
The Fellowship of Privileged Scaffolds—One Structure to Inhibit Them All


Abstract
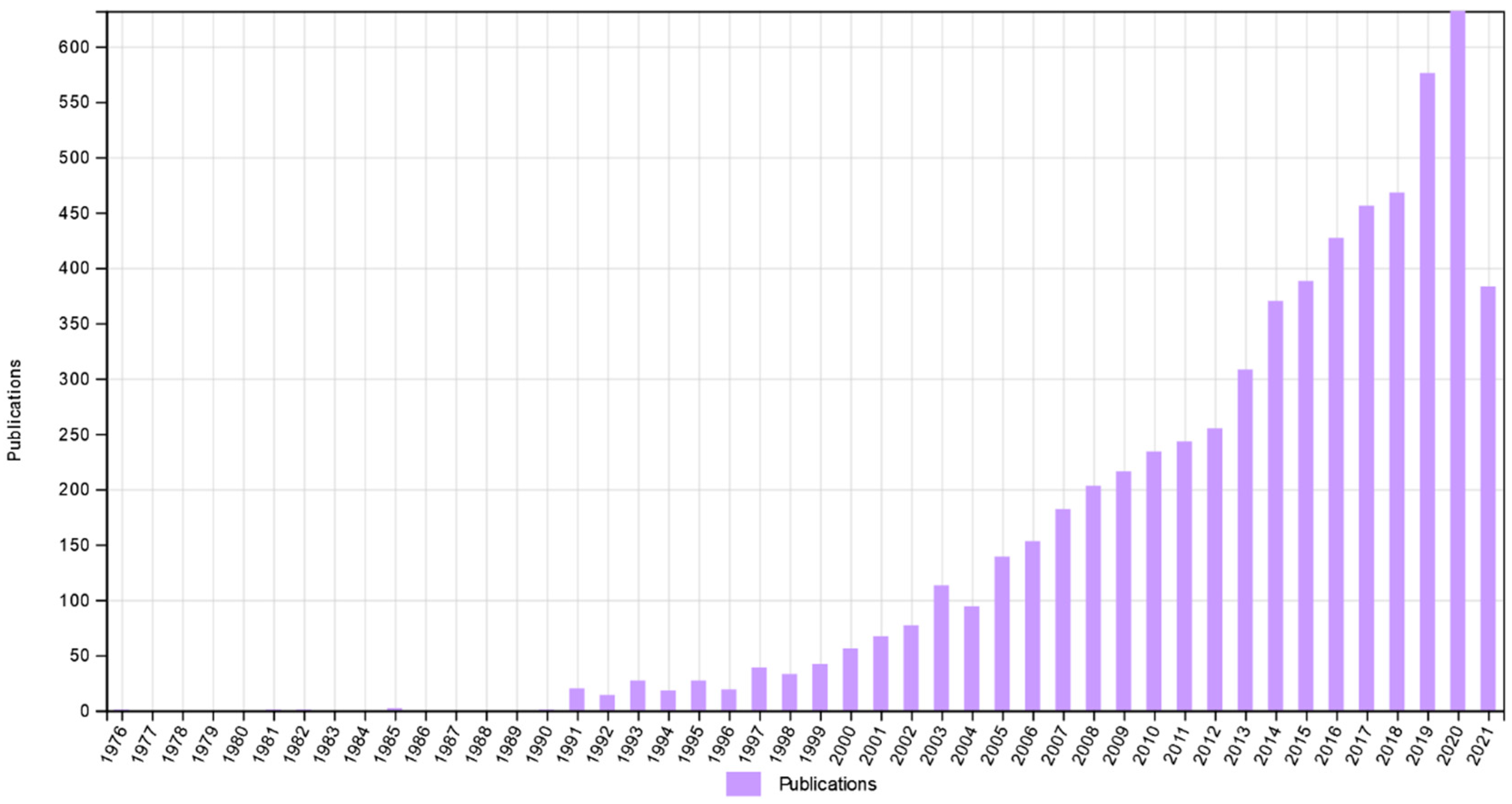
:1. Introduction
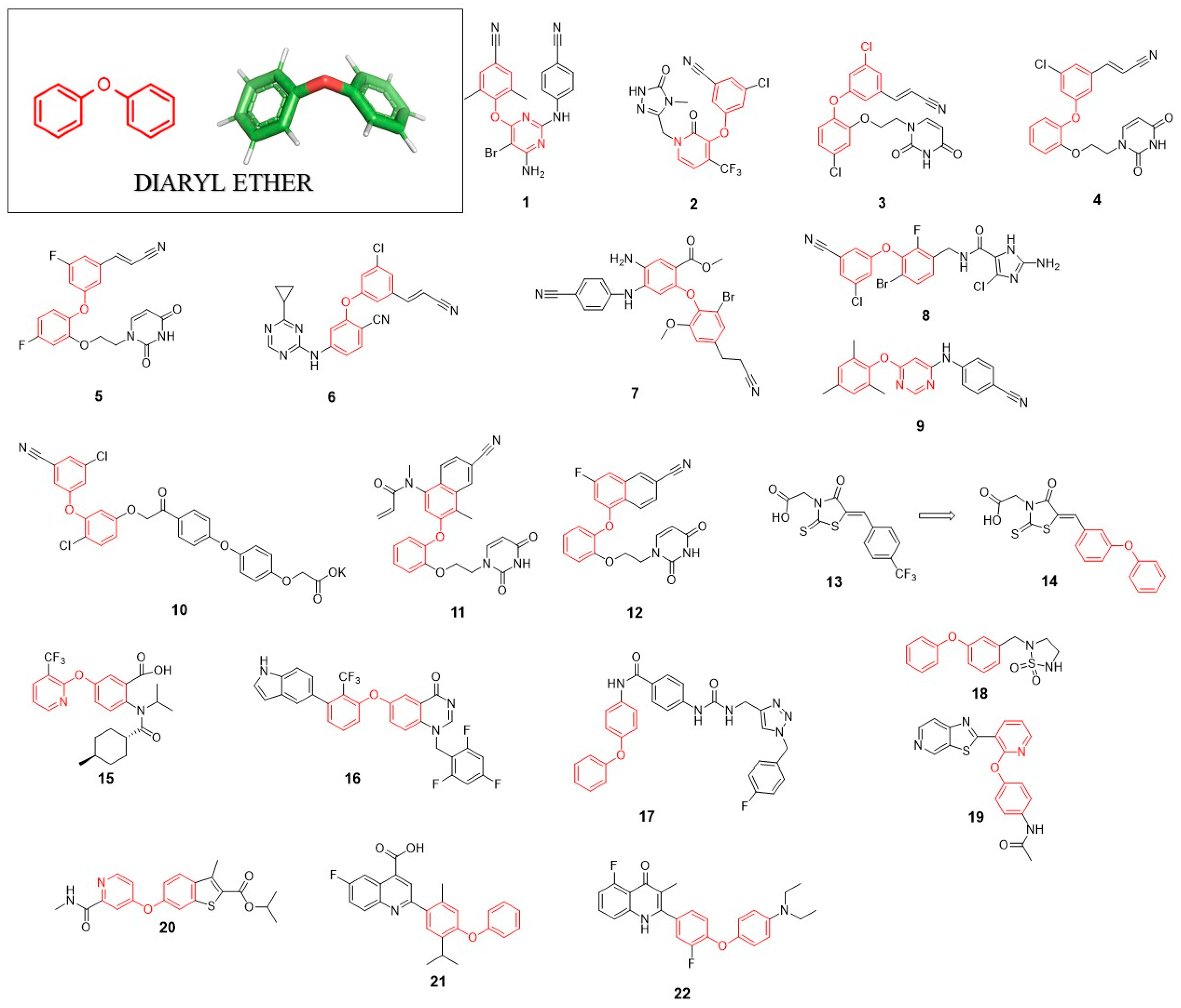
2. Diaryl Ether (DE)
2.1. Anti-HIV Agents
2.2. Anti-HCV Agents
