4.1. General Chemistry
All reagents were purchased from chemical suppliers and used without purification. Thin-layer chromatography (TLC) was performed using silica gel 60 F254 plates, with observation under UV when necessary. Melting points were recorded on an Electrothermal IA9000 melting point system. 1H NMR spectra were recorded on an AVANCE II 400 MHz Bruker spectrometer with CDCl3 or DMSO-d6 as the solvent. 13C NMR spectra were recorded at 100 MHz. All coupling constants were measured in hertz (Hz), and the chemical shifts (δH and δC) were quoted in parts per million (ppm) relative to TMS (δ0), which was used as the internal standard. Data were reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), integration, and coupling constant (Hz). High-resolution mass spectroscopy (HRMS) analyses were carried out on an LTQ-Orbitrap XL hybrid mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). Data were acquired in positive ion mode using full-scan MS with a mass range of 100–1000 m/z. The orbitrap resolution was set at 30,000 (fwhm definition). All experimental data were acquired using daily external calibration prior to data acquisition. Appropriate tuning of the electrospray ion source was done. The following electrospray inlet conditions were applied: flow rate, 100 μL/min; spray voltage, 5 kV; sheath gas (N2) flow rate, 20 arbitrary unit (au); auxiliary gas (N2) flow rate, 10 au; capillary temperature, 275 °C; capillary voltage, 45 V; tube lens, 80 V. High-performance liquid chromatography (HPLC) analyses were performed on an LC system using a YMC-Triart C-18 (250 mm × 4.6 mm, 5 μm) column as the stationary phase. Mobile phase contained water/CH3CN (30:70, v/v) and was maintained isocratically at the flow rate of 1 mL/min. The column temperature was maintained at room temperature. The peaks were monitored at a wavelength of 215 nm. The purity of all compounds tested was greater than 95%, as determined by HPLC and 1H NMR.
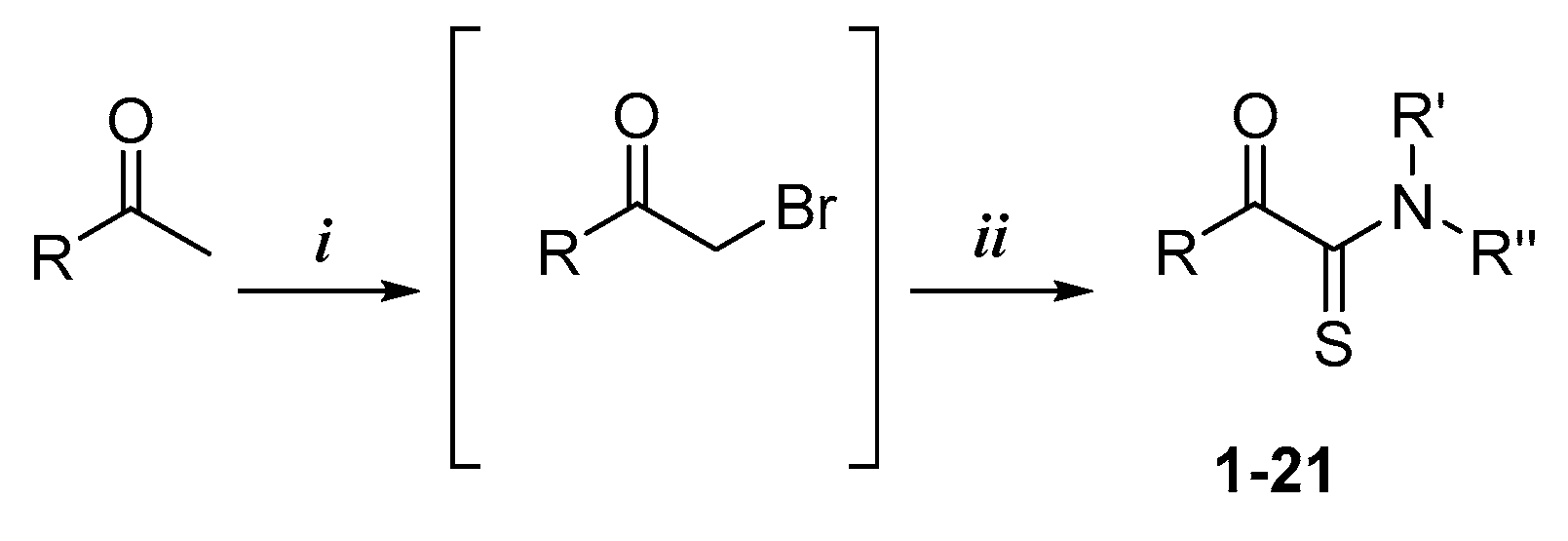
Compounds 1–11 were synthesized according to General Procedure I.
4.1.1. General Procedure I
To a stirred solution of ethanone derivative (1 equiv.) in chloroform was added dropwise a solution of dibromine (1.2 equiv.) in chloroform. After 2 h, the solvent was evaporated in vacuo to give a crude oil consisting mainly of 2-bromo-1-(substituted)-ethanone compound along with trace amounts of 2,2-dibromo-1-(substituted)-ethanone compound. The mixture was used without purification in the next step. To the synthesized or commercial 2-bromo-1-(substituted)-ethanone derivative were added, in sequence, DMF, cyclooctasulfur (1.5 equiv.), and morpholine (3 equiv.). The mixture was then stirred at room temperature. After completion, the reaction mixture was quenched with distilled water to give a precipitate, which was further washed with distilled water. The residue was recrystallized or purified by silica gel chromatography if necessary.
2-Morpholino-1-phenyl-2-thioxoethan-1-one (1). Acetophenone (2.00 g, 16.60 mmol) and dibromine (1.01 mL, 19.90 mmol) were mixed in chloroform (15 mL) to obtain the 2-bromo-1-phenyl-ethanone, and this intermediate was reacted in a second time with morpholine (4.34 mL, 49.80 mmol) and sulfur (0.79 g, 24.90 mmol) in DMF (10 mL). Methanol was used for recrystallization to afford the title compound (2.85 g, 73%). Rf 0.2 (cyclohexane/EtOAc: 8/2). M.p.: 110–112 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.59–3.62 (t, 2H, J = 4.8 Hz), 3.69–3.71 (t, 2H, J = 4.8 Hz), 3.90–3.92 (t, 2H, J = 4.8 Hz), 4.33–4.35 (t, 2H, J = 4.8 Hz), 7.48–7.52 (m, 2 ArH), 7.59–7.65 (m, 1 ArH), 7.99–8.01 (d, 2 ArH, J = 8.2 Hz). 13C NMR (100 MHz, CDCl3): δC (ppm) 47.13, 51.95, 66.40, 66.52, 128.99 (2C), 129.86 (2C), 133.26, 134.48, 187.90 (C=O), 195.70 (C=S). HRMS (ESI+): m/z calcd for C12H13NO2S [M + H]+ 236.0739, found 236.0737.
