Anti-Tick-Borne Encephalitis Virus Activity of Novel Uridine Glycoconjugates Containing Amide or/and 1,2,3-Triazole Moiety in the Linker Structure

 , , ,
, , ,  and
and
Abstract
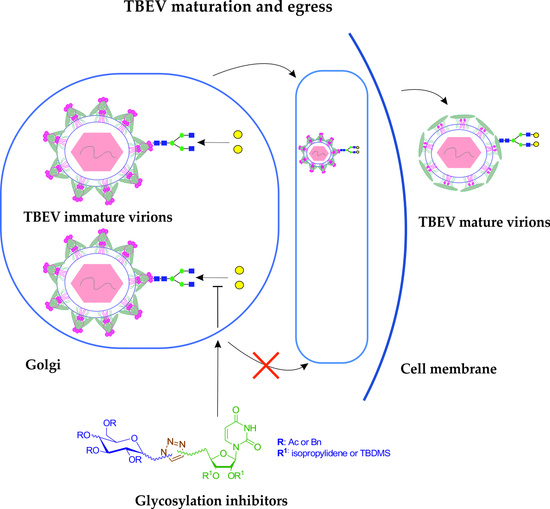
:
1. Introduction
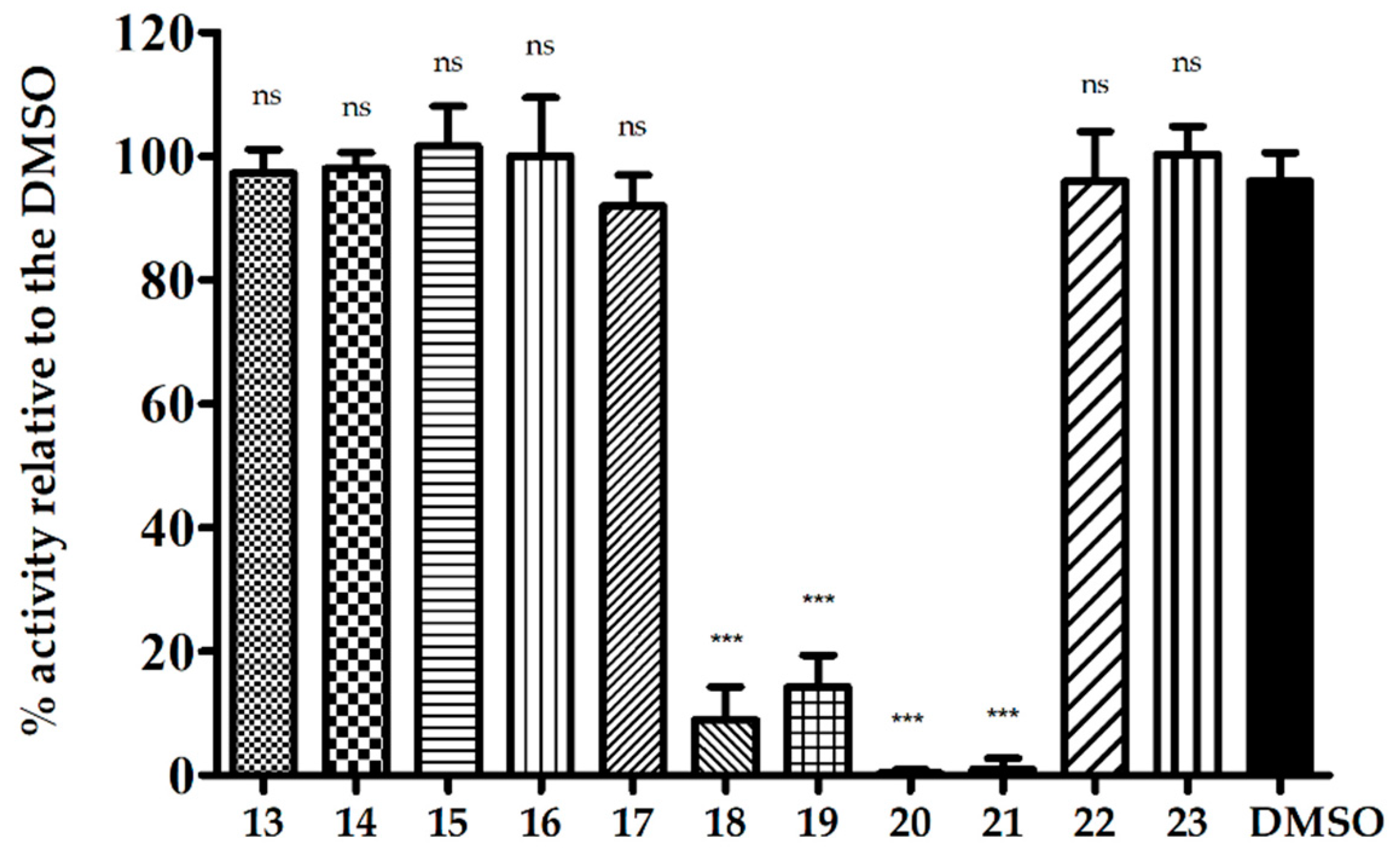
2. Results
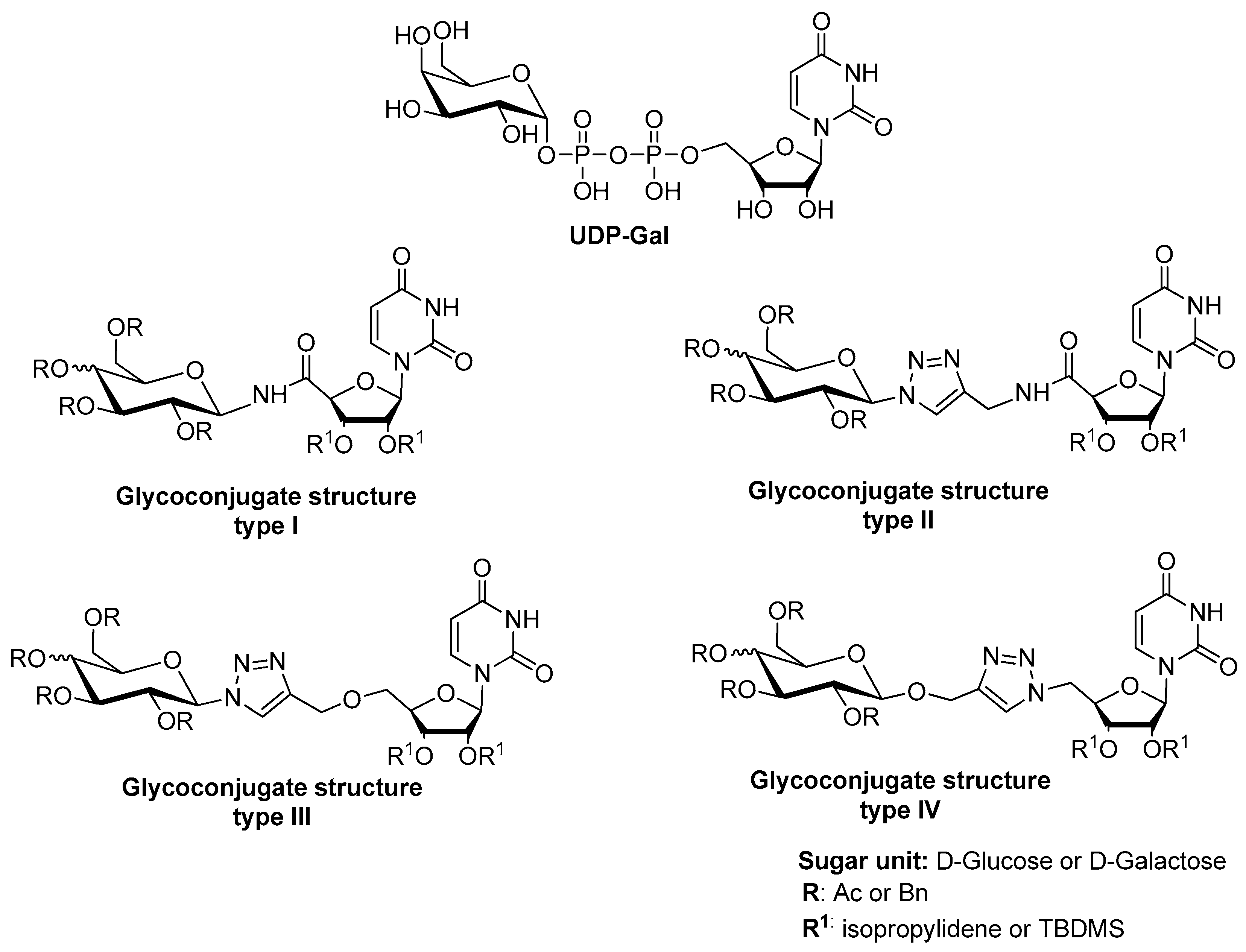
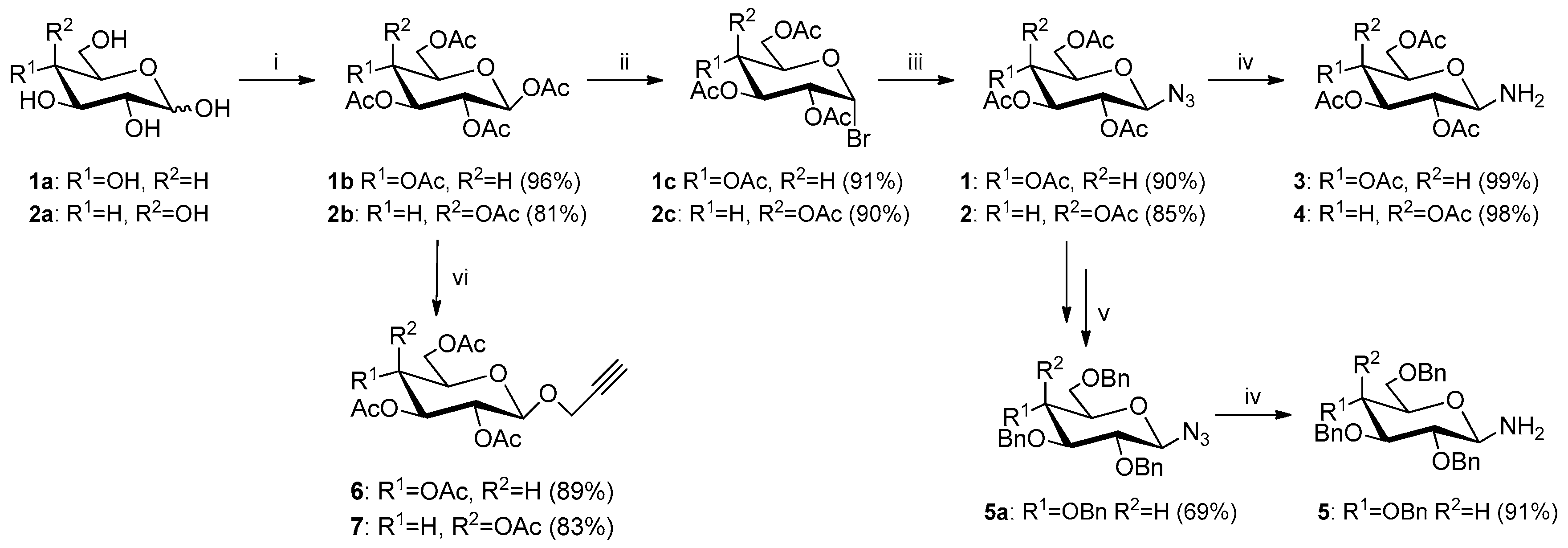
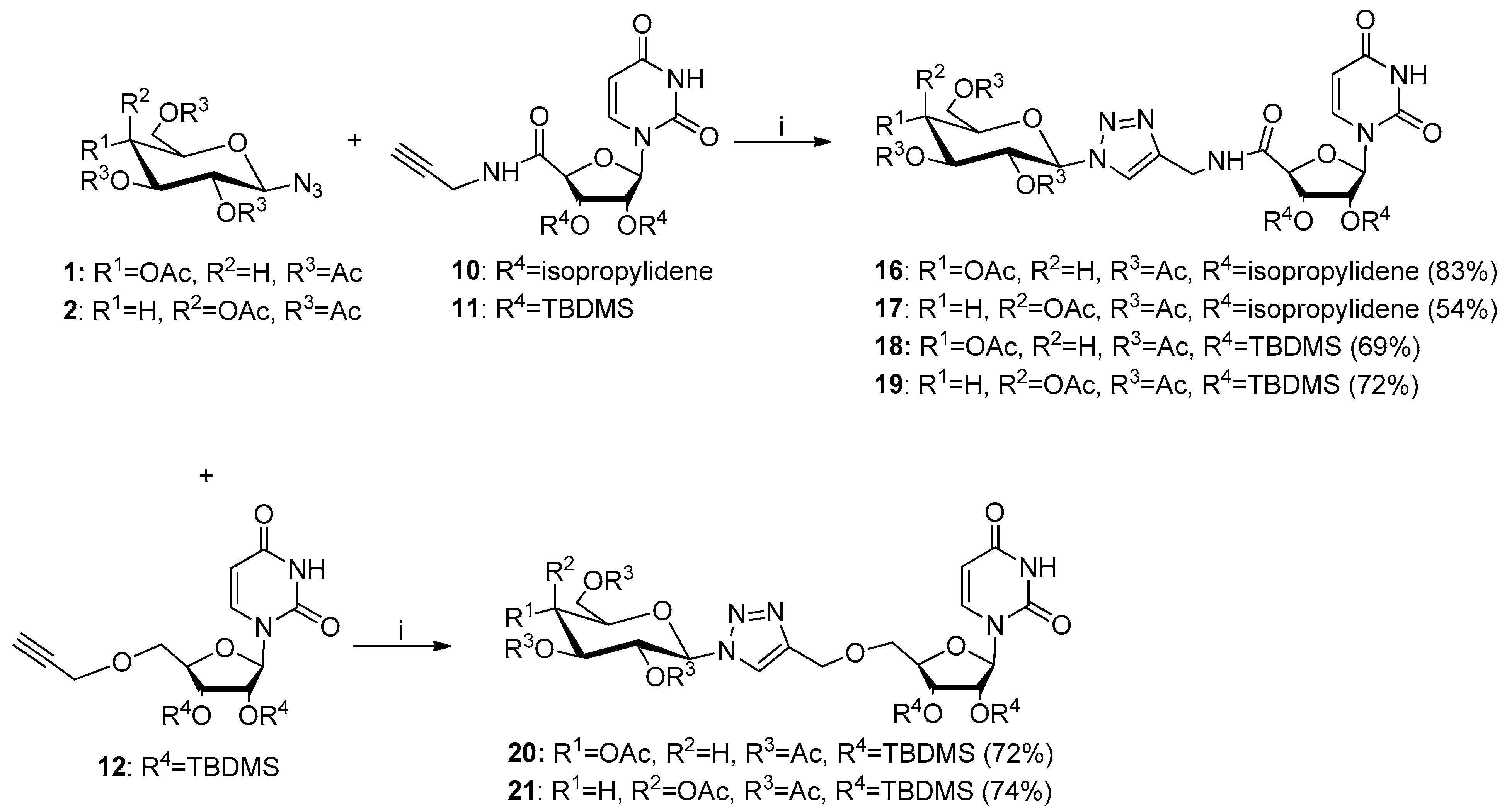
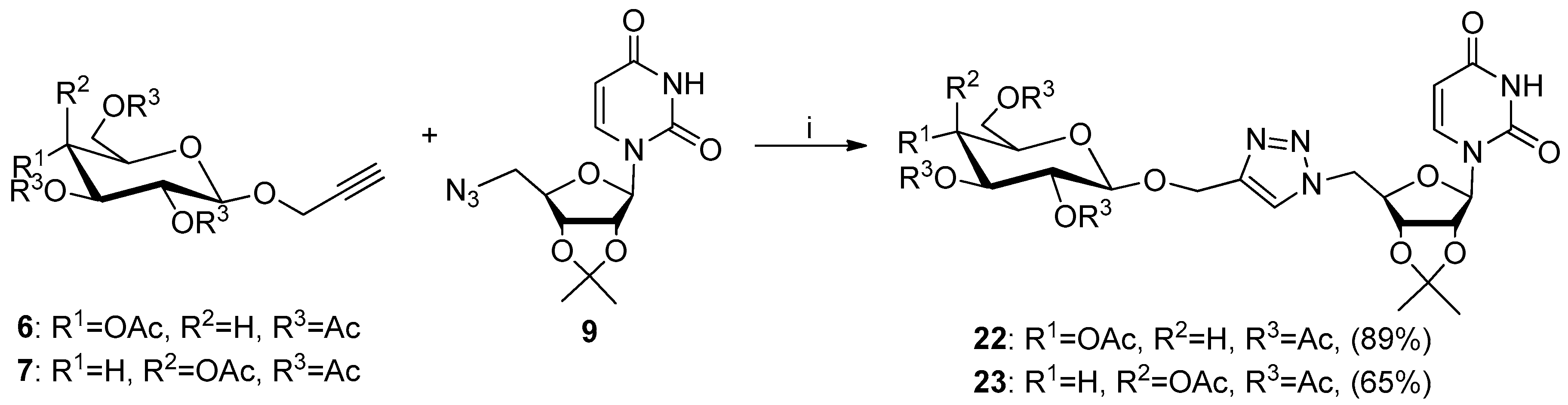
2.1. Synthesis of Glycoconjugates 13–23
