Contact System Activation in Plasma from Dengue Patients Might Harness Endothelial Virus Replication through the Signaling of Bradykinin Receptors

 ,
, 
 ,
,
Abstract
:
1. Introduction
2. Results
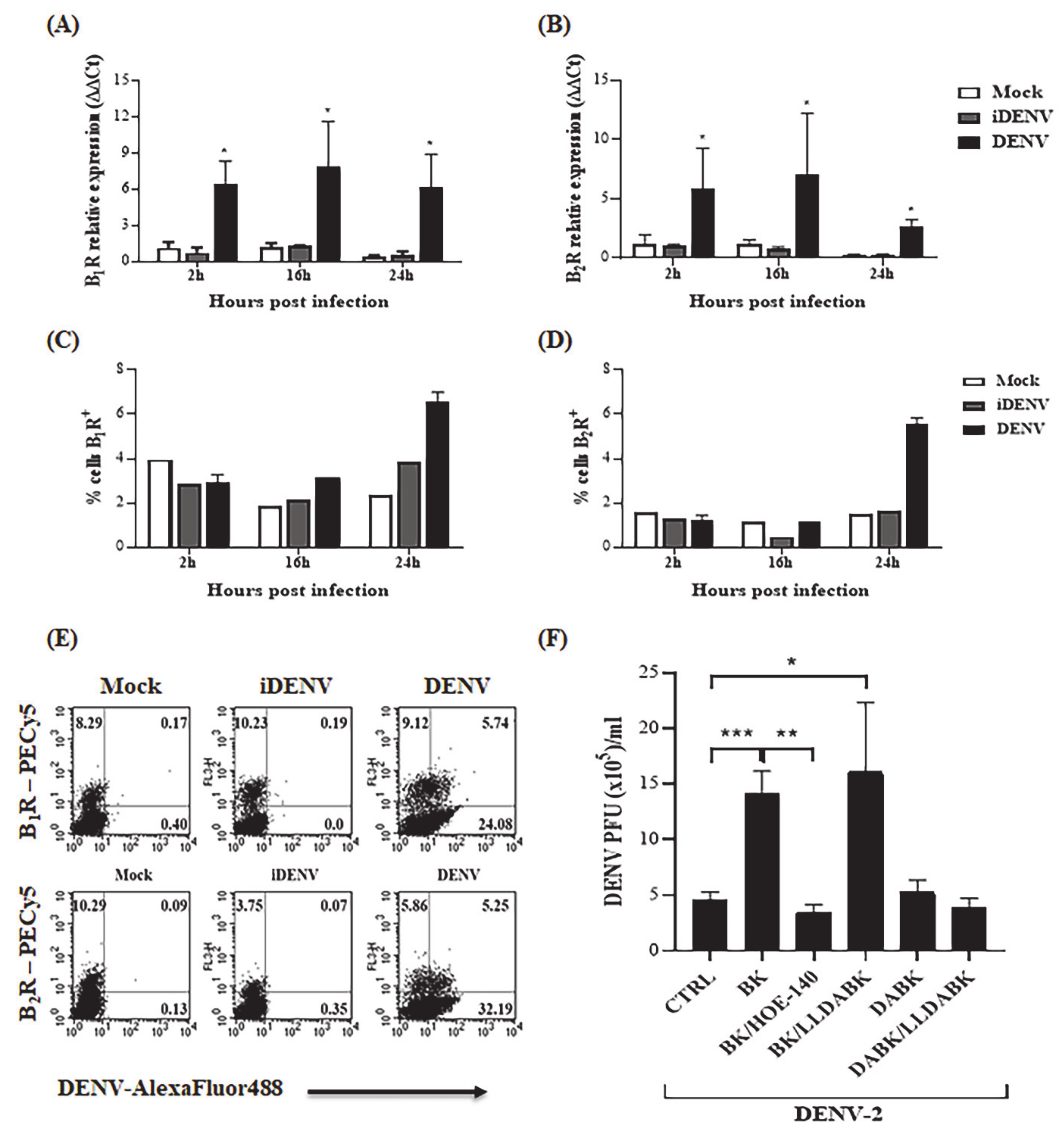
2.1. Bradykinin Increases DENV-2 Replication in Human Microvascular Endothelial Cells
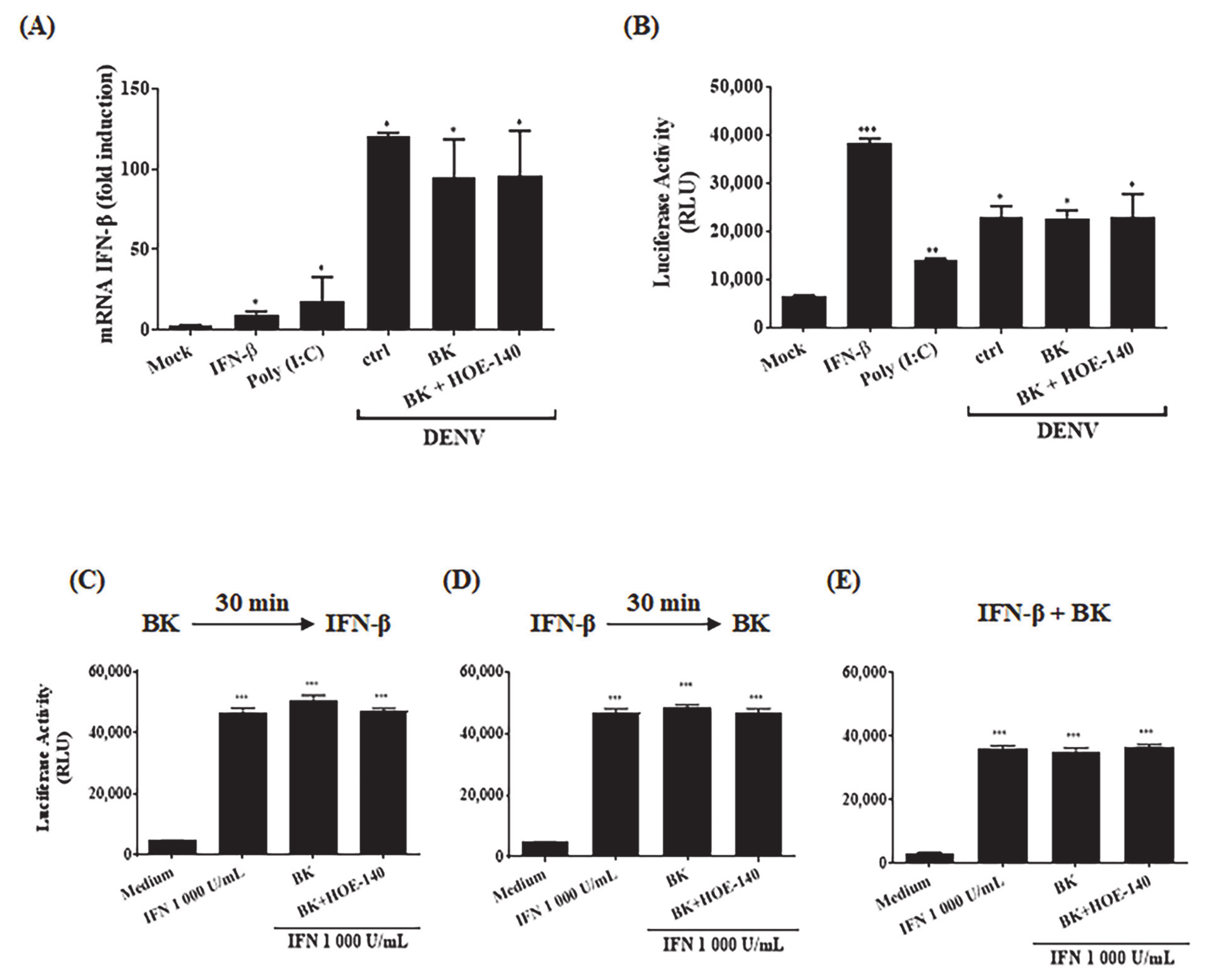
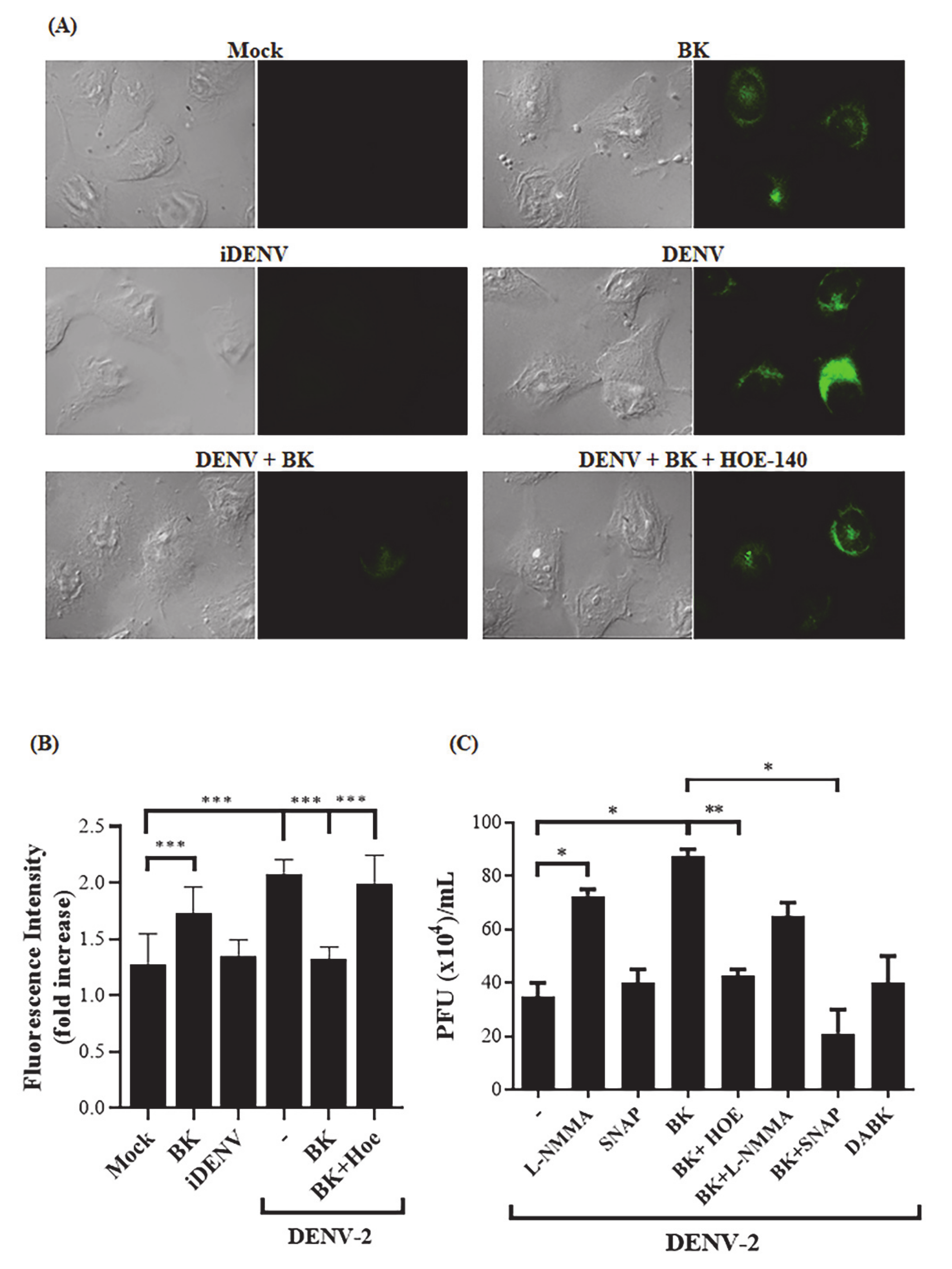
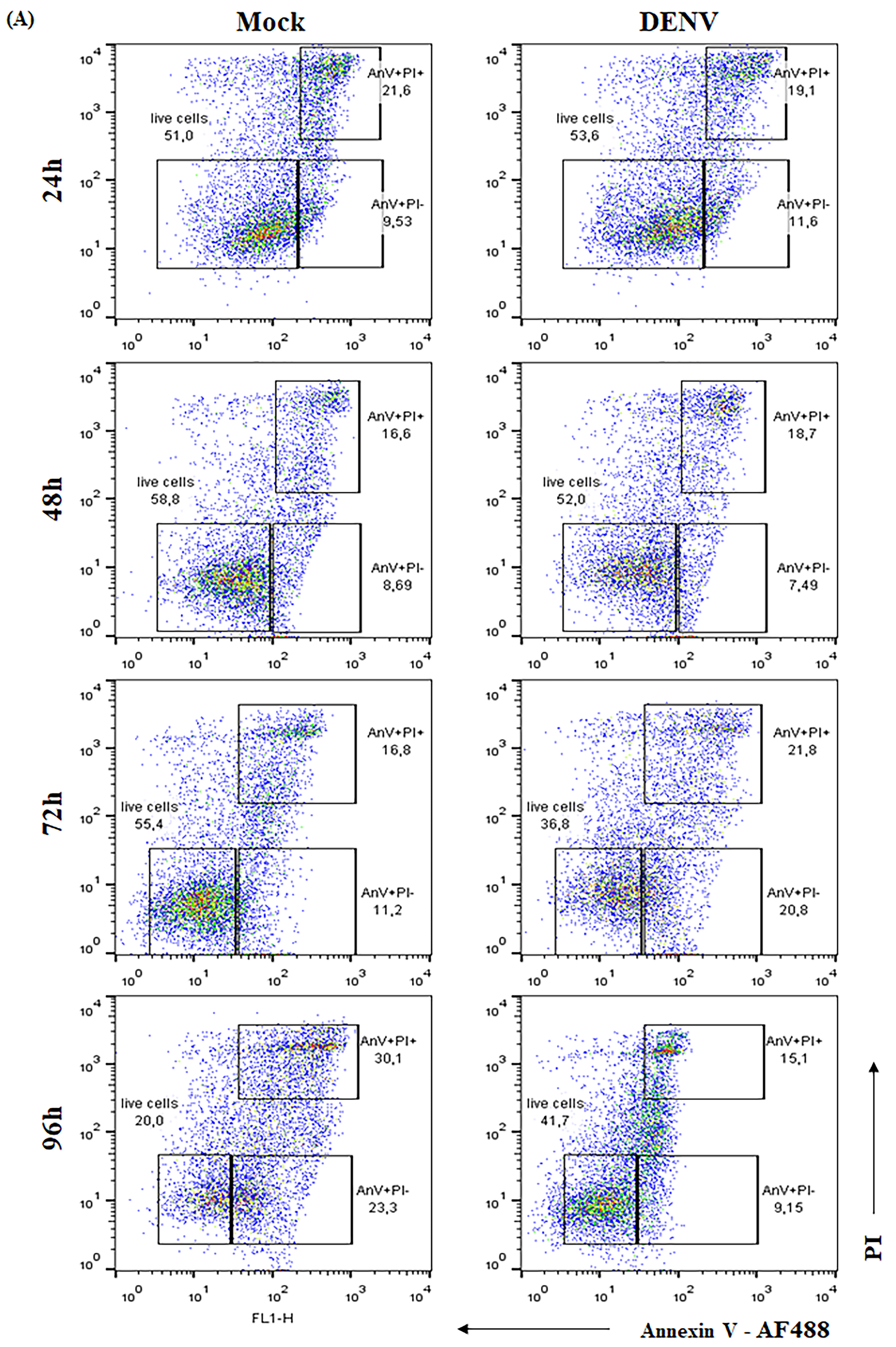
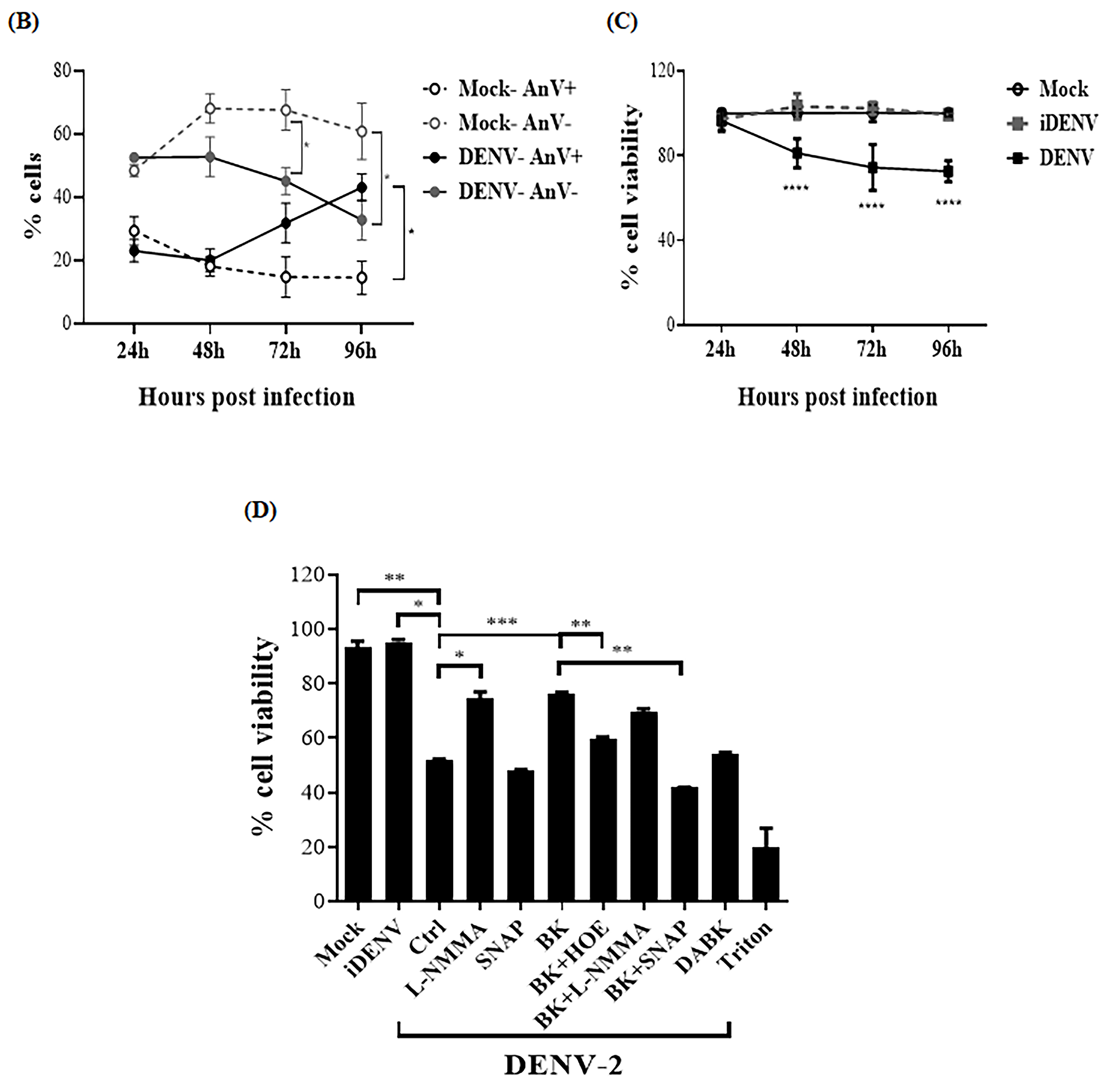
2.2. BK Decreases Nitric Oxide Production and Delays Cell Death Induced by DENV-2 in Endothelial Cells
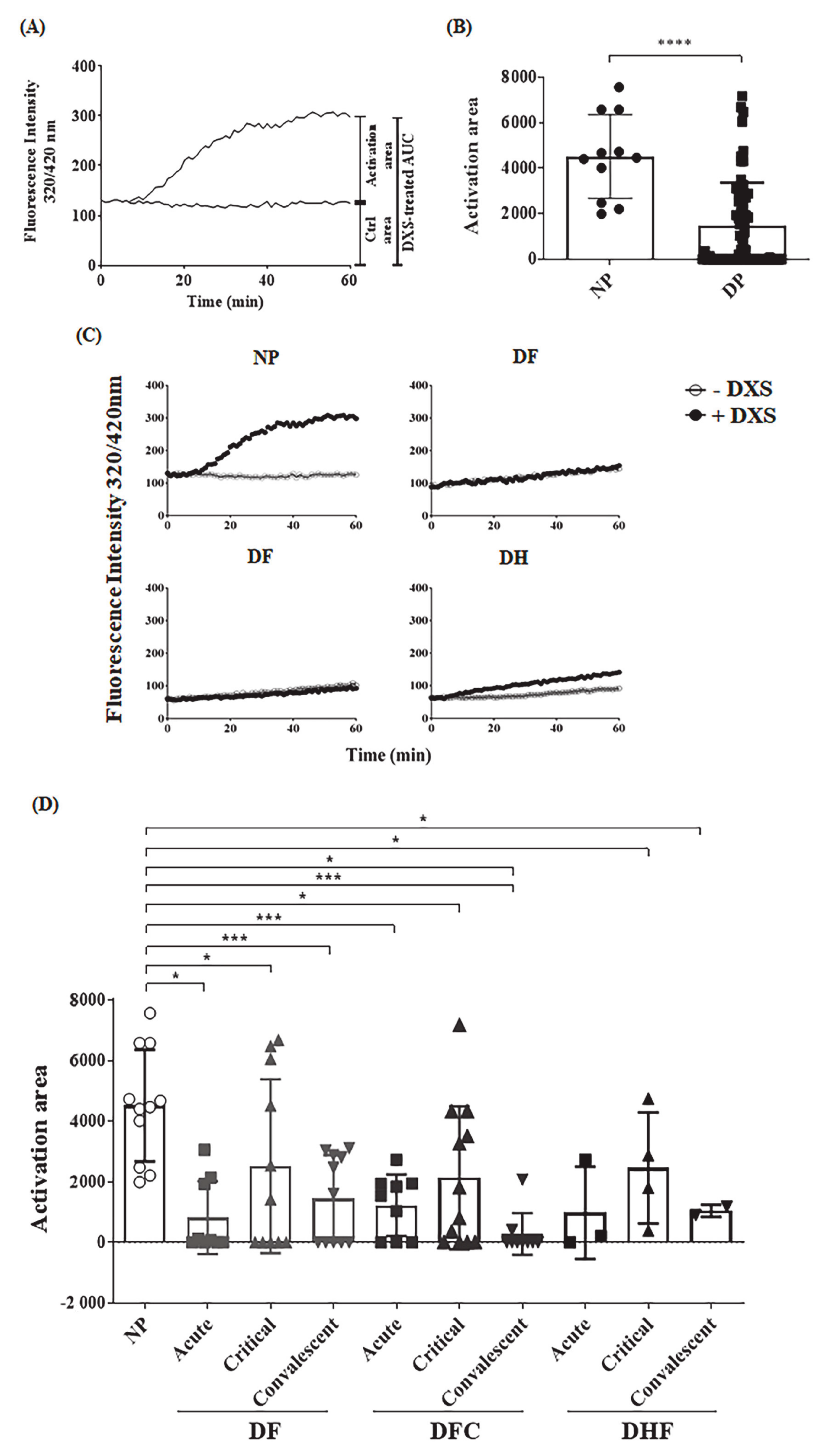
2.3. Ex Vivo Evaluation of the Contact Pathway Activation in Plasmas Obtained from Dengue Patients
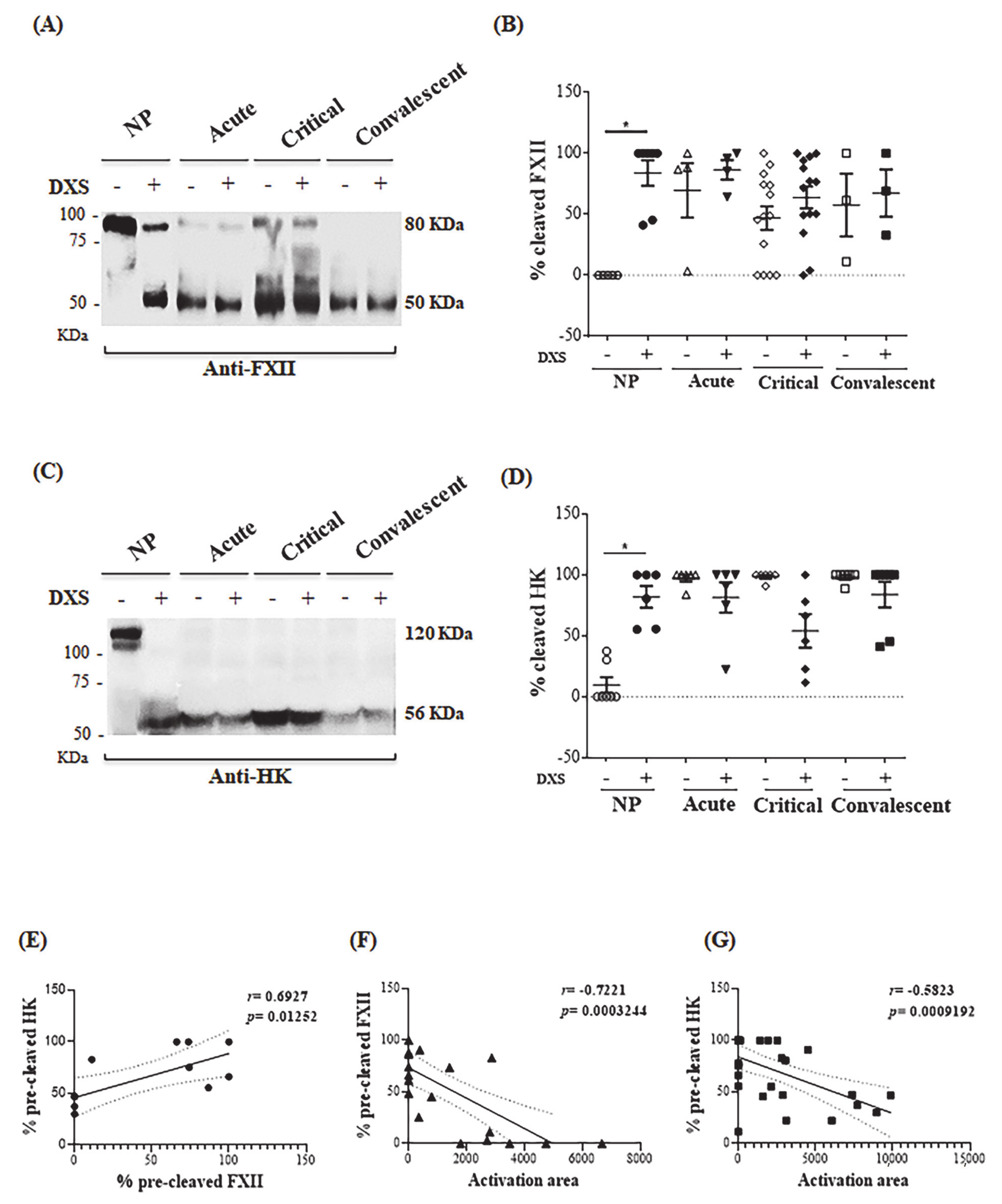
2.4. DENV Modulates the Kallikrein-Kinin Pathway by Consuming Plasma Elements Early after Infection
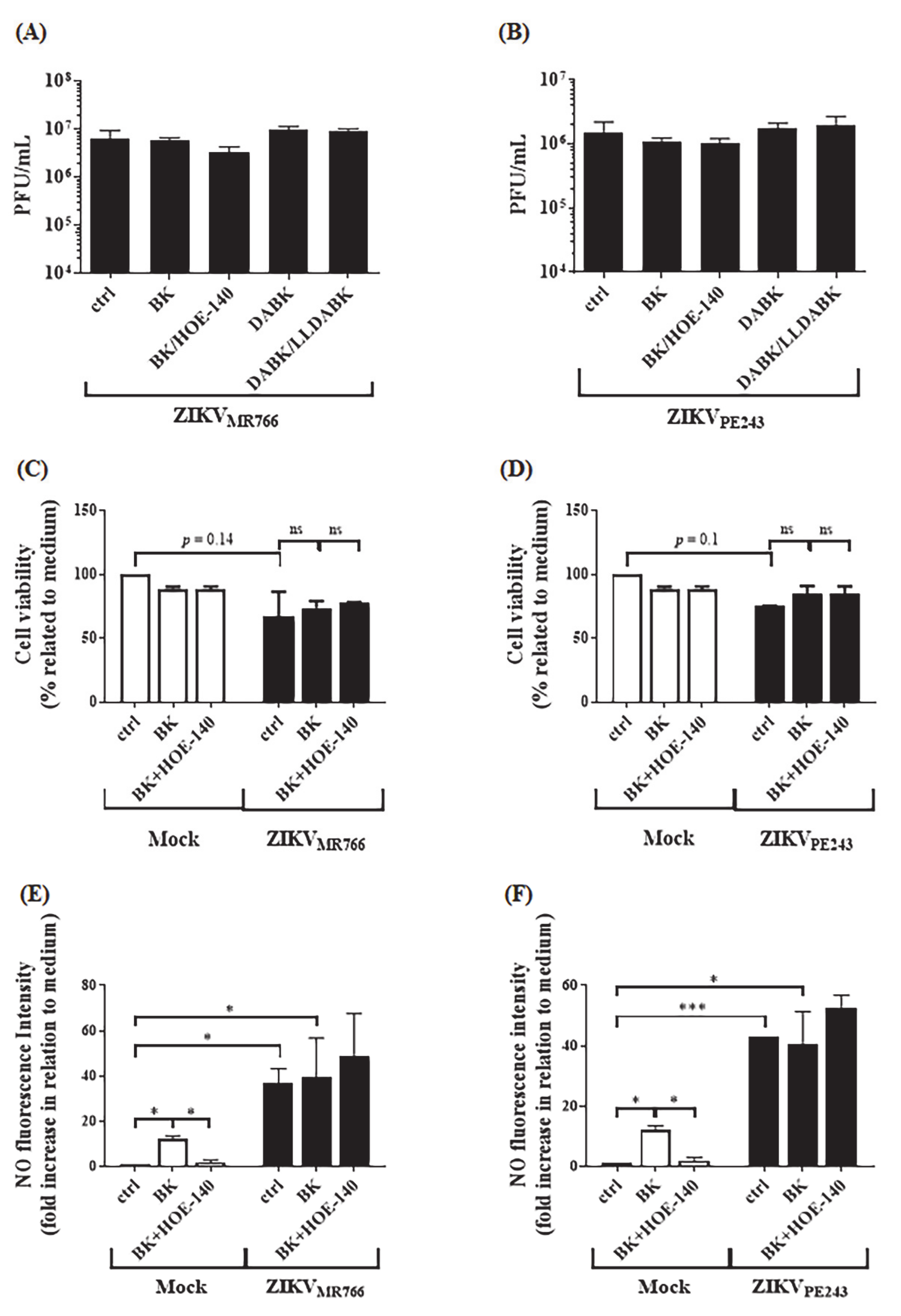
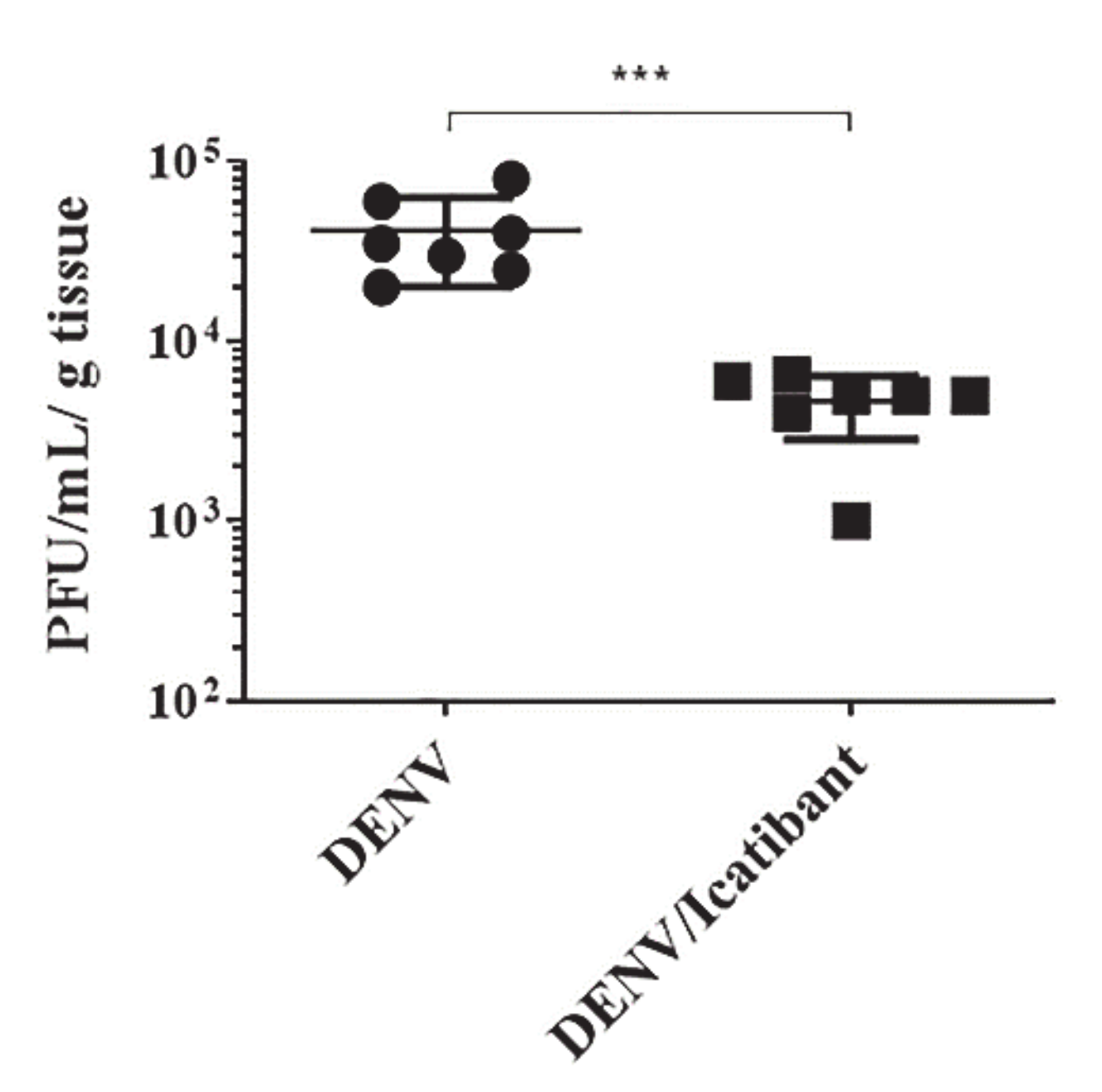
2.5. B2R Targeting in the Intracerebral Model of Dengue Reduced Viral Load in Brain Tissues
3. Discussion
4. Material and Methods
4.1. Ethical Statements
4.2. Cells and Virus
4.3. HBMEC Infection and Stimulation In Vitro
4.4. Virus Titration
4.5. Evaluation of BKR Expression by qRT-PCR and Flow Cytometry
4.6. Evaluation of IFN-β Production and IFN-Mediated Response
4.7. Nitric Oxide Production
4.8. Cell Viability
4.9. DENV Plasmas Samples Cohort