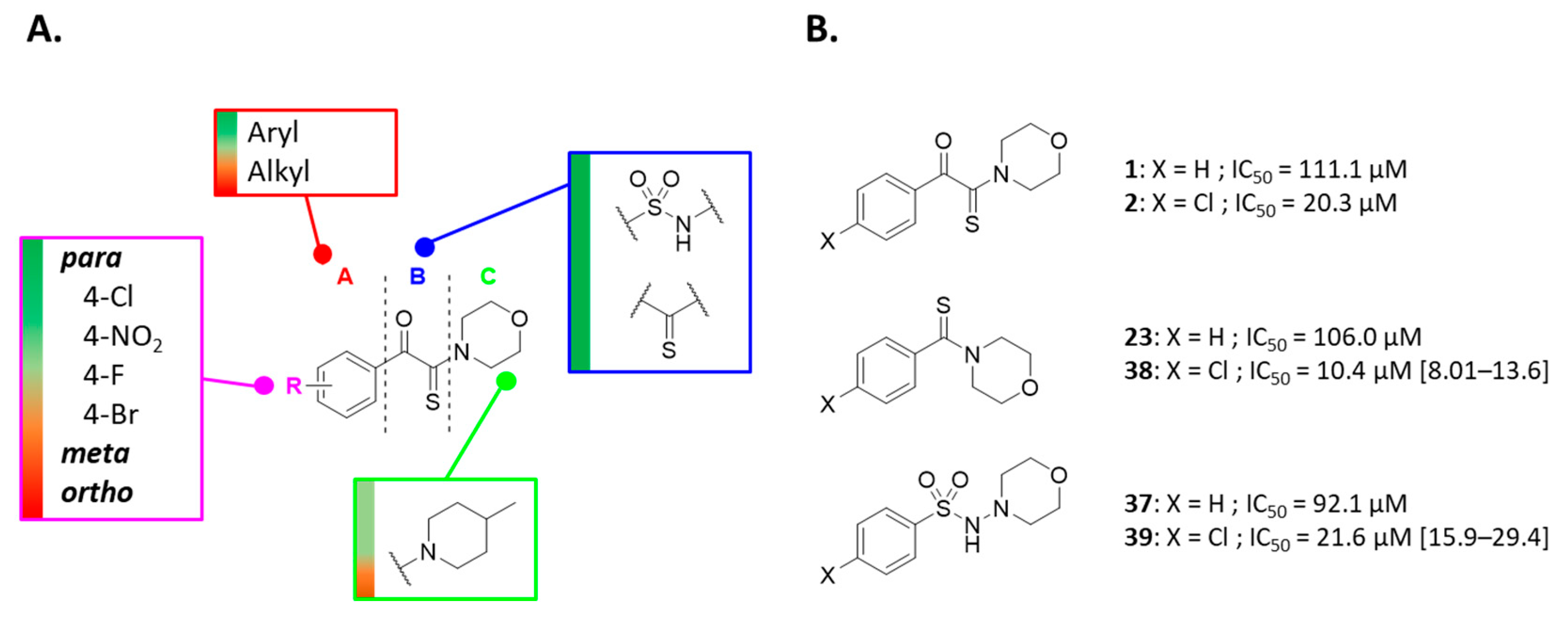
1-(4-Chlorophenyl)-2-morpholino-2-thioxoethan-1-one (2). This compound was synthesized according to General Procedure I using 2-bromo-1-(2-chlorophenyl)ethanone (0.50 g, 2.14 mmol), morpholine (0.56 mL, 6.42 mmol), and sulfur (0.10 g, 3.21 mmol) in DMF (10 mL). Acetonitrile was used for recrystallization to afford the title compound as a yellow solid (0.22 g, 38%). Rf 0.2 (cyclohexane/EtOAc: 8/2). M.p.: 135–137 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.58–3.71 (m, 4H), 3.89–3.92 (t, 2H, J = 4.8 Hz), 4.31–4.33 (t, 2H, J = 4.8 Hz), 7.46–7.48 (d, 2 ArH, J = 8.8 Hz), 7.93–7.95 (d, 2 ArH, J = 8.6 Hz). 13C NMR (100 MHz, CDCl3): δC (ppm) 47.18, 51.97, 66.39, 66.53, 129.36 (2C), 131.22 (2C), 131.74, 141.06, 186.45 (C=O), 194.94 (C=S). HRMS (ESI+): m/z calcd for C12H13ClNO2S [M + H]+ 270.0350, found 270.0350.
1-(3-Chlorophenyl)-2-morpholino-2-thioxoethan-1-one (3). This compound was synthesized according to General Procedure I. 1-(3-Chlorophenyl)ethanone (2.00 g, 12.90 mmol) and dibromine (0.78 mL, 15.50 mmol) were mixed in chloroform (15 mL) to obtain the 2-bromo-1-(3-chlorophenyl)-ethanone and this intermediate was reacted in a second time with morpholine (3.38 mL, 38.80 mmol) and sulfur (0.62 g, 19.40 mmol) in DMF (10 mL). The residue was purified by silica gel chromatography (cyclohexane/EtOAc: 8/2) and the obtained oil was collected by filtration with diethyl ether to give the title compound as a yellow solid (1.50 g, 43%). Rf 0.2 (cyclohexane/EtOAc: 8/2). M.p.: 92–94 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.59–3.61 (t, 2H, J = 4.8 Hz), 3.70–3.72 (t, 2H, J = 4.8 Hz), 3.90–3.92 (t, 2H, J = 4.8 Hz), 4.31–4.33 (t, 2H,J = 4.8 Hz), 7.42–7.46 (dd, 1 ArH, J = 7.8 Hz), 7.57–7.59 (ddd, 1 ArH, J = 8.0 and 1.0 Hz), 7.85–7.87 (ddd, 1 ArH, J = 7.8 and 1.4 Hz), 7.96–7.97 (dd, 1 ArH, J = 1.8 Hz).13C NMR (100 MHz, CDCl3): δC (ppm) 47.21, 52.00, 66.39, 66.52, 127.98, 129.61, 130.28, 134.33, 135.05, 135.30, 186.07 (C=O), 194.60 (C=S). HRMS (ESI+): m/z calcd for C12H12ClNO2S [M + H]+ 270.0277, found 270.0278.
1-(2,4-Dichlorophenyl)-2-morpholino-2-thioxoethan-1-one (4). This compound was synthesized according to General Procedure I using 2-bromo-1-(2,4-dichlorophenyl)-ethanone (1.00 g, 3.76 mmol), morpholine (0.99 mL, 11.28 mmol), and sulfur (0.18 g, 5.64 mmol) in DMF (10 mL). Acetonitrile was used for recrystallization to afford the title compound as a yellow solid (0.31 g, 28%). Rf 0.4 (cyclohexane/EtOAc: 8/2). M.p.: 116–118 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.75–3.77 (t, 2H, J = 4.8 Hz), 3.77–3.79 (t, 2H, J = 4.8 Hz), 3.89–3.91 (t, 2H, J = 4.8 Hz), 4.10–4.12 (t, 2H, J = 4.8 Hz), 7.30 (dd, 1 ArH, J = 2.6 and 8.4 Hz), 7.38 (d, 1 ArH, J = 2.6 Hz), 7.80 (d, 1 ArH, J = 8.4 Hz). 13C NMR (100 MHz, CDCl3): δC (ppm) 47.68, 52.11, 65.95, 66.03, 127.83, 130.49 (2C), 133.08, 133.56, 139.55, 183.73 (C=O), 195.19 (C=S). HRMS (ESI+): m/z calcd for C12H12Cl2NO2S [M + H]+ 303.99603, found 303.99607.
1-Cyclohexyl-2-morpholino-2-thioxoethan-1-one (5). This compound was synthesized according to General Procedure I using 1-cyclohexylethanone (2.00 g, 15.86 mmol) and dibromine (0.96 mL, 19.03 mmol) mixed in chloroform (20 mL) to obtain the 2-bromo-1-cyclohexylethanone and this intermediate was reacted in a second time with morpholine (4.18 mL, 47.58 mmol) and sulfur (0.76 g, 23.79 mmol) in DMF (10 mL). The residue was purified by silica gel chromatography (Cyclohexane/EtOAc: 8/2) to give the title compound as a brown solid (0.58 g, 15%). Rf 0.3 (Cyclohexane/EtOAc: 8/2). M.p.: 57–59 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 1.18–1.37 (m, 5H), 1.69–1.99 (m, 5H), 3.22 (m, 1H), 3.61–3.62 (m, 2H), 3.73–3.74 (t, 2H, J = 4.8 Hz), 3.82–3.84 (t, 2H, J = 4.8 Hz), 4.19–4.21 (t, 2H, J = 4.8 Hz). 13C NMR (100 MHz, CDCl3): δC (ppm) 25.52 (2C), 25.71, 28.10 (2C), 47.28, 47.49, 52.07, 66.31, 66.60, 197.85 (C=O, 201.05 (C=S). HRMS (ESI+): m/z calcd for C12H20NO2S [M + H]+ 242.1209, found 242.1208.