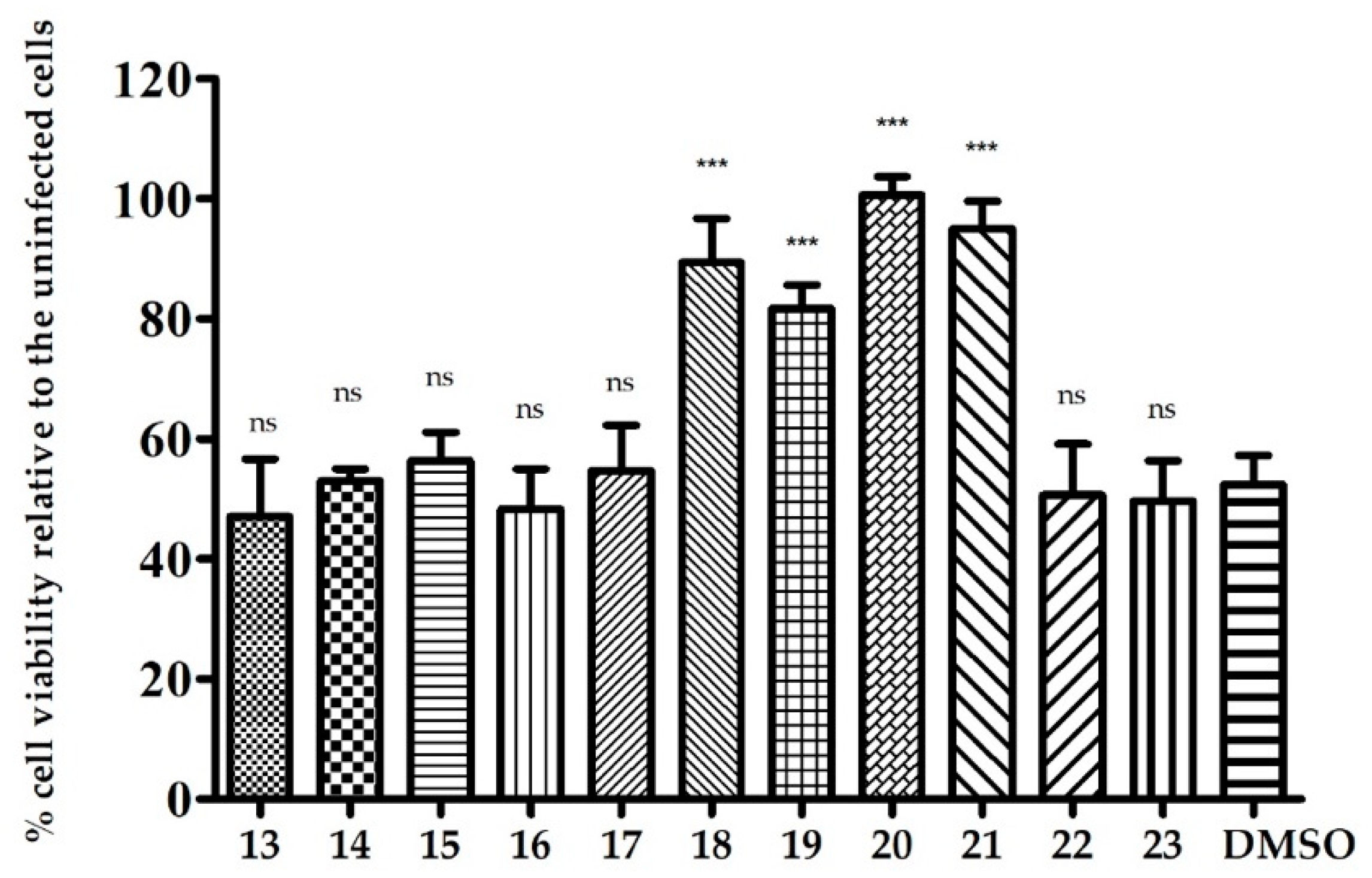
2.2. Biological Evaluation
2.2.1. Antiviral Activity of Uridine Glycoconjugates Against Tick-Borne Encephalitis Virus
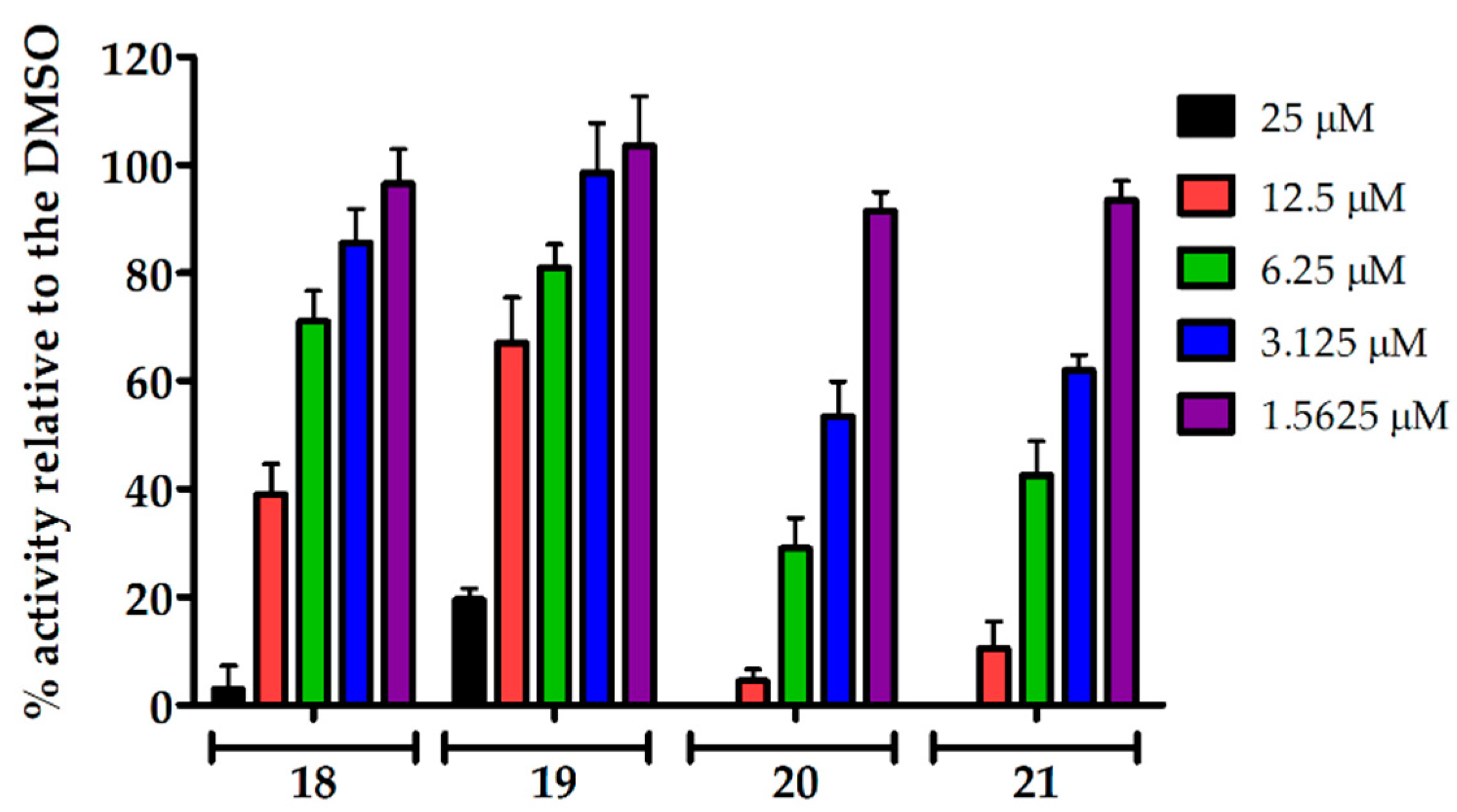
2.2.2. Dose-Response Activity of Uridine Glycoconjugates Against Tick-Borne Encephalitis Virus
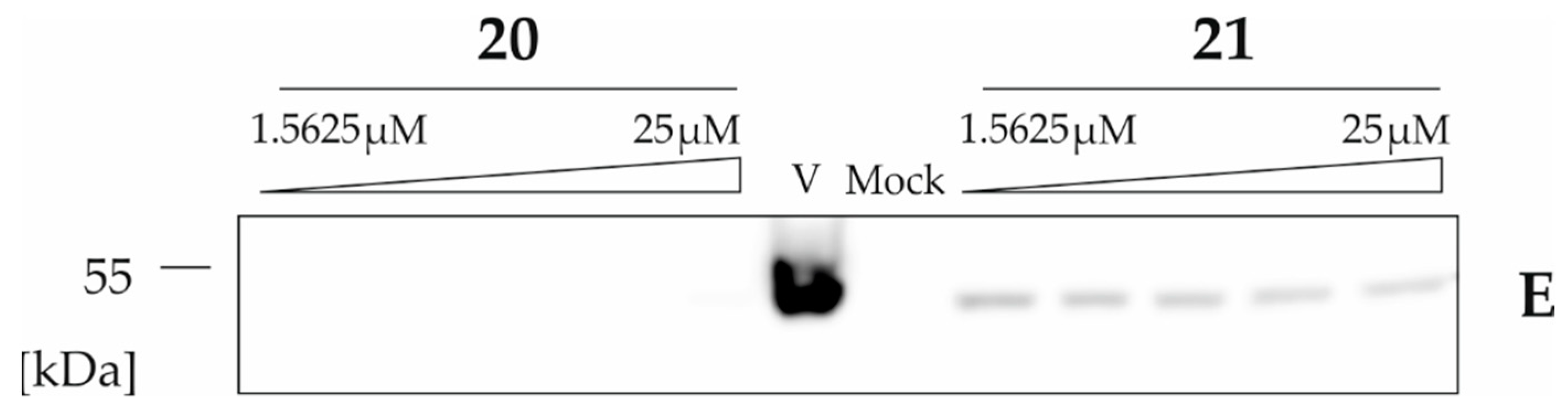
2.2.3. The Activity of Uridine Glycoconjugates on Protein Synthesis
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. Chemistry
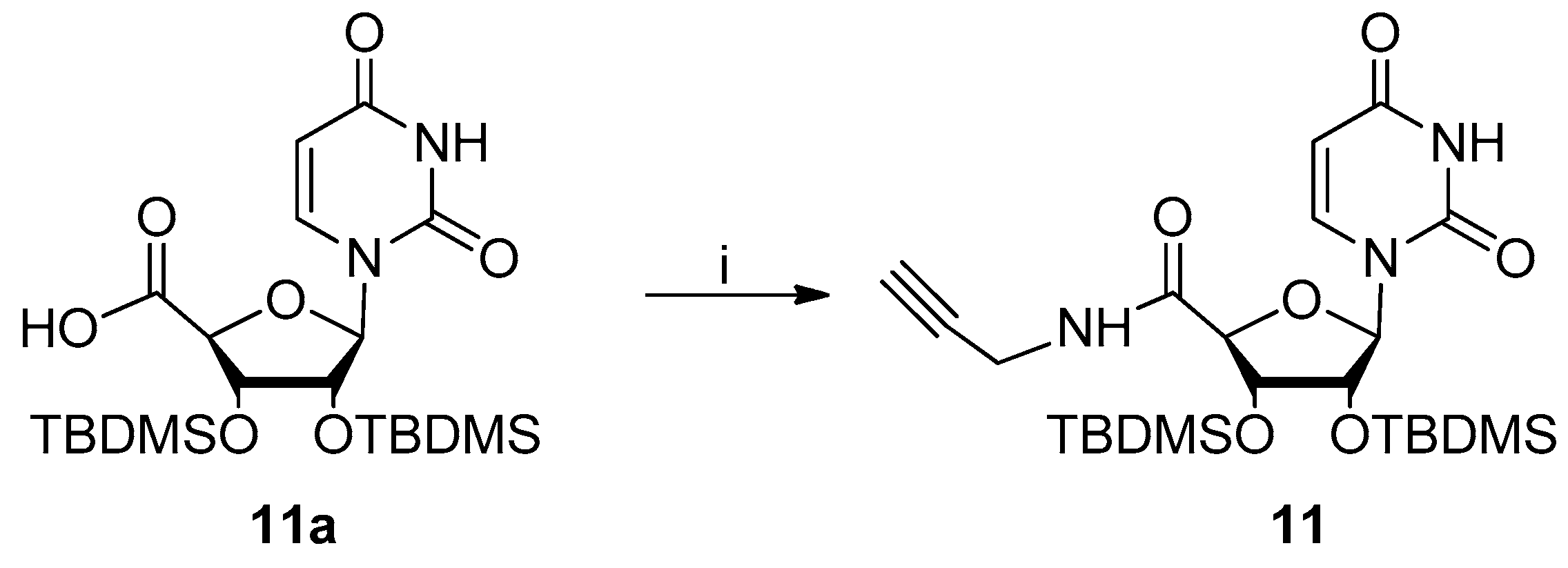