4.10. Ex Vivo Analysis of Contact System Activation
4.11. Evaluation of the Role of NS1, Apoferritin and LDL
4.12. Analysis of FXII, HK and C1INH by Western Blotting
4.13. Mouse Infection
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Broadley, K.J.; Blair, A.E.; Kidd, E.J.; Bugert, J.J.; Ford, W.R. Bradykinin-induced lung inflammation and bronchoconstriction: Role in parainfluenze-3 virus-induced inflammation and airway hyperreactivity. J. Pharmacol. Exp. Ther. 2010, 335, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.M.; Wong, R.S.; Wu, E.B.; Kong, S.L.; Wong, J.; Yip, G.W.; Soo, Y.O.; Chiu, M.L.; Chan, Y.S.; Hui, D.; et al. Cardiovascular complications of severe acute respiratory syndrome. Postgrad. Med. J. 2006, 82, 140–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Lu, H.; Zhang, W. Clinical observation and management of COVID-19 patients. Emerg. Microbes. Infect. 2020, 9, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.L.; Wahl-Jensen, V.; Copeland, A.M.; Jahrling, P.B.; Schmaljohn, C.S. Endothelial cell permeability during hantavirus infection involves factor XII-dependent increased activation of the kallikrein-kinin system. PLoS Pathog. 2013, 9, 1–14. [Google Scholar] [CrossRef]
- Azeredo, E.L.; Monteiro, R.Q.; de-Oliveira Pinto, L.M. Thrombocytopenia in Dengue: Interrelationship between Virus and the Imbalance between Coagulation and Fibrinolysis and Inflammatory Mediators. Mediators Inflamm. 2015, 2015, 1–16. [Google Scholar] [CrossRef] [Green Version]