2.3. Anti-Flaviviruses Agents
2.4. Anti-Polyomaviruses Agents
2.5. Anti-Rhinoviruses Agents
2.6. Host-Targeting Antivirals
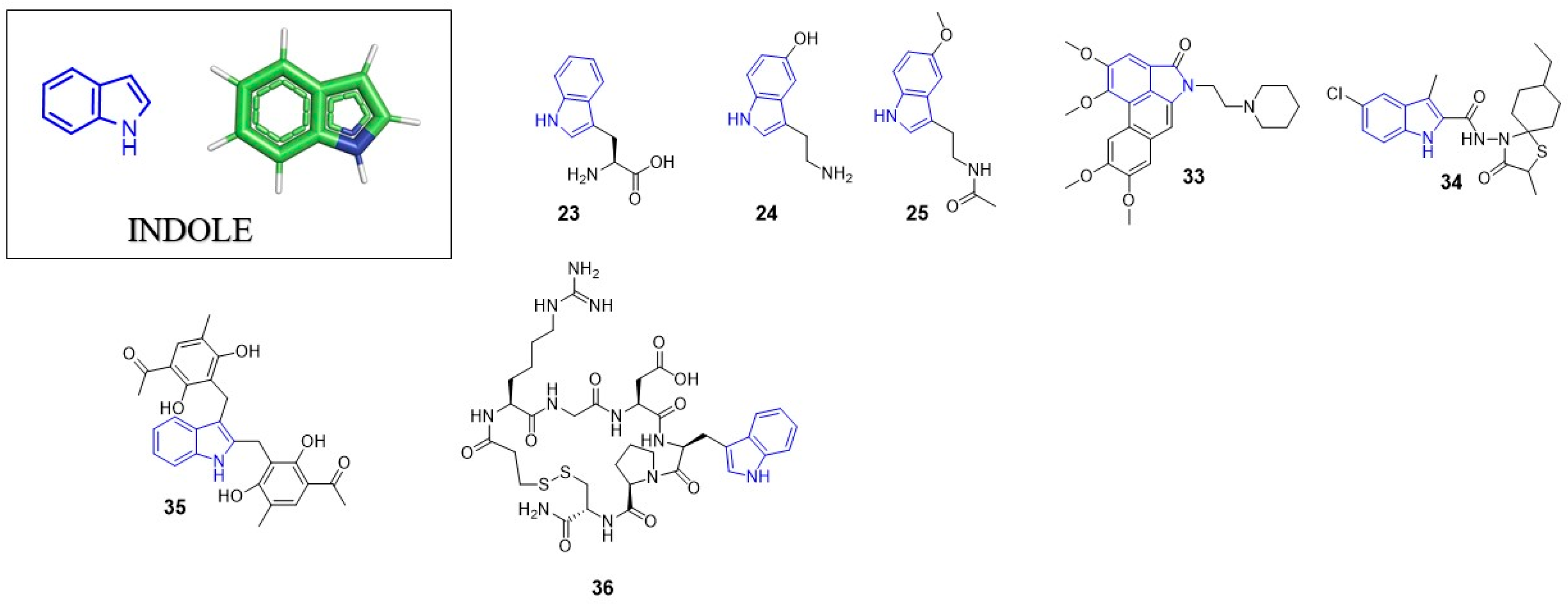
3. Indole
3.1. Anti-HIV Agents
3.2. Anti-Influenza Agents