2-Morpholino-1-(pyridin-4-yl)-2-thioxoethan-1-one (6). This compound was synthesized according to General Procedure I using 2-bromo-1-(pyridine-4-yl)ethanone (0.50 g, 1.79 mmol), morpholine (0.47 mL, 5.37 mmol), and sulfur (0.08 g, 2.68 mmol) in DMF (10 mL). The residue was purified by silica gel chromatography (CH2Cl2/EtOAc: 5/5) to give the title compound as a yellow solid (0.15 g, 36%). Rf 0.3 (CH2Cl2/EtOAc: 5/5). M.p.: 96–98 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.60–3.63 (m, 2H), 3.71–3.74 (m, 2H), 3.91–3.93 (m, 2H), 4.31–4.34 (m, 2H), 7.77–7.79 (m, 2 ArH), 8.83–8.85 (m, 2 ArH). 13C NMR (100 MHz, CDCl3): δC (ppm) 45.30, 50.05, 64.40, 64.56, 120.26 (2C), 137.86, 149.10 (2C), 183.37 (C=O), 191.58 (C=S). HRMS (ESI+): m/z calcd for C11H13N2O2S [M + H]+ 237.06922, found 237.06923.
1-(3,4-Dichlorophenyl)-2-morpholino-2-thioxoethan-1-one (7). This compound was synthesized according to General Procedure I using 2-bromo-1-(3,4-dichlorophenyl)ethan-1-one (1.00 g, 3.73 mmol), morpholine (0.96 mL, 11.2 mmol), and sulfur (0.18 g, 5.60 mmol) in DMF (15 mL). The residue was purified by silica gel chromatography (Cyclohexane/EtOAc: 3/2) to give the title compound as a yellow solid (0.32 g, 28%). Rf0.6 (Cyclohexane/EtOAc: 3/2). M.p.: 134–136 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.60–3.61 (m, 2H), 3.71–3.72 (m, 2H), 3.90–3.93 (m, 2H), 4.31–4.33 (m, 2H), 7.55–7.60 (m, 1 ArH), 7.79–7.84 (m, 1 ArH), 8.07–8.09 (m, 1 ArH). 13C NMR (100 MHz, CDCl3): δC (ppm) 44.87, 49.63, 63.99, 64.15, 126.38, 128.69, 129.11, 130.76, 131.36, 136.75, 182.57 (C=O), 191.61 (C=S). HRMS (ESI+): m/z calcd for C12H12Cl2NO2S (M + H)+ 303.99603, found 303.99549.
2-Morpholino-1-(naphthalen-2-yl)-2-thioxoethan-1-one (8). This compound was synthesized according to General Procedure I using 2-bromo-1-(naphthalene-2-yl) ethanone (0.50 g, 2.01 mmol), morpholine (0.53 mL, 6.04 mmol), and sulfur (0.09 g, 3.01 mmol) in DMF (10 mL). Cyclohexane was used for recrystallization to afford the title compound as a yellow solid (0.31 g, 28%). Rf 0.2 (Cyclohexane/EtOAc: 8/2). M.p.: 152–154 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.63–3.65 (m, 2H), 3.69–3.71 (m, 2H), 3.93–3.96 (m, 2H), 4.38–4.41 (m, 2H), 7.55–7.66 (m, 2 ArH), 7.88–7.97 (m, 3 ArH), 8.03–8.05 (dd, 1 ArH, J = 2.1 and 8.8 Hz), 8.52 (s, 1 ArH). 13C NMR (100 MHz, CDCl3): δC (ppm) 46.92, 51.70, 66.14, 66.25, 124.02, 126.87, 127.63, 128.67, 129.06, 129.54, 130.28, 132.11, 132.22, 135.88, 187.75 (C=O), 195.51 (C=S). HRMS (ESI+): m/z calcd for C16H15NO2S (M + Na)+ 308.07157, found 308.07146.
1-Morpholino-1-thioxobutan-2-one (9). This compound was synthesized according to General Procedure I using 1-bromobutan-2-one (0.34 mL, 3.33 mmol), morpholine (0.88 mL, 10.0 mmol), and sulfur (0.15 g, 4.99 mmol) in DMF (10 mL). The residue was purified by silica gel chromatography (Hexane/EtOAc: 7/3) to give the title compound as a yellow solid (0.08 g, 8%). Rf 0.5 (Cyclohexane/EtOAc: 3/2). 1H NMR (400 MHz, CDCl3): δH (ppm) 1.19 (t, 3H, J = 5.1 Hz), 2.88–2.94 (q, 2H, J = 5 Hz), 3.59–3.62 (m, 2H), 3.74–3.76 (m, 2H), 3.82–3.85 (m, 2H), 4.17–4.20 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 7.41, 33.52, 46.94, 51.45, 65.92, 66.21, 197.42 (C+O), 199.14 (C=S) HRMS (ESI+): m/z calcd for C8H14NO2S [M + H]+ 188.07398, found 188.07384.
1-((1r,3r,5r,7r)-Adamantan-2-yl)-2-morpholino-2-thioxoethan-1-one (10). This compound was synthesized according to General Procedure I using 2-bromo-1-((1 r,3r,5r,7r)-Adamantan-2-yl)-10-ethanone (1 g, 3.90 mmol), morpholine (1.02 mL, 11.70 mmol), and sulfur (0.18 g, 5.90 mmol) in DMF (10 mL). The residue was purified by silica gel chromatography (cyclohexane/EtOAc: 8/2) to give the title compound as yellow oil (0.08 g, 8%). Rf 0.3 (hexane/EtOAc 8:2). 1H NMR (400 MHz, CDCl3): δH (ppm) 1.69–1.77 (m, 6H), 2.02–2.05 (m, 9H), 3.51 (m, 2H), 3.73–3.75 (t, 2H, J = 4.8 Hz), 3.80–3.83 (t, 2H, J = 4.8 Hz), 4.18 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 27.96 (3C), 36.30 (3C), 39.79 (3C), 45.03, 46.41, 52.39, 66.30, 66.36, 196.66 (C=O), 205.54 (C=S). HRMS (ESI+): m/z calcd for C16H23NO2S [M + H]+ 294.1522, found 294.1523.
3,3-Dimethyl-1-morpholino-1-thioxobutan-2-one (11). This compound was synthesized according to General Procedure I using 1-bromo-3,3-dimethylbutan-2-one (2.0 g, 11.2 mmol), morpholine (2.96 mL, 33.7 mmol), and sulfur (0.54 g, 16.8 mmol) in DMF (10 mL). The residue was purified by silica gel chromatography (Cyclohexane/EtOAc: 8/2) to give the title compound as orange oil (0.35 g, 14%). Rf 0.2 (Cyclohexane/EtOAc: 8/2). 1H NMR (400 MHz, CDCl3): δH (ppm) 1.34 (s, 9H), 3.50–3.53 (m, 2H), 3.74–3.76 (m, 2H), 3.81–3.83 (m, 2H), 4.18 (broad, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 26.16 (3C), 40.52, 44.11, 50.03, 64.01 (2C), 194.65 (C=O), 204.21 (C=S). HRMS (ESI+): m/z calcd for C10H18NO2S [M + H]+ 216.10528, found 216.10524.
Compounds 12–21 were synthesized according to General Procedure II.
4.1.2. General Procedure II
To commercial 2-bromo-1-phenylethanone were added, in sequence, DMF, cyclooctasulfur (1.5 equiv.), and amine derivative (3 equiv.). The mixture was then stirred at room temperature. After completion, the reaction mixture was quenched with distilled water to give a precipitate, which was further washed with distilled water. The residue was recrystallized or purified by silica gel chromatography if necessary.