- St Maurice, A.; Harmon, J.; Nyakarahuka, L.; Balinandi, S.; Tumusiime, A.; Kyondo, J.; Mulei, S.; Namutebi, A.; Knust, B.; Shoemaker, T.; et al. Rift valley fever viral load correlates with the human inflammatory response and coagulation pathway abnormalities in humans with hemorrhagic manifestations. PLoS Negl. Trop. Dis. 2018, 12, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Avila-Aguero, M.L.; Avila-Aguero, C.R.; Um, S.L.; Soriano-Fallas, A.; Cañas-Coto, A.; Yan, S.B. Systemic host inflammatory and coagulation response in the Dengue virus primo-infection. Cytokine 2004, 27, 173–179. [Google Scholar] [CrossRef]
- Huerta-Zepeda, A.; Cabello-Gutiérrez, C.; Cime-Castillo, J.; Monroy-Martínez, V.; Manjarrez-Zavala, M.E.; Gutiérrez-Rodríguez, M.; Izaguirre, R.; Ruiz-Ordaz, B.H. Crosstalk between coagulation and inflammation during Dengue virus infection. Thromb. Haemost. 2008, 99, 936–943. [Google Scholar] [CrossRef]
- Chagan-Yasutan, H.; Lacuesta, T.L.; Ndhlovu, L.C.; Oguma, S.; Leano, P.S.; Telan, E.F.; Kubo, T.; Morita, K.; Uede, T.; Dimaano, E.M.; et al. Elevated levels of full-length and thrombin-cleaved osteopontin during acute dengue virus infection are associated with coagulation abnormalities. Thromb. Res. 2014, 134, 449–454. [Google Scholar] [CrossRef] [Green Version]