3.3. Anti-Alphavirus Agents
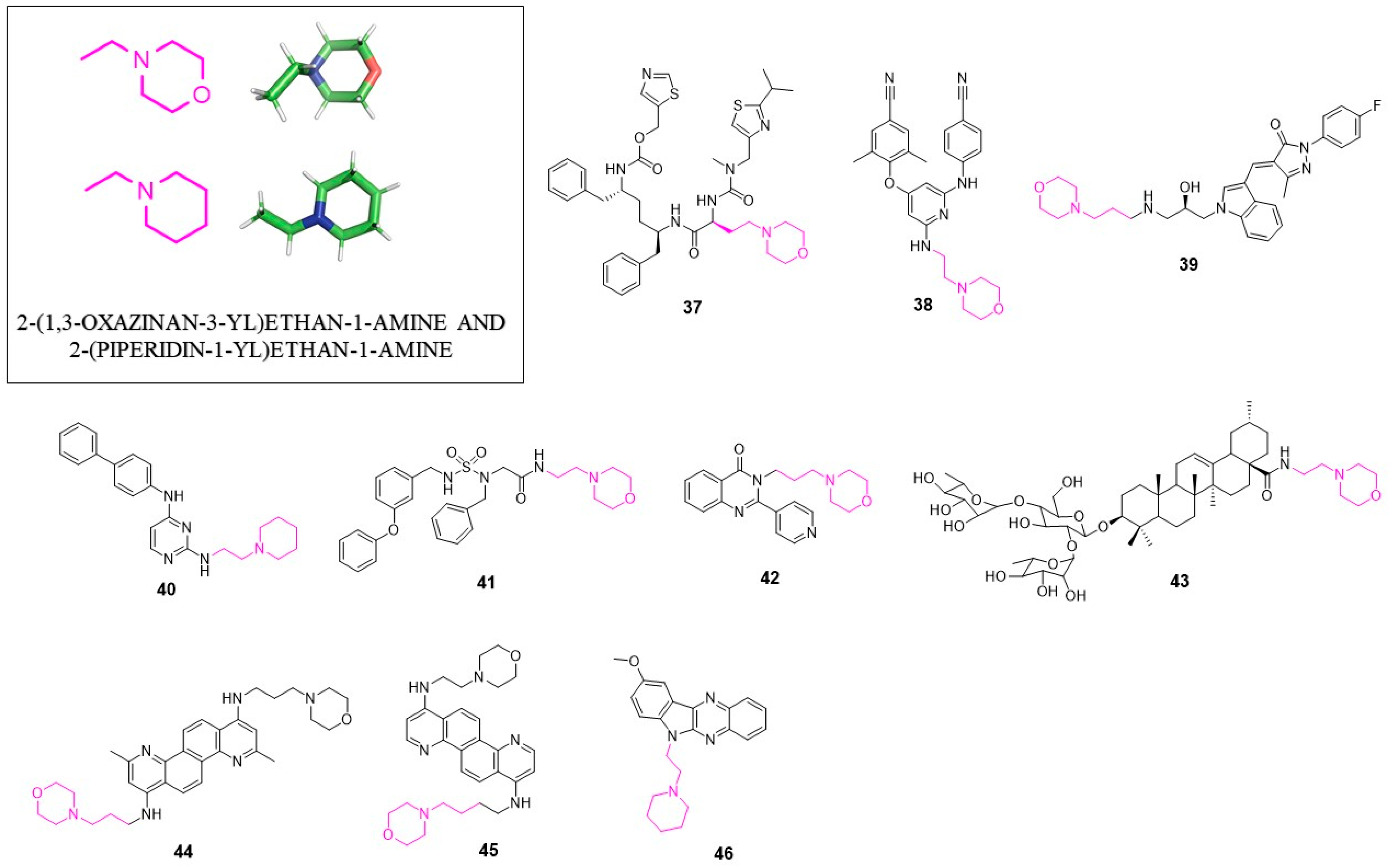
4. 2-(1,3-Oxazinan-3-yl)ethan-1-amine and 2-(Piperidin-1-yl)ethan-1-amine
4.1. Anti-HIV Agents
4.2. Anti-HCV Agents
4.3. Anti-Flavivirus Agents
4.4. Anti-Norovirus Agents