4.2.1. Procedure for Synthesis of N-Propargyl-2′,3′-O-Tert-Butyldimethylsilyluridine-5′-Amide 11
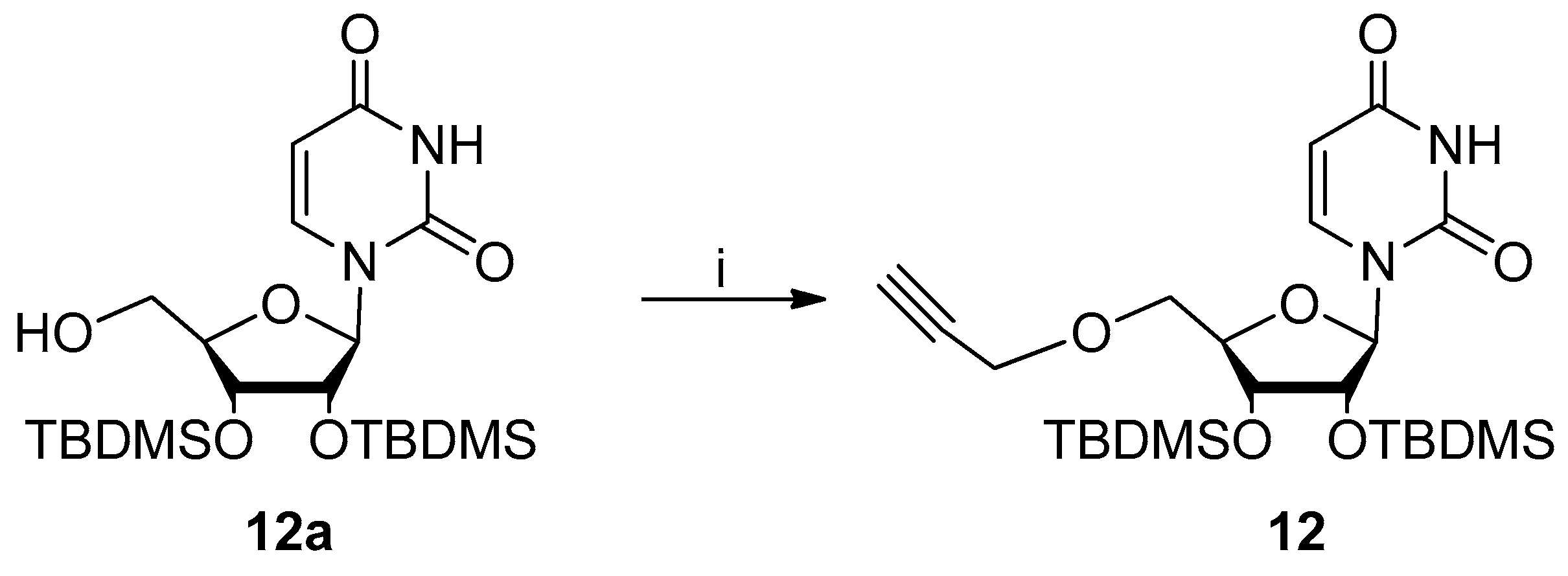
4.2.2. Procedure for Synthesis Of 2′,3′-Di-O-Tert-Butyldimethylsilyl-5′-O-Propargyl-Uridine 12
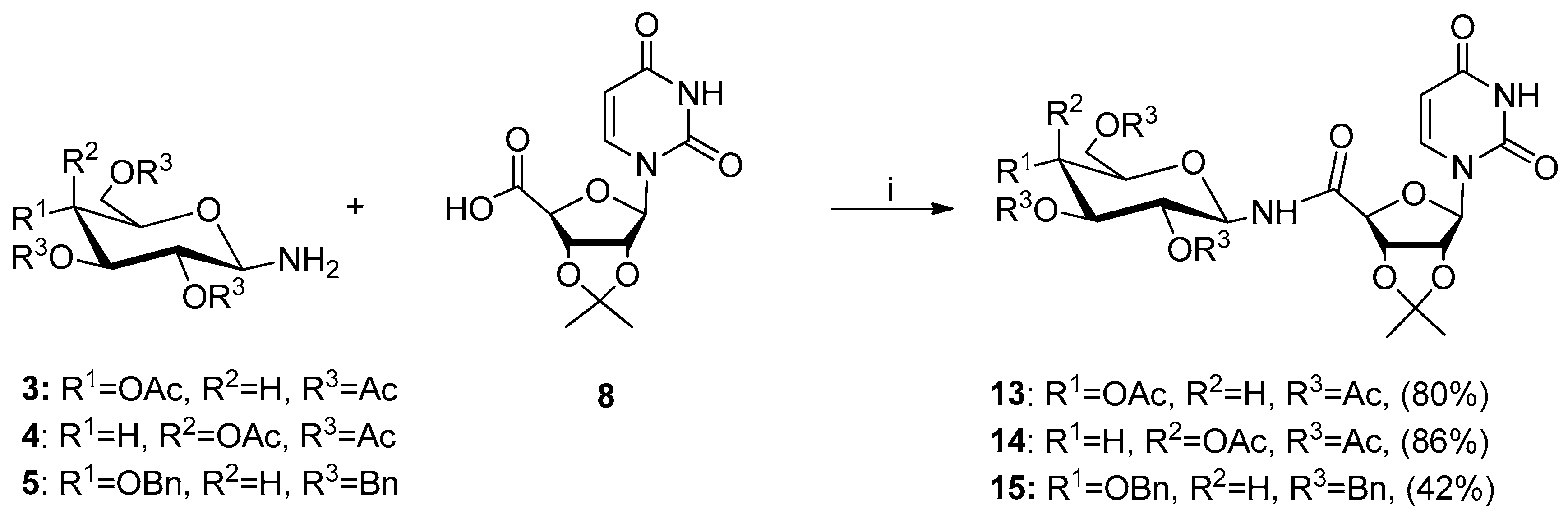
4.2.3. General Procedure for Synthesis of Glycoconjugates 13–15 By Amide Bond Formation
4.2.4. General Procedure for Synthesis of Glycoconjugates 16–23 by CuAAC Reaction
4.3. Biological Evaluation of Compounds Activity
4.3.1. Antiviral Compounds
4.3.2. Cells and Viruses