- Fried, J.R.; Gibbons, R.V.; Kalayanarooj, S.; Thomas, S.J.; Srikiatkhachorn, A.; Yoon, I.K.; Jarman, R.G.; Green, S.; Rothman, A.L.; Cummings, D.A. Serotype-specific differences in the risk of dengue hemorrhagic fever: An analysis of data collected in Bangkok, Thailand from 1994 to 2006. PLoS Negl. Trop. Dis. 2010, 4, e617. [Google Scholar] [CrossRef] [Green Version]
- Guzman, M.G.; Halstead, S.B.; Artsob, H.; Buchy, P.; Farrar, J.; Gubler, D.J.; Hunsperger, E.; Kroeger, A.; Margolis, H.S.; Martínez, E.; et al. Dengue: A continuing global threat. Nat. Rev. Microbiol. 2010, 8, S7–S16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, V.V.; Fagundes, C.T.; Valadão, D.F.; Cisalpino, D.; Dias, A.C.; Silveira, K.D.; Kangussu, L.M.; Ávila, T.V.; Bonfim, M.R.; Bonaventura, D.; et al. A model of DENV-3 infection that recapitulates severe disease and highlights the importance of IFN-γ in host resistance to infection. PLoS Negl. Trop. Dis. 2012, 6, e1663. [Google Scholar] [CrossRef] [PubMed]
- Messina, J.P.; Brady, O.J.; Golding, N.; Kraemer, M.U.G.; Wint, G.R.W.; Ray, S.E.; Pigott, D.M.; Shearer, F.M.; Johnson, K.; Earl, L.; et al. The current and future global distribution and population at risk of dengue. Nat. Microbiol. 2019, 4, 1508–1515. [Google Scholar] [CrossRef] [PubMed]
- Brazilian Ministry of Health. 2020. Available online: https://www.saude.gov.br/component/tags/tag/dengue (accessed on 15 September 2020).