4.5. Anti-Influenza Agents
4.6. Anti-Ebola Agents
4.7. Anti-Vaccinia Virus Agents
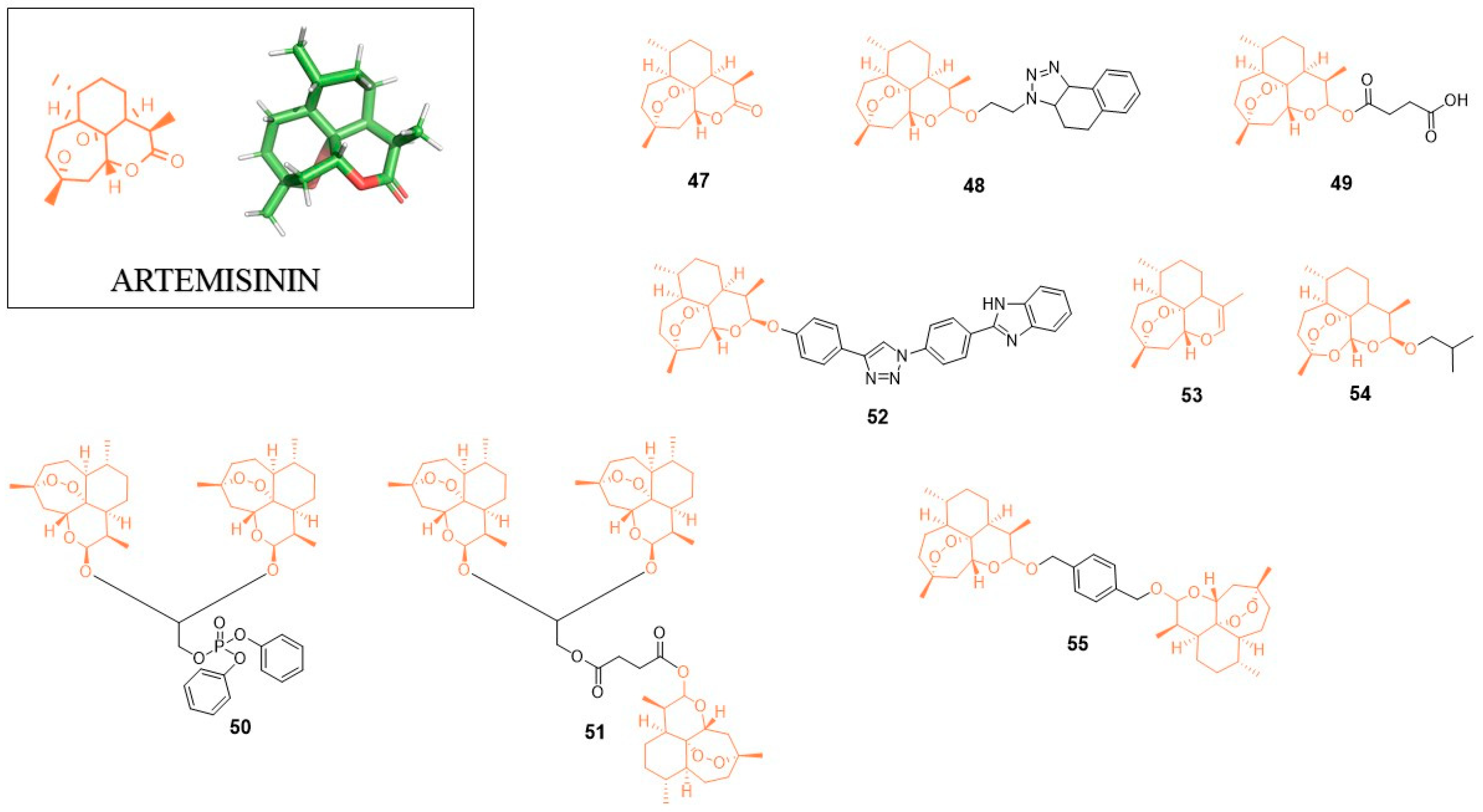
5. Artemisinin (ART)
5.1. Anti-HIV Agents
5.2. Anti-Flavivirus Agents
5.3. Anti-Herpesvirus Agents
5.4. Anti-HBV Agents
