin, G.; Zheng, Q.; Lu, Q.; Zhai, Q.; Shi, Y.; et al. CDK4/6 inhibitor palbociclib enhances the effect of pyrotinib in HER2-positive breast cancer. Cancer Lett. 2019, 447, 130–140. [Google Scholar] [CrossRef]
- Santarius, T.; Shipley, J.; Brewer, D.; Stratton, M.R.; Cooper, C.S. A census of amplified and overexpressed human cancer genes. Nat. Rev. Cancer 2010, 10, 59–64. [Google Scholar] [CrossRef]
- Yu, Q.; Geng, Y.; Sicinski, P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001, 411, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, R.; McNair, C.; Schiewer, M.J.; Neupane, N.P.; Brand, L.J.; Augello, M.A.; Li, Z.; Cheng, L.C.; Yoshida, A.; Courtney, S.M.; et al. MAPK Reliance via acquired CDK4/6 inhibitor resistance in cancer. Clin. Cancer Res. 2018, 24, 4201–4214. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicaltrials.gov Identifier. | Phase | Treatment | Drug Target | Target Disease | Primary Results |
|---|---|---|---|---|---|
| NCT03041311 | II | trilaciclib plus etoposide/carboplatin/atezolizumab | CDK4/6 plus chemotherapy/immune therapy | small cell lung cancer | N/A |
| NCT02514447 | Ib/IIa | trilaciclib plus topotecan | CDK4/6 plus chemotherapy | small cell lung cancer | better tolerability of chemotherapy |
| NCT02499770 | Ib/IIa | trilaciclib plus etoposide/carboplatin | CDK4/6 plus chemotherapy | small cell lung cancer | better tolerability of chemotherapy |
| NCT02978716 | II | trilaciclib plus gemcitabine plus carboplatin | CDK4/6 plus chemotherapy | hormone receptor negative breast cancer | N/A |
| NCT03480256 | I | SHR6390 plus pyrotinib | CDK4/6 plus ErbB | ErbB2 positive gastric cancer | partial response |
| NCT04095390 | II | SHR6390 plus pyrotinib plus letrozole/capecitabine | CDK4/6 plus ErbB plus endocrine therapy/chemotherapy | ErbB2 positive breast cancer | N/A |
| NCT03772353 | Ib/II | SHR6390 plus pyrotinib plus letrozole | CDK4/6 plus ErbB plus endocrine therapy | hormone receptor positive, ErbB2 positive breast cancer | N/A |
| NCT03481998 | Ib/II | SHR6390 plus letrozole/anastrozole/fulvestrant | CDK4/6 plus endocrine therapy | hormone receptor positive, ErbB2 negative breast cancer | N/A |
| NCT03966898 | III | SHR6390 plus letrozole/anastrozole | CDK4/6 plus endocrine therapy | hormone receptor positive, ErbB2 negative breast cancer | N/A |
| Raf Inhibitor | MEK Inhibitor | Target Disease | References |
|---|---|---|---|
| dabrafenib | trametinib | melanoma, thyroid cancer, non-small cell lung cancer | [133,134,135] |
| vemurafenib | cobimetinib | melanoma | [136] |
| encorafenib | binimetinib | melanoma, metastatic colorectal cancer with BRAFV600E mutations | [132,137] |
| trametinib | advanced melanoma with BRAFV600E and BRAFV600K mutations | [138] |
| Clinicaltrials.gov Identifier | Phase | Treatment | Drug Target | Target Disease | Primary Results |
|---|---|---|---|---|---|
| NCT03600883 | I/II | AMG 510 | KRAS G12C | advanced solid tumors with KRAS p.G12C mutation | N/A |
| NCT01875705 | I | GDC-0994 | ERK | advanced or metastatic solid tumors | partial response |