- Joob, B.; Wiwanitkit, V. COVID-19 in medical personnel: Observation from Thailand. J. Hosp. Infect. 2020, 104, 453. [Google Scholar] [CrossRef]
- Yan, G.; Lee, C.K.; Lam, L.T.; Yan, B.; Chua, Y.X.; Lim, A.Y.; Lin, L. Covert COVID-19 and false positive dengue serology in Singapore. Lancet Infect. Dis. 2020, 20, 536. [Google Scholar] [CrossRef] [Green Version]
- Estofolete, C.F.; Machado, L.F.; Zini, N.; Luckemeyer, G.D.; Moraes, M.M.; Dos Santos, T.N.I.L.; Dos Santos, B.F.; Ruiz, L.G.P.; Vasilakis, N.; Lobo, S.M.A.; et al. Fatal stroke as presentation of SARS-CoV-2 and dengue virus coinfection. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Dalrymple, N.A.; Mackow, E.R. Roles for endothelial cells in dengue virus infection. Adv. Virol. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Calderón-Peláez, M.A.; Velandia-Romero, M.L.; Bastidas-Legarda, L.Y.; Beltrán, E.O.; Camacho-Ortega, S.J.; Castellanos, J.E. Dengue Virus Infection of Blood-Brain Barrier Cells: Consequences of Severe Disease. Front. Microbiol. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Jessie, K.; Fong, M.Y.; Devi, S.; Lam, S.K.; Wong, K.T. Localization of dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. J. Med. Virol. 2004, 189, 1411–1418. [Google Scholar] [CrossRef]
- Póvoa, T.; Alves, A.; Oliveira, C.; Nuovo, G.; Chagas, V.; Paes, M. The pathology of severe dengue in multiple organs of human fatal cases: Histopathology, ultrastructure and virus replication. PLoS ONE 2014, 9, e83386. [Google Scholar] [CrossRef] [Green Version]
- Salomão, N.; Rabelo, K.; Basílio-de-Oliveira, C.; Basílio-de-Oliveira, R.; Geraldo, L.; Lima, F.; Dos Santos, F.; Nuovo, G.; Oliveira, E.R.A.; Paes, M. Fatal Dengue Cases Reveal Brain Injury and Viral Replication in Brain-Resident Cells Associated with the Local Production of Pro-Inflammatory Mediators. Viruses 2020, 12, 603. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Chaturvedi, U.C. Vascular endothelium: The battlefield of dengue viruses. FEMS Immunol. Med. Microbiol. 2008, 53, 287–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, W.; Mao, J.; Yang, Y.; Yuan, J.; Chen, J.; Luo, Y.; Lai, T.; Zuo, L. Mechanisms of mTOR and Autophagy in Human Endothelial Cell Infected with Dengue Virus-2. Viral. Immunol. 2020, 33, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, G.; Visoso-Carvajal, G.; Garcia-Cordero, J.; Leon-Juarez, M.; Chavez-Munguia, B.; Lopez, T.; Nava, P.; Villegas-Sepulveda, N.; Cedillo-Barron, L. Dengue Virus Serotype 2 and Its Non-Structural Proteins 2A and 2B Activate NLRP3 Inflammasome. Front. Immunol. 2020, 11, 352. [Google Scholar] [CrossRef]
- Ashour, J.; Morrison, J.; Laurent-Rolle, M.; Belicha-Villanueva, A.; Plumlee, C.R.; Bernal-Rubio, D.; Williams, K.L.; Harris, E.; Fernandez-Sesma, A.; Schindler, C.; et al. Mouse STAT2 restricts early dengue virus replication. Cell Host. Microbe. 2010, 8, 410–421. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, S.; Maestre, A.M.; Pagni, S.; Patel, J.R.; Savage, T.; Gutman, D.; Maringer, K.; Bernal-Rubio, D.; Shabman, R.S.; Simon, V.; et al. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. 2012, 8, e1002934. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.E.; Diamond, M.S. Dengue mouse models for evaluating pathogenesis and countermeasures. Curr. Opin. Virol. 2020, 43, 50–58. [Google Scholar] [CrossRef]
- Oliveira, E.R.; Amorim, J.F.; Paes, M.V.; Azevedo, A.S.; Gonçalves, A.J.; Costa, S.M.; Mantuano-Barradas, M.; Póvoa, T.F.; de Meis, J.; Basílio-de-Oliveira, C.A.; et al. Peripheral effects induced in BALB/c mice infected with DENV by the intracerebral route. Virology 2016, 489, 95–107. [Google Scholar] [CrossRef] [Green Version]
- Amorim, J.F.S.; Azevedo, A.S.; Costa, S.M.; Trindade, G.F.; Basílio-de-Oliveira, C.A.; Gonçalves, A.J.S.; Salomão, N.G.; Rabelo, K.; Amaral, R.; Geraldo, L.H.M.; et al. Dengue infection in mice inoculated by the intracerebral route: Neuropathological effects and identification of target cells for virus replication. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, A.S.; Gonçalves, A.J.; Archer, M.; Freire, M.S.; Galler, R.; Alves, A.M. The synergistic effect of combined immunization with a DNA vaccine and chimeric yellow fever/dengue virus leads to strong protection against dengue. PLoS ONE 2013, 8, 1–10. [Google Scholar] [CrossRef]
- Wang, R.; Gao, N.; Li, Y.; Fan, D.; Zhen, Z.; Feng, K.; Chen, H.; An, J. Cross-Protection Against Four Serotypes of Dengue Virus in Mice Conferred by a Zika DNA Vaccine. Front. Cell Infect. Microbiol. 2019, 9, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, A.P.; Joseph, K. Pathogenic mechanisms of bradykinin mediated diseases: Dysregulation of an innate inflammatory pathway. Adv. Immunol. 2014, 121, 41–89. [Google Scholar] [PubMed]
- Renné, T.; Pozgajová, M.; Grüner, S.; Schuh, K.; Pauer, H.U.; Burfeind, P.; Gailani, D.; Nieswandt, B. Defective thrombus formation in mice lacking coagulation factor XII. J. Exp. Med. 2005, 202, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinschnitz, C.; Stoll, G.; Bendszus, M.; Schuh, K.; Pauer, H.U.; Burfeind, P.; Renné, C.; Gailani, D.; Nieswandt, B.; Renné, T. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J. Exp. Med. 2006, 203, 513–518. [Google Scholar] [CrossRef] [Green Version]
- Stavrou, E.X.; Fang, C.; Merkulova, A.; Alhalabi, O.; Grobe, N.; Antoniak, S.; Mackman, N.; Schmaier, A.H. Reduced thrombosis in Klkb1-/- mice is mediated by increased Mas receptor, prostacyclin, Sirt1, and KLF4 and decreased tissue factor. Blood 2015, 125, 710–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merkulov, S.; Zhang, W.M.; Komar, A.A.; Schmaier, A.H.; Barnes, E.; Zhou, Y.; Lu, X.; Iwaki, T.; Castellino, F.J.; Luo, G.; et al. Deletion of murine kininogen gene 1 (mKng1) causes loss of plasma kininogen and delays thrombosis. Blood 2008, 111, 1274–1281. [Google Scholar] [CrossRef]
- Schmaier, A.H. The contact activation and kallikrein/kinin systems: Pathophysiologic and physiologic activities. J. Thromb. Haemost. 2016, 14, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Verhoef, J.J.; Barendrecht, A.D.; Nickel, K.F.; Dijkxhoorn, K.; Kenne, E.; Labberton, L.; McCarty, O.J.; Schiffelers, R.; Heijnen, H.F.; Hendrickx, A.P.; et al. Polyphosphate nanoparticles on the platelet surface trigger contact system activation. Blood 2017, 129, 1707–1717. [Google Scholar] [CrossRef]
- Regoli, D.; Rhaleb, N.E.; Dion, S.; Drapeau, G. New selective bradykinin receptor antagonists and bradykinin B2 receptor characterization. Trends Pharmacol. Sci. 1990, 11, 156–161. [Google Scholar] [CrossRef]
- Marceau, F.; Bachelard, H.; Bouthillier, J.; Fortin, J.P.; Morissette, G.; Bawolak, M.T.; Charest-Morin, X.; Gera, L. Bradykinin receptors: Agonists, antagonists, expression, signaling, and adaptation to sustained stimulation. Int. Immunopharmacol. 2020, 82, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Pesquero, J.B.; Bader, M. Molecular biology of the kallikrein-kinin system: From structure to function. Braz. J. Med. Biol. Res. 1998, 31, 1197–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calixto, J.B.; Cabrini, D.A.; Ferreira, J.; Campos, M.M. Kinins in pain and inflammation. Pain 2000, 87, 1–5. [Google Scholar] [CrossRef]