5.5. Anti-HCV Agents
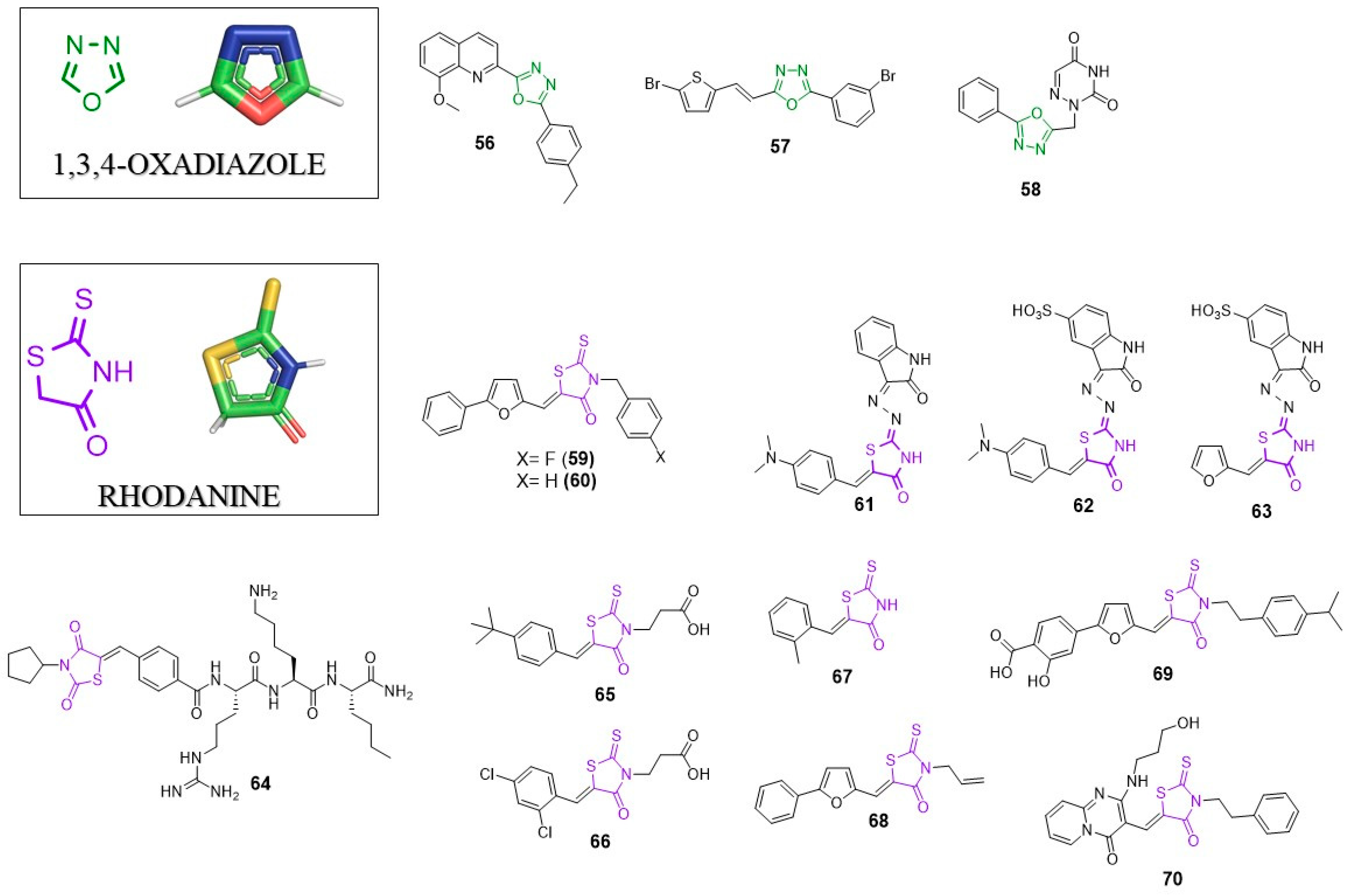
6. 1,3,4-Oxadizole
6.1. Anti-HIV Agents
6.2. Anti-Flavivirus Agents
6.3. Anti-Herpesvirus Agents
7. Rhodanine (RHO)
7.1. Anti-HIV Agents
7.2. Anti-HCV Agents
7.3. Anti-Flavivirus Agents