4.3.3. Cell Viability Assay
4.3.4. CPE Inhibition Assay
4.3.5. Plaque Assay
4.3.6. Plaque Reduction Assay
4.3.7. Dose-Response Antiviral Activity of Tested Compounds
4.3.8. Western Blot Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pierson, T.C.; Diamond, M.S. The continued threat of emerging flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Diamond, M.S. The emergence of Zika virus and its new clinical syndromes. Nature 2018, 560, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Chancey, C.; Grinev, A.; Volkova, E.; Rios, M. The global ecology and epidemiology of west Nile virus. Biomed. Res. Int. 2015, 2015, 376230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faria, N.R.; Kraemer, M.U.G.; Hill, S.C.; Goes de Jesus, J.; Aguiar, R.S.; Iani, F.C.M.; Xavier, J.; Quick, J.; du Plessis, L.; Dellicour, S.; et al. Genomic and epidemiological monitoring of yellow fever virus transmission potential. Science 2018, 361, 894–899. [Google Scholar] [CrossRef] [Green Version]
- Ruzek, D.; Avšič Županc, T.; Borde, J.; Chrdle, A.; Eyer, L.; Karganova, G.; Kholodilov, I.; Knap, N.; Kozlovskaya, L.; Matveev, A.; et al. Tick-borne encephalitis in Europe and Russia: Review of pathogenesis, clinical features, therapy, and vaccines. Antiviral Res. 2019, 164, 23–51. [Google Scholar] [CrossRef]
- Tick-Borne Encephalitis—Annual Epidemiological Report for 2018. Available online: https://www.ecdc.europa.eu/en/publications-data/tick-borne-encephalitis-annual-epidemiological-report-2018 (accessed on 17 September 2020).