- Moreau, M.E.; Bawolak, M.T.; Morissette, G.; Adam, A.; Marceau, F. Role of nuclear factor-kappaB and protein kinase C signaling in the expression of the kinin B1 receptor in human vascular smooth muscle cells. Mol. Pharmacol. 2007, 71, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Regoli, D.; Rhaleb, N.E.; Drapeau, G.; Dion, S.; Tousignant, C.; D’Orléans-Juste, P.; Devillier, P. Basic pharmacology of kinins: Pharmacologic receptors and other mechanisms. Adv. Exp. Med. Biol. 1989, 247A, 399–407. [Google Scholar]
- Skidgel, R.A.; Erdös, E.G. Angiotensin converting enzyme (ACE) and neprilysin hydrolyze neuropeptides: A brief history, the beginning and follow-ups to early studies. Peptides 2004, 25, 521–525. [Google Scholar] [CrossRef]
- Zhang, X.; Tan, F.; Brovkovych, V.; Zhang, Y.; Lowry, J.L.; Skidgel, R.A. Carboxypeptidase M augments kinin B1 receptor signaling by conformational crosstalk and enhances endothelial nitric oxide output. Biol. Chem. 2013, 394, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B., Jr.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-arg(9)bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef]
- Aliberti, J.; Viola, J.P.; Vieira-de-Abreu, A.; Bozza, P.T.; Sher, A.; Scharfstein, J. Cutting edge: Bradykinin induces IL-12 production by dendritic cells: A danger signal that drives Th1 polarization. J. Immunol. 2003, 170, 5349–5353. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, A.C.; Schmitz, V.; Morrot, A.; de Arruda, L.B.; Nagajyothi, F.; Granato, A.; Pesquero, J.B.; Müller-Esterl, W.; Tanowitz, H.B.; Scharfstein, J. Bradykinin B2 Receptors of dendritic cells, acting as sensors of kinins proteolytically released by Trypanosoma cruzi, are critical for the development of protective type-1 responses. PLoS Pathog. 2007, 3, 1730–1744. [Google Scholar] [CrossRef]
- Scharfstein, J.; Ramos, P.I.P.; Barral-Netto, M. G Protein-Coupled Kinin Receptors and Immunity against Pathogens. Adv. Immunol. 2017, 136, 29–84. [Google Scholar]
- Nascimento, C.R.; Andrade, D.; Carvalho-Pinto, C.E.; Serra, R.R.; Vellasco, L.; Brasil, G.; Ramos-Junior, E.S.; da Mota, J.B.; Almeida, L.N.; Andrade, M.V.; et al. Mast Cell Coupling to the Kallikrein-Kinin System Fuels Intracardiac Parasitism and Worsens Heart Pathology in Experimental Chagas Disease. Front. Immunol. 2017, 8, 840. [Google Scholar] [CrossRef] [PubMed]
- Scharfstein, J. Subverting bradykinin-evoked inflammation by co-opting the contact system: Lessons from survival strategies of Trypanosoma cruzi. Curr. Opin. Hematol. 2018, 25, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Barnett, J.K.; Cruse, L.W.; Proud, D. Kinins are generated in nasal secretions during influenza A infections in ferrets. Am. Rev. Respir. Dis. 1990, 142, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Bengtson, S.H.; Eddleston, J.; Christiansen, S.C.; Zuraw, B.L. Double-stranded RNA increases kinin B1 receptor expression and function in human airway epithelial cells. Int. Immunopharmacol. 2007, 7, 1880–1887. [Google Scholar] [CrossRef]
- Naclerio, R.M.; Proud, D.; Lichtenstein, L.M.; Kagey-Sobotka, A.; Hendley, J.O.; Sorrentino, J.; Gwaltney, J.M. Kinins are generated during experimental rhinovirus colds. J. Infect. Dis. 1988, 157, 133–142. [Google Scholar] [CrossRef]
- Rust, N.M.; Papa, M.P.; Scovino, A.M.; da Silva, M.M.; Calzavara-Silva, C.E.; Marques, E.T., Jr.; Peçanha, L.M.; Scharfstein, J.; Arruda, L.B. Bradykinin enhances Sindbis virus infection in human brain microvascular endothelial cells. Virology 2012, 422, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Nikolskaia, O.V.; Lima, A.P.D.A.; Kim, Y.V.; Lonsdale-Eccles, J.D.; Fukuma, T.; Scharfstein, J.; Grab, D.J. Blood-brain barrier traversal by African trypanosomes requires calcium signaling induced by parasite cysteine protease. J. Clin. Investig. 2006, 116, 2739–2747. [Google Scholar] [CrossRef] [Green Version]
- da Conceição, T.M.; Rust, N.M.; Berbel, A.C.; Martins, N.B.; do Nascimento Santos, C.A.; Da Poian, A.T.; de Arruda, L.B. Essential role of RIG-I in the activation of endothelial cells by dengue virus. Virology 2013, 435, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Seliga, A.; Lee, M.H.; Fernandes, N.C.; Zuluaga-Ramirez, V.; Didukh, M.; Persidsky, Y.; Potula, R.; Gallucci, S.; Sriram, U. Kallikrein-Kinin System Suppresses Type I Interferon Responses: A Novel Pathway of Interferon Regulation. Front. Immunol 2018, 9, 156. [Google Scholar] [CrossRef] [Green Version]
- Harris, M.B.; Ju, H.; Venema, V.J.; Liang, H.; Zou, R.; Michell, B.J.; Chen, Z.P.; Kemp, B.E.; Venema, R.C. Reciprocal phosphorylation and regulation of endothelial nitric-oxide synthase in response to bradykinin stimulation. J. Biol. Chem. 2001, 276, 16587–16591. [Google Scholar] [CrossRef] [Green Version]
- Krieg, T.; Qin, Q.; Philipp, S.; Alexeyev, M.F.; Cohen, M.V.; Downey, J.M. Acetylcholine and bradykinin trigger preconditioning in the heart through a pathway that includes Akt and NOS. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2606–H2611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.L.; Lin, Y.S.; Chen, C.L.; Wan, S.W.; Ou, Y.D.; Yu, C.Y.; Tsai, T.T.; Tseng, P.C.; Lin, C.F. Dengue Virus Infection Causes the Activation of Distinct NF-κB Pathways for Inducible Nitric Oxide Synthase and TNF-α Expression in RAW264.7 Cells. Mediators Inflamm. 2015, 2015, 1–13. [Google Scholar]
- Neves-Souza, P.C.; Azeredo, E.L.; Zagne, S.M.; Valls-de-Souza, R.; Reis, S.R.; Cerqueira, D.I.; Nogueira, R.M.; Kubelka, C.F. Inducible nitric oxide synthase (iNOS) expression in monocytes during acute Dengue Fever in patients and during in vitro infection. BMC Infect. Dis. 2005, 5, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, D.M.; Garcia, F.G.; Terra, A.P.; Lopes Tosta, A.C.; Silva, L.A.; Castellano, L.R.; Silva Teixeira, D.N. Elevated dengue virus nonstructural protein 1 serum levels and altered toll-like receptor 4 expression, nitric oxide, and tumor necrosis factor alpha production in dengue hemorrhagic Fever patients. J. Trop. Med. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Takhampunya, R.; Padmanabhan, R.; Ubol, S. Antiviral action of nitric oxide on dengue virus type 2 replication. J. Gen. Virol. 2006, 87, 3003–3011. [Google Scholar] [CrossRef]
- Fagundes, C.T.; Costa, V.V.; Cisalpino, D.; Amaral, F.A.; Souza, P.R.; Souza, R.S.; Ryffel, B.; Vieira, L.Q.; Silva, T.A.; Atrasheuskaya, A.; et al. IFN-γ production depends on IL-12 and IL-18 combined action and mediates host resistance to dengue virus infection in a nitric oxide-dependent manner. PLoS Negl. Trop. Dis. 2011, 5, e1449. [Google Scholar] [CrossRef]
- Levine, B. Apoptosis in viral infections of neurons: A protective or pathologic host response? Curr. Top Microbiol. Immunol. 2002, 265, 95–118. [Google Scholar]
- Teodoro, J.G.; Branton, P.E. Regulation of apoptosis by viral gene products. J. Virol. 1997, 71, 1739–1746. [Google Scholar] [CrossRef] [Green Version]
- Bovenzi, V.; Savard, M.; Morin, J.; Cuerrier, C.M.; Grandbois, M.; Gobeil, F., Jr. Bradykinin protects against brain microvascular endothelial cell death induced by pathophysiological stimuli. J. Cell Physiol. 2010, 222, 168–176. [Google Scholar] [CrossRef]
- Niewiarowska-Sendo, A.; Kozik, A.; Guevara-Lora, I. Influence of bradykinin B2 receptor and dopamine D2 receptor on the oxidative stress, inflammatory response, and apoptotic process in human endothelial cells. PLoS ONE 2018, 13, 1–22. [Google Scholar] [CrossRef]