1-Phenyl-2-(pyrrolidin-1-yl)-2-thioxoethan-1-one (12). This compound was synthesized according to General Procedure II using 2-bromo-1-phenylethanone (0.5 g, 2.52 mmol), pyrrolidine (0.62 mL, 7.56 mmol), and sulfur (0.12 g, 3.78 mmol) in DMF (10 mL). The residue was purified by silica gel chromatography (Cyclohexane/EtOAc: 8/2) to give the title compound as a yellow solid (0.12 g, 22%). Rf 0.4 (Cyclohexane/EtOAc: 8/2). M.p.: 57–59 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 1.61 (m, 4H), 3.53–3.56 (m, 2H), 3.94–3.98 (m, 2H), 7.46–7.48 (m, 2H), 7.50–7.62 (m, 1H), 7.99–8.01 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 22.97, 25.24, 50.22, 50.40, 127.97 (2C), 129.24 (2C), 131.96, 133.37, 187.92 (C=O), 191.87 (C=S). HRMS (ESI+): m/z calcd for C12H14NOS [M + H]+ 220.07906, found 220.07877.
2-(4,4-Difluoropiperidin-1-yl)-1-phenyl-2-thioxoethan-1-one (13). This compound was synthesized according to General Procedure II using 2-bromo-1-phenylethanone (0.5 g, 2.52 mmol), 4,4-difluoropiperidine HCl (1.19 g, 7.56 mmol), and sulfur (0.12 g, 3.78 mmol) in DMF (10 mL). Cyclohexane was used for recrystallization to afford the title compound as a white solid (0.12 g, 18%). Rf 0.8 (Cyclohexane/EtOAc: 3/2). M.p.: 117–119 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 1.42–1.58 (m, 2H), 2.08–2.25 (m, 2H), 3.68 (m, 2H), 4.44 (m, 2H), 7.48–7.52 (m, 2H), 7.61–7.65 (m, 1H), 7.98–8.00 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 31.25 (t), 32.32 (t), 41.73 (t), 46.22 (t), 118.84, and 121.26 (C-F), 127.18 (2C), 128.02 (2C), 131.23, 132.77, 186.10 (C=O), 194.87 (C=S). HRMS (ESI+): m/z calcd for C13H14NOSF2 [M + H]+ 270.07587, found 270.07568.
2-(4-Methylpiperidin-1-yl)-1-phenyl-2-thioxoethan-1-one (14). This compound was synthesized according to General Procedure II using 2-bromo-1-phenylethanone (1.0 g, 5.05 mmol), 4-methylpiperidine (3.82 g, 15.15 mmol), and sulfur (0.24 g, 7.57 mmol) in DMF (10 mL). The residue was purified by silica gel chromatography (Hexane/EtOAc: 9/1) to give the title compound as a yellow solid (0.47 g, 38%). Rf 0.3 (Hexane/EtOAc: 7/3). M.p.: 71–73 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 1.00 (s, 3H), 1.15–1.25 (m, 1H), 1.30 (m, 1H), 1.78–1.92 (m, 3H), 3.08–3.14 (m, 1H) 1.15–1.20 (m, 3H), 3.26–3.32 (m, 1H), 3.74–3.76 (m, 1H), 5.39 (m, 1H), 7.47–7.49 (m, 2H), 7.59 (m, 1H), 7.98 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 18.86, 28.42, 30.95, 31.97, 45.03, 49.83, 126.48 (2C), 127.41 (2C), 131.01, 131.82, 185.65 (C=O), 192.08 (C=S). HRMS (ESI+): m/z calcd for C14H17NOSNa (M + Na)+ 270.09231, found 270.09219.
1-Phenyl-2-(piperidin-1-yl)-2-thioxoethan-1-one (15). This compound was synthesized according to General Procedure II using 2-bromo-1-phenylethanone (2.0 g, 10.01 mmol), piperidine (2.99 mL, 30.30 mmol), and sulfur (0.48 g, 15.10 mmol) in DMF (10 mL). The residue was purified by silica gel chromatography (Cyclohexane/EtOAc: 8/2) to give the title compound as a yellow solid (0.47 g, 20%). Rf 0.6 (Cyclohexane/EtOAc: 3/2). M.p.: 67–69 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 1.56–1.63 (m, 2H), 1.77–1.84 (m, 4H), 3.53–3.55 (m, 2H), 4.24–4.27 (m, 2H), 7.47–7.50 (m, 2H), 7.58–7.62 (m, 1H), 7.98–8.00 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 23.79, 25.13, 26.20, 47.88, 52.79, 52.79, 128.63 (2C), 129.52 (2C), 133.14, 133.97, 187.76 (C=O), 194.03 (C=S). 18.86, 28.42, 30.95, 31.97, 45.03, 49.83, 126.48 (2C), 127.41 (2C), 131.01, 131.82, 185.65 (C=O), 192.08 (C=S). HRMS (ESI+): m/z calcd for C13H15NOSNa (M + Na)+ 256.07666, found 256.07661.
N,N-Diethyl-2-oxo-2-phenylethanethioamide (16). This compound was synthesized according to General Procedure II using 2-bromo-1-phenylethanone (1.0 g, 5.05 mmol), diethylamine (1.56 mL, 15.15 mmol), and sulfur (0.24 g, 7.57 mmol) in DMF (10 mL). The residue was purified by silica gel chromatography (Hexane/EtOAc: 7/3) to give the title compound as brown oil (0.09 g, 8%). Rf 0.3 (Hexane/EtOAc: 7/3).1H NMR (400 MHz, CDCl3): δH (ppm) 1.15–1.20 (m, 3H), 1.26–1.34 (m, 3H), 3.37–3.42 (m, 2H), 3.94–3.99 (m, 2H), 7.36–7.40 (m, 2H), 7.48–7.52 (m, 1H), 7.87–7.89 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 11.29, 13.63, 44.55, 47.99, 128.80 (2C), 129.90 (2C), 133.51, 134.09, 187.51 (C=O), 195.58 (C=S). HRMS (ESI+): m/z calcd for C12H16NOS [M + H]+ 222.09471, found 222.09456.
1-Phenyl-2-thiomorpholino-2-thioxoethan-1-one (17). This compound was synthesized according to General Procedure II using 2-bromo-1-phenylethanone (1.0 g, 5.05 mmol), thiomorpholine (1.52 mL, 15.15 mmol), and sulfur (0.24 g, 7.57 mmol) in DMF (10 mL). The residue was purified by silica gel chromatography (Hexane/EtOAc: 7/3) to give the title compound as a yellow solid (0.40 g, 43%). Rf 0.7 (Cyclohexane/EtOAc: 3/2). M.p.: 127–129 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 2.69 (m, 2H), 2.88–2.90 (t, 2H, J = 5.1 Hz), 3.83–3.86 (t, 2H, J = 5.1 Hz), 4.59 (m, 2H), 7.47–7.51 (m, 2H), 7.59–7.63 (m, 1H), 7.97–7.99 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 27.31, 28.25, 49.71, 54.64, 128.96 (2C), 129.90 (2C), 133.23, 134.44, 187.73 (C=O), 196.06 (C=S). HRMS (ESI+): m/z calcd for C12H14NOS2 [M + H]+ 252.05113, found 252.05117.
2-(4-Methylpiperazin-1-yl)-1-phenyl-2-thioxoethan-1-one (18). This compound was synthesized according to General Procedure II using 2-bromo-1-phenylethanone (2 g, 10.10 mmol), N-methylpiperazine (3.37 mL, 30.30 mmol), and sulfur (0.48 g, 15.15 mmol) in DMF (10 mL). The residue was purified by silica gel chromatography (Cyclohexane/EtOAc: 8/2) to give the title compound as brown solid (0.58 g, 23%). Rf 0.2 (Cyclohexane/EtOAc: 8/2). M.p.: 93–95 °C 1H NMR (400 MHz, CDCl3): δH (ppm) 2.34 (s, 3H, CH3), 2.41–2.44 (m, 2H), 2.62–2.64 (m, 2H), 3.58–3.60 (m, 2H), 4.31–4.34 (m, 2H), 7.47–7.51 (m, 2H), 7.59–7.63 (m, 1H), 7.98–8.00 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 45.53, 46.61, 51.31, 53.95, 54.57, 128.78 (2C), 129.71 (2C), 133.18, 134.21, 187.80 (C=O), 195.17 (C=S). HRMS (ESI+): m/z calcd for C13H17N2OS [M + H]+ 249.10561, found 249.10561.