- Dai, X.; Shang, G.; Lu, S.; Yang, J.; Xu, J. A new subtype of eastern tick-borne encephalitis virus discovered in Qinghai-Tibet Plateau, China. Emerg. Microbes Infect. 2018, 7, 74. [Google Scholar] [CrossRef]
- Kovalev, S.Y.; Mukhacheva, T.A. Reconsidering the classification of tick-borne encephalitis virus within the Siberian subtype gives new insights into its evolutionary history. Infect. Genet. Evol. 2017, 55, 159–165. [Google Scholar] [CrossRef]
- Růžek, D.; Dobler, G.; Mantke, O.D. Tick-borne encephalitis: Pathogenesis and clinical implications. Travel Med. Infect. Dis. 2010, 8, 223–232. [Google Scholar] [CrossRef]
- Barrett, P.N.; Portsmouth, D.; Ehrlich, H.J. 34—Tick-borne encephalitis virus vaccines. In Vaccines, 6th ed.; Plotkin, S.A., Orenstein, W.A., Offit, P.A., Eds.; W.B. Saunders: London, UK, 2013; pp. 773–788. [Google Scholar]
- Eyer, L.; Valdés, J.J.; Gil, V.A.; Nencka, R.; Hřebabecký, H.; Šála, M.; Salát, J.; Černý, J.; Palus, M.; De Clercq, E.; et al. Nucleoside inhibitors of tick-borne encephalitis virus. Antimicrob. Agents Chemother. 2015, 59, 5483–5493. [Google Scholar] [CrossRef] [Green Version]
- Lenz, N.; Engler, O.; Grandgirard, D.; Leib, S.L.; Ackermann-Gäumann, R. Evaluation of antivirals against tick-borne encephalitis virus in organotypic brain slices of rat cerebellum. PLoS ONE 2018, 13, e0205294. [Google Scholar] [CrossRef]
- Eyer, L.; Šmídková, M.; Nencka, R.; Neča, J.; Kastl, T.; Palus, M.; De Clercq, E.; Růžek, D. Structure-activity relationships of nucleoside analogues for inhibition of tick-borne encephalitis virus. Antiviral Res. 2016, 133, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Osolodkin, D.I.; Kozlovskaya, L.I.; Dueva, E.V.; Dotsenko, V.V.; Rogova, Y.V.; Frolov, K.A.; Krivokolysko, S.G.; Romanova, E.G.; Morozov, A.S.; Karganova, G.G.; et al. Inhibitors of tick-borne flavivirus reproduction from structure-based virtual screening. ACS Medicinal Chem. Lett. 2013, 4, 869–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krol, E.; Brzuska, G.; Szewczyk, B. Production and biomedical application of flavivirus-like particles. Trends Biotechnol. 2019, 0. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinz, F.X.; Allison, S.L. Flavivirus structure and membrane fusion. Adv. Virus Res. 2003, 59, 63–97. [Google Scholar] [CrossRef]
- Yoshii, K.; Yanagihara, N.; Ishizuka, M.; Sakai, M.; Kariwa, H. N-linked glycan in tick-borne encephalitis virus envelope protein affects viral secretion in mammalian cells, but not in tick cells. J. Gen. Virol. 2013, 94, 2249–2258. [Google Scholar] [CrossRef]
- Pastuch-Gawolek, G.; Chaubey, B.; Szewczyk, B.; Krol, E. Novel thioglycosyl analogs of glycosyltransferase substrates as antiviral compounds against classical swine fever virus and hepatitis C virus. Eur. J. Med. Chem. 2017, 137, 247–262. [Google Scholar] [CrossRef]
- Krol, E.; Wandzik, I.; Brzuska, G.; Eyer, L.; Růžek, D.; Szewczyk, B. Antiviral activity of uridine derivatives of 2-deoxy sugars against tick-borne encephalitis Virus. Molecules 2019, 24, 1129. [Google Scholar] [CrossRef] [Green Version]
- Krol, E.; Pastuch-Gawolek, G.; Chaubey, B.; Brzuska, G.; Erfurt, K.; Szewczyk, B. Novel uridine glycoconjugates, derivatives of 4-aminophenyl 1-thioglycosides, as potential antiviral compounds. Molecules 2018, 23, 1435. [Google Scholar] [CrossRef] [Green Version]
- Krol, E.; Wandzik, I.; Gromadzka, B.; Nidzworski, D.; Rychlowska, M.; Matlacz, M.; Tyborowska, J.; Szewczyk, B. Anti-influenza A virus activity of uridine derivatives of 2-deoxy sugars. Antiviral Res. 2013, 100, 90–97. [Google Scholar] [CrossRef]
- Rajaram, H.; Palanivelu, M.K.; Arumugam, T.V.; Rao, V.M.; Shaw, P.N.; McGeary, R.P.; Ross, B.P. ‘Click’ assembly of glycoclusters and discovery of a trehalose analogue that retards Aβ40 aggregation and inhibits Aβ40-induced neurotoxicity. Bioorg. Med. Chem. Lett. 2014, 24, 4523–4528. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk, M.; Pastuch-Gawolek, G.; Mrozek-Wilczkiewicz, A.; Kuczak, M.; Skonieczna, M.; Musiol, R. Synthesis of 8-hydroxyquinoline glycoconjugates and preliminary assay of their β1,4-GalT inhibitory and anti-cancer properties. Bioorg. Chem. 2019, 84, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Pastuch-Gawolek, G.; Plesniak, M.; Komor, R.; Byczek-Wyrostek, A.; Erfurt, K.; Szeja, W. Synthesis and preliminary biological assay of uridine glycoconjugate derivatives containing amide and/or 1,2,3-triazole linkers. Bioorg. Chem. 2017, 72, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Xavier, N.M.; Lucas, S.D.; Jorda, R.; Schwarz, S.; Loesche, A.; Csuk, R.; Oliveira, M.C. Synthesis and Evaluation of the Biological Profile of Novel Analogues of Nucleosides and of Potential Mimetics of Sugar Phosphates and Nucleotides. Synlett. 2015, 26, 2663–2672. [Google Scholar] [CrossRef]
- Rowan, A.S.; Nicely, N.I.; Cochrane, N.; Wlassoff, W.A.; Claiborne, A.; Hamilton, C.J. Nucleoside triphosphate mimicry: A sugar triazolyl nucleoside as an ATP-competitive inhibitor of B. anthracis pantothenate kinase. Org. Biomol. Chem. 2009, 7, 4029–4036. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M. Organic carbamates in drug design and medicinal chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.-R.; Qiao, W.-H.; Zhang, S.-M.; Qu, J.-P.; Liu, D.-L. Synthesis and characterization of a new cationic galactolipid with carbamate for gene delivery. Tenside Surfactants Deterg. 2010, 47, 294–299. [Google Scholar] [CrossRef]
- Shin, I.; Jung, H.; Lee, M. Chemoselective ligation of maleimidosugars to peptides/protein for the preparation of neoglycopeptides/neoglycoprotein. Tetrahedron Lett. 2001, 42, 1325–1328. [Google Scholar] [CrossRef]
- Ghirardello, M.; Ledru, H.; Sau, A.; Galan, M.C. Chemo-selective Rh-catalysed hydrogenation of azides into amines. Carbohydr. Res. 2020, 489, 107948. [Google Scholar] [CrossRef]
- Pastuch-Gawołek, G.; Malarz, K.; Mrozek-Wilczkiewicz, A.; Musioł, M.; Serda, M.; Czaplinska, B.; Musiol, R. Small molecule glycoconjugates with anticancer activity. Eur. J. Med. Chem. 2016, 112, 130–144. [Google Scholar] [CrossRef]