- Fogaça, S.E.; Melo, R.L.; Pimenta, D.C.; Hosoi, K.; Juliano, L.; Juliano, M.A. Differences in substrate and inhibitor sequence specificity of human, mouse and rat tissue kallikreins. Biochem. J. 2004, 380, 775–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albuquerque, L.M.; Trugilho, M.R.; Chapeaurouge, A.; Jurgilas, P.B.; Bozza, P.T.; Bozza, F.A.; Perales, J.; Neves-Ferreira, A.G. Two-dimensional difference gel electrophoresis (DiGE) analysis of plasmas from dengue fever patients. J. Proteome. Res. 2009, 8, 5431–5441. [Google Scholar] [CrossRef] [PubMed]
- Houghton-Triviño, N.; Martín, K.; Giaya, K.; Rodríguez, J.A.; Bosch, I.; Castellanos, J.E. Comparison of the transcriptional profiles of patients with dengue fever and dengue hemorrhagic fever reveals differences in the immune response and clues in immunopathogenesis. Biomedica 2010, 30, 587–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puerta-Guardo, H.; Glasner, D.R.; Espinosa, D.A.; Biering, S.B.; Patana, M.; Ratnasiri, K.; Wang, C.; Beatty, P.R.; Harris, E. Flavivirus NS1 Triggers Tissue-Specific Vascular Endothelial Dysfunction Reflecting Disease Tropism. Cell Rep. 2019, 26, 1598–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libraty, D.H.; Young, P.R.; Pickering, D.; Endy, T.P.; Kalayanarooj, S.; Green, S.; Vaughn, D.W.; Nisalak, A.; Ennis, F.A.; Rothman, A.L. High circulating levels of the dengue virus nonstructural protein NS1 early in dengue illness correlate with the development of dengue hemorrhagic fever. J. Infect. Dis. 2002, 186, 1165–1168. [Google Scholar] [CrossRef]
- Avirutnan, P.; Punyadee, N.; Noisakran, S.; Komoltri, C.; Thiemmeca, S.; Auethavornanan, K.; Jairungsri, A.; Kanlaya, R.; Tangthawornchaikul, N.; Puttikhunt, C.; et al. Vascular leakage in severe dengue virus infections: A potential role for the nonstructural viral protein NS1and complement. J. Infect. Dis. 2006, 193, 1078–1088. [Google Scholar] [CrossRef] [Green Version]
- van de Weg, C.A.; Huits, R.M.; Pannuti, C.S.; Brouns, R.M.; van den Berg, R.W.; van den Ham, H.J.; Martina, B.E.; Osterhaus, A.D.; Netea, M.G.; Meijers, J.C.; et al. Hyperferritinaemia in dengue virus infected patients is associated with immune activation and coagulation disturbances. PLoS Negl. Trop. Dis. 2014, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lima, W.G.; Souza, N.A.; Fernandes, S.O.A.; Cardoso, V.N.; Godói, I.P. Serum lipid profile as a predictor of dengue severity: A systematic review and meta-analysis. Rev. Med. Virol. 2019, 29, e2056. [Google Scholar] [CrossRef]
- Shibayama, Y.; Skoner, D.; Suehiro, S.; Konishi, J.; Fireman, P.; Kaplan, A.P. Bradykinin levels during experimental nasal infection with rhinovirus and attenuated influenza virus. Immunopharmacology 1996, 33, 311–313. [Google Scholar] [CrossRef]
- Edelman, R.; Nimmannitya, S.; Colman, R.W.; Talamo, R.C.; Top, F.H., Jr. Evaluation of the plasma kinin system in dengue hemorrhagic fever. J. Lab. Clin. Med. 1975, 86, 410–421. [Google Scholar]
- Scharfstein, J.K. Compendium of Inflammatory Diseases, 1st ed.; Parnham, M.J., Ed.; Springer: Basel, Switzerland, 2016; pp. 815–836. [Google Scholar]
- Zhang, X.; Brovkovych, V.; Zhang, Y.; Tan, F.; Skidgel, R.A. Downregulation of kinin B1 receptor function by B2 receptor heterodimerization and signaling. Cell Signal. 2015, 27, 90–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Macrae, F.L.; Duval, C.; Papareddy, P.; Baker, S.R.; Yuldasheva, N.; Kearney, K.J.; McPherson, H.R.; Asquith, N.; Konings, J.; Casini, A.; et al. A fibrin biofilm covers blood clots and protects from microbial invasion. J. Clin. Investig. 2018, 128, 3356–3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charnsilpa, W.; Takhampunya, R.; Endy, T.P.; Mammen, M.P., Jr.; Libraty, D.H.; Ubol, S. Nitric oxide radical suppresses replication of wild-type dengue 2 viruses in vitro. J. Med. Virol. 2005, 77, 89–95. [Google Scholar] [CrossRef]
- Ubol, S.; Chareonsirisuthigul, T.; Kasisith, J.; Klungthong, C. Clinical isolates of dengue virus with distinctive susceptibility to nitric oxide radical induce differential gene responses in THP-1 cells. Virology 2008, 376, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Kuhr, F.; Lowry, J.; Zhang, Y.; Brovkovych, V.; Skidgel, R.A. Differential regulation of inducible and endothelial nitric oxide synthase by kinin B1 and B2 receptors. Neuropeptides 2010, 44, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Golser, R.; Gorren, A.C.; Leber, A.; Andrew, P.; Habisch, H.J.; Werner, E.R.; Schmidt, K.; Venema, R.C.; Mayer, B. Interaction of endothelial and neuronal nitric-oxide synthases with the bradykinin B2 receptor. Binding of an inhibitory peptide to the oxygenase domain blocks uncoupled NADPH oxidation. J. Biol. Chem. 2000, 275, 5291–5296. [Google Scholar] [CrossRef] [Green Version]
- Michel, T.; Li, G.K.; Busconi, L. Phosphorylation and subcellular translocation of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1993, 90, 6252–6256. [Google Scholar] [CrossRef] [Green Version]
- Venema, R.C. Post-translational mechanisms of endothelial nitric oxide synthase regulation by bradykinin. Int. Immunopharmacol. 2002, 2, 1755–1762. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Ding, Y.; Ni, Z.; Barton, C.H. Bradykinin down-regulates, whereas arginine analogs up-regulates, endothelial nitric-oxide synthase expression in coronary endothelial cells. J. Pharmacol. Exp. Ther. 2005, 313, 121–126. [Google Scholar] [CrossRef] [Green Version]
- Schleicher, M.; Brundin, F.; Gross, S.; Müller-Esterl, W.; Oess, S. Cell cycle-regulated inactivation of endothelial NO synthase through NOSIP-dependent targeting to the cytoskeleton. Mol. Cell Biol. 2005, 25, 8251–8258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Lumry, W.R.; Li, H.H.; Levy, R.J.; Potter, P.C.; Farkas, H.; Moldovan, D.; Riedl, M.; Li, H.; Craig, T.; Bloom, B.J.; et al. Randomized placebo-controlled trial of the bradykinin B₂ receptor antagonist icatibant for the treatment of acute attacks of hereditary angioedema: The FAST-3 trial. Ann. Allergy Asthma. Immunol. 2011, 107, 529–537. [Google Scholar] [CrossRef]
- Hakl, R.; Kuklínek, P.; Krčmová, I.; Králíčková, P.; Freiberger, T.; Janků, P.; Vlková, M.; Litzman, J. Treatment of Hereditary Angioedema Attacks with Icatibant and Recombinant C1 Inhibitor During Pregnancy. J. Clin. Immunol. 2018, 38, 810–815. [Google Scholar] [CrossRef] [PubMed]
- van de Veerdonk, F.L.; Kouijzer, I.J.E.; de Nooijer, A.H.; van der Hoeven, H.G.; Maas, C.; Netea, M.G.; Brüggemann, R.J.M. Outcomes associated with use of a kinin b2 receptor antagonist among patients with COVID-19. JAMA Netw. Ope. 2020, 3, 1–4. [Google Scholar]
- Sikpa, D.; Whittingstall, L.; Savard, M.; Lebel, R.; Côté, J.; McManus, S.; Chemtob, S.; Fortin, D.; Lepage, M.; Gobeil, F. Pharmacological Modulation of Blood-Brain Barrier Permeability by Kinin Analogs in Normal and Pathologic Conditions. Pharmaceuticals 2020, 13, 279. [Google Scholar] [CrossRef]
- Syenina, A.; Saron, W.A.A.; Jagaraj, C.J.; Bibi, S.; Arock, M.; Gubler, D.J.; Rathore, A.P.S.; Abraham, S.N.; St John, A.L. Th1-Polarized, Dengue Virus-Activated Human Mast Cells Induce Endothelial Transcriptional Activation and Permeability. Viruses 2020, 12, 1379. [Google Scholar] [CrossRef]
- Cordeiro, M.T.; Silva, A.M.; Brito, C.A.; Nascimento, E.J.; Magalhaes, M.C.; Guimarães, G.F.; Lucena-Silva, N.; Carvalho, E.M.F.; Marques, E.T.A. Characterization of a dengue patient cohort in Recife, Brazil. Am. J. Trop. Med. Hyg. 2007, 77, 1128–1134. [Google Scholar] [CrossRef] [Green Version]