2-(4-(2-Oxo-2-phenylethyl)piperazin-1-yl)-1-phenyl-2-thioxoethan-1-one (19). This compound was synthesized according to General Procedure I using 2-bromo-1-phenylethanone (1.00 g, 5.05 mmol), piperazine (1.30 g, 15.15 mmol), and sulfur (0.24 g, 7.57 mmol) in DMF (10 mL). The residue was purified by silica gel chromatography (hexane/EtOAc: 9/1) to give the title compound as orange oil (0.40 g, 22%). Rf 0.2 (cyclohexane/EtOAc: 8/2). 1H NMR (400 MHz, CDCl3): δH (ppm) 2.67–2.70 (t, 2H, J = 4.8 Hz), 2.87–2.90 (t, 2H, J = 4.8 Hz), 3.66–3.69 (t, 2H, J = 4.8 Hz), 4.40–4.43 (t, 2H, J = 4.8 Hz), 7.42–7.53 (m, 4 ArH), 7.58–7.64 (m, 2 ArH), 7.94–8.15 (m, 4 ArH). HRMS (ESI+): m/z calcd for C20H20N2O2S [M + H]+ 353.1318, found 353.1318.
1-Phenyl-2-(4-phenylpiperazin-1-yl)-2-thioxoethan-1-one (20). This compound was synthesized according to General Procedure II using 2-bromo-1-phenylethanone (1.0 g, 5.05 mmol), 1-phenyl-piperazine (2.31 mL, 15.15 mmol), and sulfur (0.24 g, 7.57 mmol) in DMF (10 mL). The residue was purified by silica gel chromatography (Cyclohexane/EtOAc: 8/2) to give the title compound as a yellow solid (0.20 g, 13%). Rf 0.4 (Cyclohexane/EtOAc: 8/2). M.p.: 89–91 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.20–3.22 (m, 2H), 3.40–3.43 (m, 2H), 3.73–3.76 (m, 2H), 4.46–4.48 (m, 2H), 6.92–6.96 (m, 3H), 7.28–7.31 (m, 2H), 7.48–7.52 (m, 2H), 7.60–7.64 (m, 1H), 8.00–8.02 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 45.06, 47.45, 48.10, 49.66, 115.28 (2C), 119.55, 126.78, 127.30 (2C), 127.74 (2C), 128.24 (2C), 131.65, 132.77, 148.47, 186.30 (C=O), 193.88 (C=S). HRMS (ESI+): m/z calcd for C18H19N2OS [M + H]+ 311.12181, found 311.12068.
2-(4-Benzhydrylpiperazin-1-yl)-1-phenyl-2-thioxoethan-1-one (21). This compound was synthesized according to General Procedure II using 2-bromo-1-phenylethanone (1.0 g, 5.05 mmol), 1-(diphenylmethyl)piperazine (3.82 g, 15.15 mmol) and sulfur (0.24 g, 7.57 mmol) in DMF (10 mL). Cyclohexane was used for recrystallization to afford the title compound as a white solid (0.60 g, 30%). Rf 0.5 (Hexane/EtOAc: 8/2). M.p.: 130–132 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 2.40 (m, 2H), 2.62 (m, 2H), 3.57 (m, 2H), 4.29 (m, 3H), 7.17–7.25 (m, 6H), 7.39–7.47 (m, 6H), 7.56–7.59 (m, 1H), 7.95–7.96 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 45.65, 49.77, 50.33, 50.41, 74.22, 126.09 (2C), 126.41 (4C), 127.44 (4C), 127.55 (2C), 128.48 (2C), 131.99, 132.94, 140.26 (2C), 186.66 (C=O), 193.69 (C=S). HRMS (ESI+): m/z calcd for C25H25N2OS [M + H]+ 401.16821, found 401.16826.
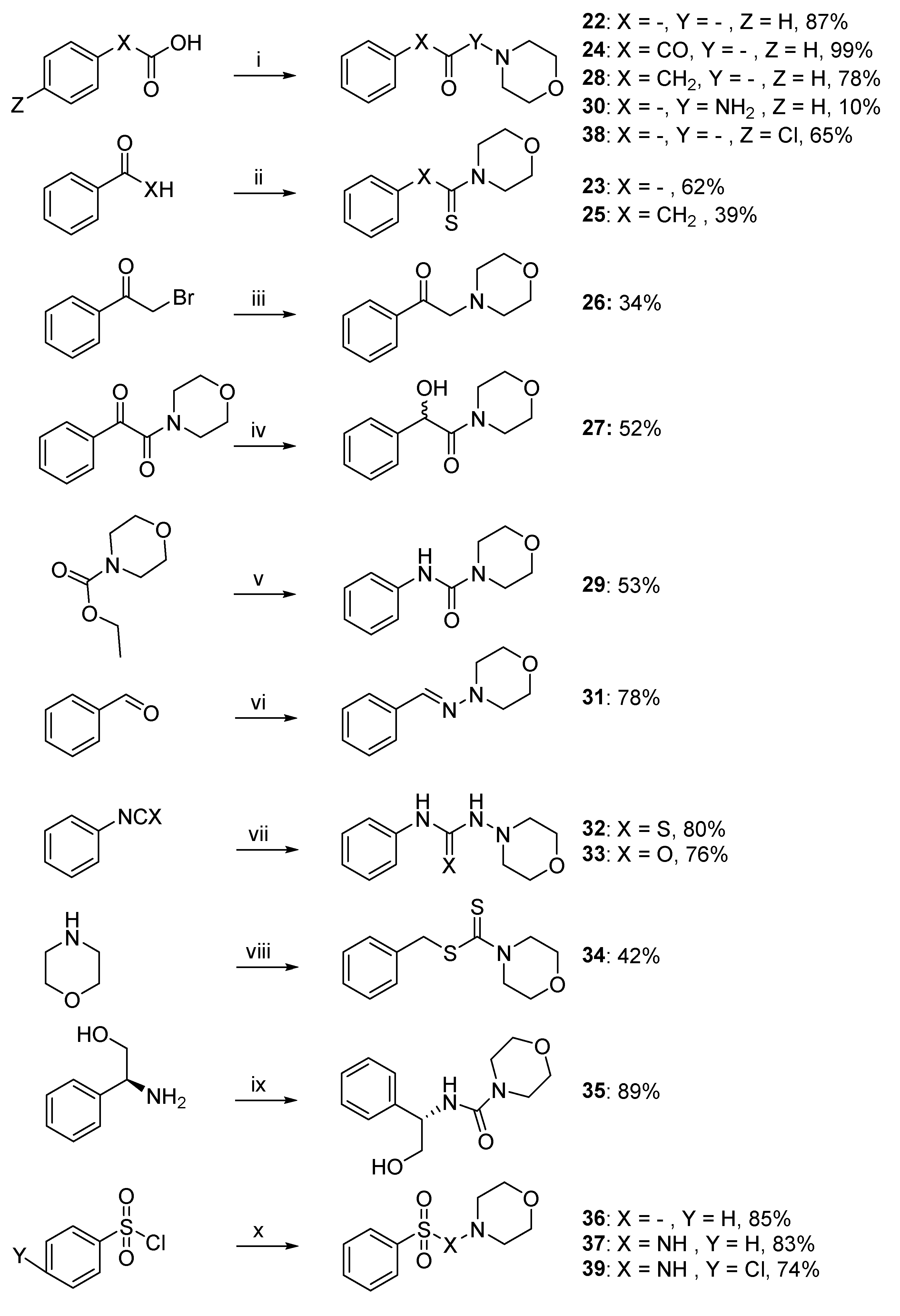
4.1.3. Synthesis of Compounds 22–39
Morpholino(phenyl)methanone (22). To a stirring solution of benzoic acid (2.50 g, 20.47 mmol, 1 equiv.) in CH2Cl2 (50 mL) were added catalytic amount of DMF and oxalyl chloride (2.60 g, 20.47 mmol, 1 equiv.). The reaction mixture was stirred at room temperature until the generation of gasses was stopped (HCl), cooled to 0 °C, and a solution of morpholine (2.67 g, 30.7 mmol, 1.5 equiv.) and DIPEA (5.29 g, 40.94 mmol, 2 equiv.) was slowly added. The mixture was stirred at room temperature for 6 h. The reaction was quenched by adding water (50 mL) following by extraction with CH2Cl2 (75 mL). The combined organic layer were washed with brine and dried over Na2SO4. The solvent was removed in vacuo, and the residue was purified by silica gel chromatography (Cyclohexane/EtOAc: 2/3) to give the title compound as a white solid (17.81 mmol, 3.40 g, 87%). Rf 0.45 (Cyclohexane/EtOAc: 3/2). M.p.: 62–63 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.41–3.50 (m, 4H), 3.64–3.79 (m, 4H), 7.39–7.44 (m, 5H). 13C NMR (100 MHz, CDCl3): δC (ppm) 26.84, 42.60, 48.19, 66.74, 127.20 (2C), 128.67 (2C), 129.99, 135.24, 170.32 (C=O). HRMS (ESI+): m/z calcd for C11H14NO2 [M + H]+ 192.1024, found 192.1014.