| NCT02457793 | I | GDC-0994 plus cobimetinib | ERK plus MEK | advanced or metastatic solid tumors | overlapping adverse events |
| NCT01781429 | I | BVD-523 (ulixertinib) | ERK | advanced solid tumors | partial response |
| NCT02296242 | I/II | BVD-523 (ulixertinib) | ERK | AML, MDS | N/A |
| NCT04198818 | I/II | HH2710 | ERK | advanced solid tumors | N/A |
| NCT04081259 | I | LY3214996 | ERK | AML | N/A |
| NCT01393990 | I | LY2228820 (ralimetinib) monotherapy or LY2228820 plus midazolam/tamoxifen | p38/sedative/endocrine therapy | advanced tumors | N/A |
| NCT01663857 | Ib/II | LY2228820 (ralimetinib) plus gemcitabine plus carboplatin | p38 plus chemotherapy | recurrent platinum-sensitive ovarian cancer | improvement of progression free survival |
| NCT02860780 | I | LY2228820 (ralimetinib) plus prexasertib | p38 plus CHK1 | advanced or metastatic tumors | N/A |
| NCT00095680 | II | SCIO-469 monotherapy or SCIO-469 plus bortezomib | p38/proteasome | multiple myeloma | N/A |
| NCT00113893 | II | SCIO-469 | p38 | MDS, hematologic diseases | N/A |
| Clinicaltrials.gov Identifier | Phase | Treatment | Drug Target | Target Disease | Primary Results |
|---|---|---|---|---|---|
| NCT02159066 | II | LGX818 (encorafenib) plus binimetinib plus ribociclib | BRAF plus MEK1/2 plus CDK4/6 | BRAF-mutated melanoma | N/A |
| NCT01543698 | Ib/II | LGX818 (encorafenib) plus binimetinib plus ribociclib | BRAF plus MEK1/2 plus CDK4/6 | tumors with BRAFV600 mutations | N/A |
| NCT03981614 | II | binimetinib plus palbociclib | MEK1/2 plus CDK4/6 | advanced stage colorectal carcinoma with NRAS or KRAS mutations | N/A |
| NCT03170206 | I/II | binimetinib plus palbociclib | MEK1/2 plus CDK4/6 | advanced stage NSCLC with KRAS mutations | N/A |
| NCT02780128 | I | ceritinib plus trametinib plus ribociclib | ALK plus MEK1/2 plus CDK4/6 | relapsed neuroblastoma with ALK or MAPK mutations | N/A |
| NCT03454035 | I | BVD-523 (ulixertinib) plus palbociclib | ERK plus CDK4/6 | pancreatic cancer and other solid tumors | N/A |
| NCT02065063 | I/II | palbociclib plus trametinib | CDK4/6 plus MEK1/2 | solid tumors | N/A |
| NCT03132454 | I | palbociclib plus sorafenib/decitabine/dexamethasone | CDK4/6 plus Raf-1/chemotherapy | recurrent or refractory leukemia | N/A |
| NCT01781572 | Ib/II | ribociclib plus binimetinib | CDK4/6 plus MEK1/2 | advanced NRAS-mutant melanoma | partial response |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheiblecker, L.; Kollmann, K.; Sexl, V. CDK4/6 and MAPK—Crosstalk as Opportunity for Cancer Treatment. Pharmaceuticals 2020, 13, 418. https://doi.org/10.3390/ph13120418
Scheiblecker L, Kollmann K, Sexl V. CDK4/6 and MAPK—Crosstalk as Opportunity for Cancer Treatment. Pharmaceuticals. 2020; 13(12):418. https://doi.org/10.3390/ph13120418
Chicago/Turabian StyleScheiblecker, Lisa, Karoline Kollmann, and Veronika Sexl. 2020. "CDK4/6 and MAPK—Crosstalk as Opportunity for Cancer Treatment" Pharmaceuticals 13, no. 12: 418. https://doi.org/10.3390/ph13120418
APA StyleScheiblecker, L., Kollmann, K., & Sexl, V. (2020). CDK4/6 and MAPK—Crosstalk as Opportunity for Cancer Treatment. Pharmaceuticals, 13(12), 418. https://doi.org/10.3390/ph13120418