- Coelho, S.V.A.; Neris, R.L.S.; Papa, M.P.; Schnellrath, L.C.; Meuren, L.M.; Tschoeke, D.A.; Leomil, L.; Verçoza, B.R.F.; Miranda, M.; Thompson, F.L.; et al. Development of standard methods for Zika virus propagation, titration, and purification. J. Virol. Methods 2017, 246, 65–74. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Papa, M.P.; Meuren, L.M.; Coelho, S.V.A.; Lucas, C.G.O.; Mustafá, Y.M.; Matassoli, F.L.; Silveira, P.P.; Frost, P.S.; Pezzuto, P.; Ribeiro, M.R.; et al. Zika Virus Infects, Activates, and Crosses Brain Microvascular Endothelial Cells, without Barrier Disruption. Front. Microbiol. 2017, 8, 2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Dengue Haemorrhagic Fever: Diagnosis, Treatment, Prevention and Control, 2nd ed.; World Health Organization: Geneva, Switzerland, 1997. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Code | Gender | Age | Phase of Infection | Days of Symptoms | IgM | IgG | Leukocytes | Platelets | Hematocrit | Albumin | Denv Clinical Classification | Infection |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | Acute | 4 | + | − | 3420 | 337,000 | 33.4 | 4.3 | ||||
| F | 8 | Critical | 8 | + | + | 6190 | 362,000 | 31.95 | 4.3 | DF | Primary | |
| Convalescent | 29 | + | + | 9840 | 497,000 | 34.9 | 4.6 | |||||
| Patient 2 | Acute | 4 | − | + | nd | nd | nd | nd | ||||
| M | 47 | Critical | 7 | − | + | 6530 | 189,000 | 41.7 | nd | DF | Secondary | |
| Convalescent | 36 | + | + | nd | nd | nd | nd | |||||
| Patient 3 | Acute | 3 | − | − | nd | nd | nd | nd | ||||
| M | 38 | Critical | 5 | − | − | nd | nd | nd | nd | DF | Primary | |
| Convalescent | 48 | + | + | nd | nd | nd | nd | |||||
| Patient 4 | Acute | 4 | + | + | nd | nd | nd | nd | ||||
| F | 37 | Critical | 6 | + | + | 5100 | 248,000 | 38 | 4.1 | DHF | Secondary | |
| Convalescent | 18 | + | + | nd | nd | nd | nd | |||||
| Patient 5 | ||||||||||||
| F | 12 | Critical | 6 | − | − | nd | nd | nd | nd | DF | Primary | |
| Convalescent | 26 | + | + | nd | nd | nd | nd | |||||
| Patient 6 | ||||||||||||
| M | 35 | Critical | 8 | + | − | 6270 | 125,000 | 45.8 | 3.8 | DHF | Primary | |
| Patient 7 | ||||||||||||
| F | 33 | Critical | 5 | − | − | 1760 | 110,000 | 36.4 | 4.2 | DFC | Primary | |
| Convalescent | 37 | + | + | nd | nd | nd | nd | |||||
| Patient 8 | Acute | 3 | − | + | 4900 | 134,000 | 39.4 | 3.3 | ||||
| M | 41 | Critical | 9 | + | + | 4420 | 131,000 | 46.8 | 3.4 | DHF | Secondary | |
| Patient 9 | Acute | 4 | + | − | 2400 | 52,000 | 43.5 | 3.9 | ||||
| F | 15 | Critical | 6 | + | − | 2500 | 66,000 | 37.9 | nd | DFC | Primary | |
| Patient 10 | Acute | 3 | − | + | 3080 | 105,000 | 38.8 | nd | ||||
| M | 48 | Critical | 5 | − | + | 2580 | 92,000 | 42.4 | 4.2 | DFC | Secondary | |
| Convalescent | 38 | + | + | 4970 | 152,000 | 40.9 | nd | |||||
| Patient 11 | Acute | 3 | − | − | nd | nd | nd | nd | ||||
| M | 23 | Critical | 5 | − | − | nd | nd | nd | nd | DF | Primary | |
| Convalescent | 30 | + | + | nd | nd | nd | nd | |||||
| Patient 12 | Acute | 3 | − | + | nd | nd | nd | nd | ||||
| F | 30 | Critical | 5 | − | + | nd | nd | nd | nd | DF | Secondary | |
| Convalescent | 31 | − | + | 6410 | 166,000 | 38.2 | 4.4 | |||||
| Patient 13 | Acute | 3 | − | + | nd | nd | nd | nd | ||||
| F | 40 | Critical | 5 | − | + | 3000 | 120,000 | 41.4 | 4.13 | DFC | Secondary | |
| Convalescent | 33 | − | + | nd | nd | nd | nd | |||||
| Patient 14 | Acute | 3 | − | + | 3700 | 124,000 | 47.3 | nd | ||||
| M | 31 | Critical | 5 | − | + | 2900 | 120,000 | 46 | 4.48 | DFC | Secondary | |
| Convalescent | 18 | − | + | 9700 | 283,000 | 43.4 | 4.54 | |||||
| Patient 15 | Acute | 4 | − | + | 2360 | 143,000 | 35.7 | 3.8 | ||||
| F | 40 | Critical | 7 | + | + | 3810 | 143,000 | 34.3 | 4.0 | DF | Secondary | |
| Convalescent | 30 | − | + | 5040 | 327,000 | 32.6 | nd | |||||
| Patient 16 | Acute | 4 | + | + | 5750 | 14,000 | 39.1 | nd | ||||
| M | 45 | Critical | 6 | + | + | 5200 | 110,000 | 41.7 | 4.2 | DFC | Secondary | |
| Convalescent | 34 | + | + | nd | nd | nd | nd | |||||
| Patient 17 | Acute | 3 | − | + | 4440 | 125,000 | 45.2 | |||||
| M | 44 | Critical | 5 | + | + | 3060 | 107,000 | 44.9 | 4.5 | DFC | Primary | |
| Convalescent | 34 | + | + | 7290 | 185,000 | 45.9 | 4.7 | |||||
| Patient 18 | Acute | 4 | − | + | 2600 | 116,000 | 41.2 | nd | ||||
| F | 47 | Critical | 6 | + | + | 3500 | 92,000 | 38 | nd | DFC | Secondary | |
| Convalescent | 33 | + | + | 6700 | 505,000 | 38.4 | nd | |||||
| Patient 19 | Acute | 3 | − | − | nd | nd | nd | nd | ||||
| M | 42 | Critical | 5 | − | nd | nd | nd | nd | DF | Primary | ||
| Convalescent | 31 | + | + | nd | nd | nd | nd | |||||
| Patient 20 | Acute | 3 | + | + | 2770 | 211,000 | 41.3 | nd | ||||
| F | 53 | Critical | 5 | + | + | 3260 | 164,000 | 39 | 4.2 | DF | Secondary | |
| Convalescent | 41 | + | + | nd | nd | nd | nd | |||||
| Patient 21 | ||||||||||||
| F | 27 | Critical | 6 | + | − | nd | nd | nd | nd | DF | Primary | |
| Convalescent | 33 | + | + | nd | nd | nd | nd | |||||
| Patient 22 | Acute | 4 | − | + | 2100 | 90,000 | 34.6 | 3.7 | ||||
| F | 21 | Critical | 6 | + | + | nd | nd | nd | nd | DF | Secondary | |
| Convalescent | 48 | + | + | nd | nd | nd | nd | |||||
| Patient 23 | ||||||||||||
| M | 18 | Critical | 6 | − | − | nd | nd | nd | nd | DFC | Primary | |
| Patient 24 | ||||||||||||
| F | 49 | Critical | 8 | − | + | 5910 | 178,000 | 33.6 | 3.9 | DFC | Secondary | |
| Convalescent | 30 | − | + | nd | nd | nd | nd | |||||
| Patient 25 | Acute | 4 | − | + | 3560 | 131,000 | 40.2 | 3.9 | ||||
| M | 48 | Critical | 7 | − | + | 8260 | 114,000 | 40.3 | 3.6 | DFC | Secondary | |
| Patient 26 | Acute | 4 | − | + | 1260 | 133,000 | 34.7 | 3.5 | ||||
| F | 31 | Critical | 8 | + | + | 3600 | 141,000 | 34.4 | nd | DFC | Secondary | |
| Convalescent | 52 | + | + | nd | nd | nd | nd | |||||
| Patient 27 | Acute | 3 | − | + | 4770 | 122,000 | 42.8 | 4.2 | ||||
| M | 36 | Critical | 6 | + | + | 2170 | 71,000 | 47.2 | 3.9 | DHF | Secondary | |
| Convalescent | 37 | + | + | nd | nd | nd | nd |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, S.V.A.; Rust, N.M.; Vellasco, L.; Papa, M.P.; Pereira, A.S.G.; da Silva Palazzo, M.F.; Juliano, M.A.; Costa, S.M.; Alves, A.M.B.; Cordeiro, M.T.; et al. Contact System Activation in Plasma from Dengue Patients Might Harness Endothelial Virus Replication through the Signaling of Bradykinin Receptors. Pharmaceuticals 2021, 14, 56. https://doi.org/10.3390/ph14010056
Coelho SVA, Rust NM, Vellasco L, Papa MP, Pereira ASG, da Silva Palazzo MF, Juliano MA, Costa SM, Alves AMB, Cordeiro MT, et al. Contact System Activation in Plasma from Dengue Patients Might Harness Endothelial Virus Replication through the Signaling of Bradykinin Receptors. Pharmaceuticals. 2021; 14(1):56. https://doi.org/10.3390/ph14010056
Chicago/Turabian StyleCoelho, Sharton V. A., Naiara M. Rust, Lucas Vellasco, Michelle P. Papa, Aline S. G. Pereira, Matheus Ferreira da Silva Palazzo, Maria Aparecida Juliano, Simone M. Costa, Ada M. B. Alves, Marli T. Cordeiro, and et al. 2021. "Contact System Activation in Plasma from Dengue Patients Might Harness Endothelial Virus Replication through the Signaling of Bradykinin Receptors" Pharmaceuticals 14, no. 1: 56. https://doi.org/10.3390/ph14010056
APA StyleCoelho, S. V. A., Rust, N. M., Vellasco, L., Papa, M. P., Pereira, A. S. G., da Silva Palazzo, M. F., Juliano, M. A., Costa, S. M., Alves, A. M. B., Cordeiro, M. T., Marques, E. T. A., Scharfstein, J., & de Arruda, L. B. (2021). Contact System Activation in Plasma from Dengue Patients Might Harness Endothelial Virus Replication through the Signaling of Bradykinin Receptors. Pharmaceuticals, 14(1), 56. https://doi.org/10.3390/ph14010056