Morpholino(phenyl)methanethione (23). Benzaldehyde (1.00 g, 9.42 mmol, 1 equiv.), morpholine (2.05 g, 23.5 mmol, 2.5 equiv.), sulfur (0.30 g, 9.42 mmol, 1 equiv.), and para-toluene sulfonic acid (0.05 g, 0.26 mmol, 0.028 equiv.) were mixed together and the solution stirred at 85 °C for 12 h. The brown solid formed was recovered with 50 mL of CH2Cl2. The solution was then filtered and the organic layer was extracted 3 times with water (80 mL). The organic layer was evaporated under vacuo and the desired product was recrystallized in EtOH to give the title compound as a yellow solid (5.84 mmol, 1.21 g, 62%). Rf 0.7 (Cyclohexane/EtOAc: 2/3). M.p.: 128–129 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.59–3.62 (m, 2H), 3.64–3.66 (m, 2H), 3.88–3.91 (m, 2H), 4.44–4.46 (m, 2H), 7.27–7.30 (m, 2H), 7.34–7.39 (m, 3H). 13C NMR (100 MHz, CDCl3): δC (ppm) 47.32, 50.29, 64.33, 64.54, 123.57 (2C), 126.45 (2C), 126.68, 140.13, 198.90 (C=S). HRMS (ESI+): m/z calcd for C11H14NOS [M + H]+ 208.0796, found 208.0791.
1-Morpholino-2-phenylethane-1,2-dione (24). To a solution of 2-oxo-2-phenylacetic acid (2.50 g, 16.6 mmol, 1 equiv.) in CH2Cl2 were added catalytic amount of DMF and oxalyl chloride (2.11 g, 16.6 mmol, 1 equiv.). The reaction was stirred at room temperature until the generation of gasses (HCl) was stopped, cooled to 0 °C and a solution of morpholine (2.17 g, 24.9 mmol, 1.5 equiv.) with DIPEA (4.29 g, 33.2 mmol, 2 equiv.) was slowly added. The mixture was stirred at room temperature for 14 h, quenched by adding water and extracted with CH2Cl2. The combined organic layer were washed with brine and dried over Na2SO4. The solvents were removed in vacuo, and the resulting crude product was purified by silica gel chromatography (Cyclohexane/EtOAc: 3/2) to give the title compound as a white solid (16.4 mmol, 3.60 g, 99%). Rf 0.35 (Cyclohexane/EtOAc: 3/2). M.p.: 50–52 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.38–3.40 (m, 2H), 3.65–3.68, (m, 2H), 3.80 (m, 4H), 7.51–7.55 (m, 2H), 7.65–7.69 (m, 1H), 7.96–7.98 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 41.64, 46.28, 66.70, 66.77, 129.14 (2C), 129.71 (2C), 133.06, 134.99, 165.48 (C=O), 191.19 (C=O). HRMS (ESI+): m/z calcd for C12H14NO3 [M + H]+ 220.0973, found 220.0968.
1-Morpholino-2-phenylethane-1-thione (25). To a solution of acetophenone (1.50 g, 12.48 mmol, 1 equiv.) and sulfur (0.60 g, 18.73 mmol, 1.5 equiv.) in DMF (20 mL), morpholine (3.26 g, 12.48 mmol, 3 equiv.) was slowly added and the reaction was stirred at reflux for 10 h. After the completion of the reaction, water was added on the mixture and the solution was extracted with EtOAc (150 mL). The organic layers were dried and evaporated under vacuo to afford the crude mixture. The residue was purified by silica gel chromatography (Cyclohexane/EtOAc: 2/3) to give the title compound as a yellow solid (4.87 mmol, 1.08 g, 39%). Rf 0.67 (EtOAc/Cyclohexane: 3/2). M.p.: 66–67 °C. 1H NMR (400 MHz, (CD3)2SO): δH (ppm) 3.34 (s, 2H), 3.40–3.42 (m, 2H), 3.63–3.66 (m, 2H), 3.69–3.72 (m, 2H), 4.22–4.25 (m, 2H), 7.23–7.28 (m, 1H), 7.32–7.35 (m, 4H). 13C NMR (100 MHz, (CD3)2SO): δC (ppm) 48.66, 49.40, 50.22, 65.23, 65.33, 126.27, 127.53 (2C), 128.19 (2C), 135.95, 198.39 (C=S). HRMS (ESI+): m/z calcd for C12H16NOS [M + H]+ 222.0952, found 222.0941.
2-Morpholino-1-phenylethan-1-one (
26). 2-bromo-1-phenylethanone (1.00 g, 5.02 mmol, 1 equiv.) and morpholine (1.71 g, 19.59 mmol, 3.9 equiv.) were mixed in a solution of K
2CO
3 (3.12 g, 22.59 mmol, 4.5 equiv.) in acetonitrile and stirred for 3 h. Then the solution was filtered to remove K
2CO
3 and evaporated under reduced pressure. The resulting crude product was purified by silica gel chromatography (Cyclohexane/EtOAc: 3/2) to give the title compound as red oil (1.71 mmol, 0.35 g, 34%). R
f 0.4 (Cyclohexane/EtOAc: 3/2).
1H NMR (400 MHz, CDCl
3):
δH (ppm) 261–2.63 (m, 4H), 3.78–3.80 (m, 4H), 3.83 (s, 2H), 7.45–7.49 (m, 2H), 7.56–7.60 (m, 1H), 7.99–8.01 (m, 2H).
13C NMR (100 MHz, CDCl
3):
δC (ppm) 53.91 (2C), 64.72, 66.84 (2C), 128.09 (2C), 128.61 (2C), 133.37, 135.95, 196.10 (C=O). HRMS (ESI
+):
m/z calcd for C
11H
16NO
2 [M + H]
+ 206.11810, found 206.11425. Data are in agreement with the literature [
12].
2-Hydroxy-1-morpholino-2-phenylethan-1-one (27). To a solution of 1-morpholino-2-phenylethane-1,2-dione (0.25 g, 1.14 mmol, 1 equiv.) in 10 mL of MeOH, NaBH4 (0.11 g, 2.85 mmol, 2.5 equiv.) was added and stirred during 5 h. The reaction mixture was then evaporated, diluted in water, and extracted with CH2Cl2. The organic layer was evaporated and purified by silica gel chromatography (Cyclohexane/EtOAc: 8/2) to give the title compound as a white solid (0.59 mmol, 0.13 g, 52%). Rf 0.53 (Cyclohexane/EtOAc: 8/2). M.p.: 85–87 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.02–3.09 (m, 1H), 3.13–3.18 (m, 1H), 3.44–3.49 (m, 1H), 3.52–3.63 (m, 2H), 3.67–3.71 (m, 1H), 3.76–3.81 (m, 1H), 4.73 (s, 1H, OH), 5.19 (s, 1H), 7.29–7.40 (m, 5H). 13C NMR (100 MHz, CDCl3): δC (ppm) 43.15, 45.31, 65.81, 66.56, 71.54, 127.39 (2C), 128.74, 129.21 (2C), 139.13, 171.03 (C=O). HRMS (ESI+): m/z calcd for C12H16NO3 [M + H]+ 222.1130, found 222.1125.