- Mereyala, H.B.; Gurrala, S.R. A highly diastereoselective, practical synthesis of allyl, propargyl 2,3,4,6-tetra-O-acetyl-β-d-gluco, β-d-galactopyranosides and allyl, propargyl heptaacetyl-β-d-lactosides. Carbohydr. Res. 1998, 307, 351–354. [Google Scholar] [CrossRef]
- Sheehan, J.C.; Hess, G.P. A new method of forming peptide bonds. J. Am. Chem. Soc. 1955, 77, 1067–1068. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Liang, L.; Astruc, D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Krol, E.; Wandzik, I.; Szeja, W.; Grynkiewicz, G.; Szewczyk, B. In vitro antiviral activity of some uridine derivatives of 2-deoxy sugars against classical swine fever virus. Antiviral Res. 2010, 86, 154–162. [Google Scholar] [CrossRef]
- Krol, E.; Wandzik, I.; Krejmer-Rabalska, M.; Szewczyk, B. Biological evaluation of uridine derivatives of 2-deoxy sugars as potential antiviral compounds against influenza A virus. Int. J. Mol. Sci. 2017, 18, 1700. [Google Scholar] [CrossRef] [Green Version]
- Routhu, N.K.; Lehoux, S.D.; Rouse, E.A.; Bidokhti, M.R.M.; Giron, L.B.; Anzurez, A.; Reid, S.P.; Abdel-Mohsen, M.; Cummings, R.D.; Byrareddy, S.N. Glycosylation of Zika virus is important in host–virus interaction and pathogenic potential. Int. J. Mol. Sci. 2019, 20, 5206. [Google Scholar] [CrossRef] [Green Version]
- Martina, B.E.E.; Koraka, P.; van den Doel, P.; Rimmelzwaan, G.F.; Haagmans, B.L.; Osterhaus, A.D.M.E. DC-SIGN enhances infection of cells with glycosylated West Nile virus in vitro and virus replication in human dendritic cells induces production of IFN-alpha and TNF-alpha. Virus Res. 2008, 135, 64–71. [Google Scholar] [CrossRef]
- Goo, L.; DeMaso, C.R.; Pelc, R.S.; Ledgerwood, J.E.; Graham, B.S.; Kuhn, R.J.; Pierson, T.C. The Zika virus envelope protein glycan loop regulates virion antigenicity. Virology 2018, 515, 191–202. [Google Scholar] [CrossRef]
- Davis, C.W.; Mattei, L.M.; Nguyen, H.-Y.; Ansarah-Sobrinho, C.; Doms, R.W.; Pierson, T.C. The location of asparagine-linked glycans on West Nile virions controls their interactions with CD209 (dendritic cell-specific ICAM-3 grabbing nonintegrin). J. Biol. Chem. 2006, 281, 37183–37194. [Google Scholar] [CrossRef] [Green Version]
- Hacker, K.; White, L.; de Silva, A.M. N-linked glycans on dengue viruses grown in mammalian and insect cells. J. Gen. Virol. 2009, 90, 2097–2106. [Google Scholar] [CrossRef]
- Khoo, U.-S.; Chan, K.Y.K.; Chan, V.S.F.; Lin, C.L.S. DC-SIGN and L-SIGN: The SIGNs for infection. J. Mol. Med. (Berl) 2008, 86, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Carbaugh, D.L.; Baric, R.S.; Lazear, H.M. Envelope protein glycosylation mediates Zika virus pathogenesis. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, A.; Yoshii, K.; Obara, M.; Ueki, T.; Mizutani, T.; Kariwa, H.; Takashima, I. Role of the N-linked glycans of the prM and E envelope proteins in tick-borne encephalitis virus particle secretion. Vaccine 2005, 23, 3043–3052. [Google Scholar] [CrossRef] [PubMed]
- Mossenta, M.; Marchese, S.; Poggianella, M.; Slon Campos, J.L.; Burrone, O.R. Role of N-glycosylation on Zika virus E protein secretion, viral assembly and infectivity. Biochem. Biophys. Res. Commun. 2017, 492, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Hanna, S.L.; Pierson, T.C.; Sanchez, M.D.; Ahmed, A.A.; Murtadha, M.M.; Doms, R.W. N-linked glycosylation of West Nile Virus envelope proteins influences particle assembly and infectivity. J. Virol. 2005, 79, 13262–13274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, I.C.; Kartenbeck, J.; Mezzacasa, A.; Allison, S.L.; Heinz, F.X.; Helenius, A. Intracellular assembly and secretion of recombinant subviral particles from tick-borne encephalitis Virus. J. Virol. 2003, 77, 4370–4382. [Google Scholar] [CrossRef] [Green Version]
- Lattová, E.; Straková, P.; Pokorná-Formanová, P.; Grubhoffer, L.; Bell-Sakyi, L.; Zdráhal, Z.; Palus, M.; Ruzek, D. Comprehensive N -glycosylation mapping of envelope glycoprotein from tick-borne encephalitis virus grown in human and tick cells. Sci. Rep. 2020, 10, 13204. [Google Scholar] [CrossRef]
- Epp, J.B.; Widlanski, T.S. Facile preparation of nucleoside-5′-carboxylic acids. J. Org. Chem. 1999, 64, 293–295. [Google Scholar] [CrossRef]
- Winans, K.A.; Bertozzi, C.R. An Inhibitor of the human UDP-GlcNAc 4-epimerase identified from a uridine-based library: A Strategy to inhibit O-linked glycosylation. Chem. Biol. 2002, 9, 113–129. [Google Scholar] [CrossRef] [Green Version]
- Hwu, J.R.; Jain, M.L.; Tsai, F.-Y.; Tsay, S.-C.; Balakumar, A.; Hakimelahi, G.H. Ceric ammonium nitrate on silica gel for efficient and selective removal of trityl and silyl groups. J. Org. Chem. 2000, 65, 5077–5088. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Uridine Glycoconjugate Structure | Compound | CC50 (µM) a | IC50 (µM) b | SI c |
|---|---|---|---|---|
| II | 18 | 69 | 9.3 | 7.4 |
| 19 | 77 | 15.1 | 5.1 | |
| III | 20 | 184 | 3.7 | 49.7 |
| 21 | 116 | 4.7 | 24.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brzuska, G.; Pastuch-Gawolek, G.; Krawczyk, M.; Szewczyk, B.; Krol, E. Anti-Tick-Borne Encephalitis Virus Activity of Novel Uridine Glycoconjugates Containing Amide or/and 1,2,3-Triazole Moiety in the Linker Structure. Pharmaceuticals 2020, 13, 460. https://doi.org/10.3390/ph13120460
Brzuska G, Pastuch-Gawolek G, Krawczyk M, Szewczyk B, Krol E. Anti-Tick-Borne Encephalitis Virus Activity of Novel Uridine Glycoconjugates Containing Amide or/and 1,2,3-Triazole Moiety in the Linker Structure. Pharmaceuticals. 2020; 13(12):460. https://doi.org/10.3390/ph13120460
Chicago/Turabian StyleBrzuska, Gabriela, Gabriela Pastuch-Gawolek, Monika Krawczyk, Boguslaw Szewczyk, and Ewelina Krol. 2020. "Anti-Tick-Borne Encephalitis Virus Activity of Novel Uridine Glycoconjugates Containing Amide or/and 1,2,3-Triazole Moiety in the Linker Structure" Pharmaceuticals 13, no. 12: 460. https://doi.org/10.3390/ph13120460
APA StyleBrzuska, G., Pastuch-Gawolek, G., Krawczyk, M., Szewczyk, B., & Krol, E. (2020). Anti-Tick-Borne Encephalitis Virus Activity of Novel Uridine Glycoconjugates Containing Amide or/and 1,2,3-Triazole Moiety in the Linker Structure. Pharmaceuticals, 13(12), 460. https://doi.org/10.3390/ph13120460