1-Morpholino-2-phenylethan-1-one (28). To a solution of 2-phenylacetic-acid (0.50 g, 3.70 mmol, 1 equiv.) in CH2Cl2 were added catalytic amounts of DMF and oxalyl chloride (0.47 g, 3.7 mmol, 1 equiv.). The reaction was stirred at room temperature until the generation of gasses (HCl) was stopped. The resulting solution was cooled to 0 °C and a solution of morpholine (0.48 g, 5.55 mmol, 1.5 equiv.) and DIPEA (0.96 g, 7.40 mmol, 2 equiv.) was slowly added. The mixture was stirred at room temperature for 14 h, quenched by adding water (100 mL), and the mixture was extracted with CH2Cl2 (150 mL). The combined organic layers were washed with brine and dried over Na2SO4. The residue was purified by silica gel chromatography (Cyclohexane/EtOAc: 2/3) to give the title compound as a white solid (2.90 mmol, 0.59 g, 78%). Rf 0.29 (Cyclohexane/EtOAc: 2/3). M.p.: 60–62 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.42–3.45 (m, 2H), 3.47–3.49 (m, 2H), 3.65 (s, 4H), 3.74 (s, 2H), 7.23–7.27 (m, 3H), 7.31–7.35 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 39.10, 40.36, 44.75, 64.69, 65.03, 125.23, 126.75 (2C), 127.06 (2C), 133.02. HRMS (ESI+): m/z calcd for C12H16NO2 [M + H]+ 206.1181, found 206.1176.
N-Phenylmorpholine-4-carboxamide (29). To a MeCN solution of ethyl morpholine-4-carboxylate (0.40 g, 2.50 mmol, 1 equiv.) were added POCl3 (1.15 g, 7.50 mmol, 3 equiv.) under Ar. The reaction mixture was stirred at reflux and monitored by TLC. After completion, the solution was diluted in water and extracted with CH2Cl2. The organic layer was evaporated, diluted in dry CH2Cl2, cooled to 0 °C, and a solution of aniline (0.35 g, 3.75 mmol, 1.5 equiv.) and DIPEA (0.65 g, 3.75 mmol, 2 equiv.) in CH2Cl2 was slowly added. The reaction mixture was stirred at room temperature for 50 h. The reaction was quenched by adding water and the mixture was extracted with CH2Cl2. The combined organic layers were washed with brine and dried over Na2SO4. The resulting crude product was purified by silica gel chromatography (Cyclohexane/EtOAc: 8/2) to give the title compound as a green solid (1.32 mmol, 0.27 g, 53%). Rf 0.14 (Cyclohexane/EtOAc: 3/2). M.p.: 156–158 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.48–3.50 (m, 4H), 3.74–3.77 (m, 4H), 6.69 (s, 1H, NH), 7.04–7.08 (m, 1H), 7.28–7.37 (m, 4H). 13C NMR (100 MHz, CDCl3): δC (ppm) 44.26 (2C), 66.51 (2C), 120.14 (2C), 123.40, 128.95 (2C), 138.71, 155.19 (C=O). HRMS (ESI+): m/z calcd for C11H15N2O2 [M + H]+ 207.1133, found 207.1128.
N-Morpholinobenzamide (30). To a solution of benzoic acid (2.50 g, 20.47 mmol, 1 equiv.) in CH2Cl2, a catalytic amount of DMF and oxalyl chloride were added (2.60 g, 20.47 mmol, 1 equiv.) and the mixture was stirred at room temperature until the generation of gasses (HCl) was stopped. The reaction was cooled to 0 °C before adding a solution of morpholin-4-amine (3.13 g, 30.7 mmol, 1.5 equiv.) and DIPEA (4.53 g, 40.94 mmol, 2 equiv.) and the reaction was stirred at room temperature for 6 h. The solution was quenched by water (30 mL) and the mixture was extracted with CH2Cl2 (100 mL). The combined organic layers were washed with brine and dried over Na2SO4. The solvent was removed in vacuo, and the resulting crude product was purified by silica gel chromatography (Cyclohexane/EtOAc: 3/2) to give the title compound as yellow oil (2.05 mmol, 0.42 g, 10%). Rf 0.25 (Cycloxane/EtOAc: 3/2). 1H NMR (400 MHz, CDCl3): δH (ppm) 2.96–2.97 (m, 4H), 3.87–3.89 (m, 4H), 6.84 (s, 1H), 7.40–7.56 (m, 2H), 7.52–7.54 (m, 1H), 7.74–7.75 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 56.12 (2C), 66.44 (2C), 127.05, 128.69 (2C), 128.69 (2C), 144.76, 151.61(C=O). HRMS (ESI+): m/z calcd for C11H15N2O2 [M + H]+ 207.11335, found 207.11221.
(E)-N-Morpholino-1-phenylmethanimine (31). To a solution of MgSO4 (2.27 g, 18.86 mmol, 2 equiv.) in CH2Cl2 was added benzaldehyde (0.96 mL, 9.43 mmol, 1 equiv.) under Ar and the solution was stirred overnight at room temperature. The solution was filtered to remove MgSO4 and evaporated under reduced pressure. The resulting crude product was purified by silica gel chromatography (CH2Cl2) to give the title compound as a white solid (7.35 mmol, 1.39 g, 78%). Rf 0.2 (CH2Cl2). M.p.: 86–88 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.17–319 (m, 4H), 3.88–3.91 (m, 4H), 7.26–730 (m, 1H), 7.33–7.37 (m, 2H), 7.60–7.61 (m, 3H). 13C NMR (100 MHz, CDCl3): δC (ppm) 51.88 (2C), 66.50 (2C), 126.23 (2C), 128.40, 128.58 (2C), 135.92, 136.31(C=N). HRMS (ESI+): m/z calcd for C11H15N2O [M + H]+ 191.11844, found 191.11733.
1-Morpholino-3-phenylthiourea (32). To a solution of isothiocyanatobenzene (1.00 g, 7.40 mmol, 1 equiv.) in MeOH, 4-amino-morpholin (0.91 g, 8.9 mmol, 1.2 equiv.) was slowly added and the reaction was monitored with TLC until complete disappearance of the starting material and the formation of a white solid. The solution was then filtered and residue recovered was recrystallized in MeOH to afford the desired product as a white solid (5.92 mmol, 1.40 g, 80%). Rf 0.35 (Cyclohexane/EtOAc: 3/2). M.p.: 160–161 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 2.99–3.02 (m, 4H), 3.74–3.76 (m, 4H), 7.54–7.59 (m, 2H) 7.62–7.66 (m, 1H), 7.75–7.78 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 45.54 (2C), 65.74 (2C), 127.45 (2C), 128.80 (2C), 132.64, 134.60, 157.40 (C=S). HRMS (ESI+): m/z calcd for C11H16N3OS [M + H]+ 238.1114, found 238.1009.
1-Morpholino-3-phenylurea (33). To a stirring solution of phenylisocyanate (1.00 g, 8.4 mmol, 1 equiv.) in CH2Cl2, 4-amino-morpholin (1.03 g, 10.1 mmol, 1.2 equiv.) was slowly added and the reaction was monitored with TLC until complete disappearance of the starting material. A white solid appeared in the mixture. The solution was filtered and the residue was purified by silica gel chromatography (EtOAc) to give the title compound as a white solid (6.64 mmol, 1.47 g, 79%). Rf 0.4 (EtOAc). M.p.: 147–148 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 2.67–3.05 (m, 4H), 3.71–3.91 (m, 4H), 5.53 (s, 1H), 7.04–7.08 (m, 1H), 7.29–7.33 (m, 2H), 7.47–7.49 (m, 2H), 8.05 (s, 1H). 13C NMR (100 MHz, CDCl3): δC (ppm) 56.81 (2C), 66.75 (2C), 119.21 (2C), 123.26, 129.02 (2C), 138.07, 154.75 (C=O). HRMS (ESI+): m/z calcd for C11H14NO2 [M + H]+ 222.1242, found 222.1231.
Benzyl morpholine-4-carbodithioate (34). To a solution of CS2 (0.5 g, 6.6 mmol, 1 equiv.) in diethyl ether, morpholine (1.13 mL, 13.2 mmol, 2 equiv.) was slowly added. The resulting mixture was stirred for 1 h at room temperature. The precipitated salt was recovered by filtration and dried over vacuum. The morpholinium salt (0.5 g, 2.0 mmol, 1 equiv.) was then mixed with (thiocyanatomethyl)benzene (0.3 g, 2.0 mmol, 1 equiv.) in acetonitrile and stirred at room temperature during 2 h. The solution was evaporated under vacuum, the resulting powder was diluted in 15 mL of water and extracted with EtOAc. The combined organic layers were washed with brine and dried over Na2SO4. The solvents were removed in vacuo, and the resulting crude product was purified by silica gel chromatography (Cyclohexane/EtOAc: 3/2) to give the title compound as a white solid (0.8 mmol, 0.2 g, 42%). Rf 0.7 (Cyclohexane/EtOAc: 3/2). M.p.: 70–71 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.38–3.40 (m, 2H), 3.65–3.68, (m, 2H), 3.80 (m, 4H), 3.76 (bs, 4H), 3.93–4.34 (m, 4H), 4.58 (s, 2H), 7.26–7.34 (m, 3H), 7.38–7.39 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 42.03 (2C), 66.43 (2C), 127.66, 128.66 (2C), 129.41 (2C), 135.69, 197.20 (C=S). HRMS (ESI+): m/z calcd for C12H16NOS2 [M + H]+ 254.06733, found 254.06602.
(S)-N-(2-Hydroxy-1-phenylethyl)morpholine-4-carboxamide (35). To a solution of (S)-(+)-2phenylglycinol (1.00 g, 7.29 mmol, 1.5 equiv.) in anhydrous CH2Cl2 under Ar, DIPEA (1.71 mL, 9.71 mmol, 2 equiv.) and 4-morpholinylcarbonyl chloride (0.57 mL, 4.86 mmol, 1 equiv.) were added at 0 °C. The resulting solution was stirred at room temperature for 12 h. The solvent was removed in vacuo, and the resulting crude product was purified by silica gel chromatography (CH2Cl2/MeOH: 9/1) to give the title compound as a yellow solid (4.32 mmol, 1.62 g, 89%). Rf 0.2 (EtOAc). M.p.: 90–91 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.10–3.21 (m, 4H), 3.45–3.54 (m, 4H), 3.57–3.67 (m, 2H), 4.31 (s, 1H), 4.76–4.81 (m, 1H), 5.83 (d, 1H, J = 6.2 Hz), 7.20–7.25 (m, 3H), 7.29–7.32 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 43.89 (2C), 57.11, 66.02, 66.38 (2C), 126.58 (2C), 127.43, 128.54 (2C), 140.67, 158.06 (C=O). HRMS (ESI+): m/z calcd for C13H19N2O3 [M + H]+ 251.13957, found 251.13835.
4-(Phenylsulfonyl)morpholine (36). To a solution of benzenesulfonyl chloride (1.65 g, 9.60 mmol, 1.2 equiv.) in CH2Cl2, morpholine (1.00 g, 11.5 mmol, 1 equiv.) and Et3N (0.97 mL, 9.60 mmol, 1.2 equiv.) were added and the reaction was stirred during 3 h. A white solid slowly appeared in the mixture. The residue was filtered and recrystallized in EtOH to afford the desired compound as a white solid (8.16 mmol, 1.85 g, 85%). Rf 0.27 (Cyclohexane/EtOAc: 3/2). M.p.: 107–109 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 2.99–3.02 (m, 4H), 3.74–3.76 (m, 4H), 7.54–7.59 (m, 2H), 7.62–7.66 (m, 1H), 7.75–7.78 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 45.63 (2C), 65.74 (2C), 127.50 (2C), 128.80 (2C), 132.75, 134.70. HRMS (ESI+): m/z calcd for C10H14NO3S [M + H]+ 228.0694, found 228.0688.
N-Morpholinobenzenesulfonamide (37). To a solution of benzenesulfonyl chloride (1.65 g, 9.60 mmol, 1.2 equiv.) in CH2Cl2 was added 4-amino-morpholine (1.17 g, 11.5 mmol, 1 equiv.) and Et3N (0.97 mL, 9.60 mmol, 1.2 equiv.). The reaction was stirred for 3 h and followed by TLC. After complete disappearance of the starting material, 100 mL of water was added on the mixture and the solution was extracted with CH2Cl2 (75 mL). The organic layers were dried and evaporated under vacuum. The solid recovered was recrystallized in EtOH to afford the desired compound as a white solid (7.97 mmol, 1.93 g, 83%). Rf 0.29 (Cyclohexane/EtOAc: 3/2). M.p.: 110–112 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 2.60–2.63 (m, 4H), 3.60–3.62 (m, 4H), 5.34 (s, 1H, NH), 7.51–7.55 (m, 2H), 7.60–7.64 (m, 1H), 7.97–7.99 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 56.74 (2C), 66.64 (2C), 128.16 (2C), 128.89 (2C), 133.21, 138.61. HRMS (ESI+): m/z calcd for C10H15N2O3S [M + H]+ 243.0803, found 243.0798.
(4-Chlorophenyl)(morpholino)methanethione (38). 4-chloro-benzaldehyde (1.00 g, 7.10 mmol, 1 equiv.), morpholine (1.86 g, 21.30 mmol, 3.0 equiv.), and sulfur (0.34 g, 10.70 mmol, 1 equiv.) were mixed in DMF and the solution stirred at 85 °C for 12 h. The solution was filtered, H2O (100 mL) was added and the organic layer was extracted 3 times with EtOAc. The organic layer was evaporated under vacuo, the residue was purified by silica gel chromatography (Cyclohexane/EtOAc: 4/1) to give a yellow powder. This powder was recrystallized in EtOH to give the title compound as a yellow solid (4.59 mmol, 1.11 g, 65%). Rf 0.5 (Cyclohexane/EtOAc: 3/2). M.p.: 140–142 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.60–3.65 (m, 4H), 3.87–3.89 (m, 2H), 4.41–4.43 (m, 2H), 7.23–7.25 (m, 2H), 7.33–7.35 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 49.55, 52.53, 66.46, 66.62, 126.97 (2C), 128.95 (2C), 134.93, 140.60, 199.54 (C=S). HRMS (ESI+): m/z calcd for C11H13ClNOS [M + H]+ 242.04064, found 242.03942.
4-Chloro-N-morpholinobenzenesulfonamide (39). To a solution of 4-chlorobenzenesulfonyl chloride (1.24 g, 5.87 mmol, 1.2 equiv.) in CH2Cl2 was added 4-amino-morpholine (0.5 g, 4.89 mmol, 1 equiv.) and Et3N (0.82 mL, 5.87 mmol, 1.2 equiv.). The reaction was stirred for 12 h. After complete disappearance of the starting material, 100 mL of water was added on the mixture and the solution was extracted with CH2Cl2 (75 mL). The organic layers were dried and evaporated under vacuum. The solid recovered was recrystallized in EtOH to afford the desired compound as a white solid (4.34 mmol, 1.20 g, 74%). Rf 0.55 (Cyclohexane/EtOAc: 3/2). M.p.: 161–163 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 2.64–2.66 (m, 4H), 3.61–3.64 (m, 4H), 5.34 (s, 1H, NH), 7.50–7.52 (m, 2H), 7.90–7.92 (m, 2H). 13C NMR (100 MHz, CDCl3): δC (ppm) 54.65 (2C), 64.46 (2C), 127.04 (2C), 127.44 (2C), 134.95, 137.64. HRMS (ESI+): m/z calcd for C10H15NO3S [M + H]+ 277.0414, found 277.0408.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





