7.4. Anti-Alphavirus Agents
7.5. Broad Spectrum Antivirals
7.6. Host Factors–Targeting Antivirals
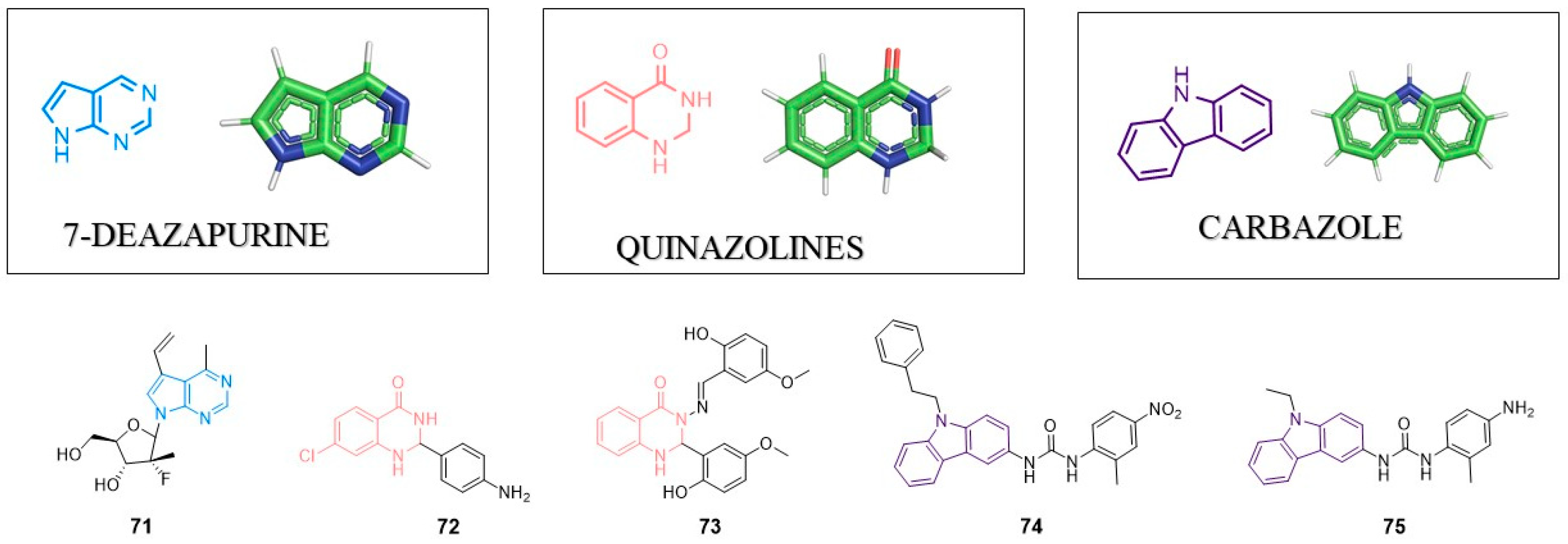
8. Pyrrolo[2,3-d]pyrimidine (7-deazopurine) Nucleoside
9. Quinazolines
9.1. Anti-HIV Agents
9.2. Anti-HCV Agents
10. Carbazole
11. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; Dipardo, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, G.F.; Veber, D.F.; Anderson, P.S.; Chang, R.S.L.; et al. Methods for Drug Discovery: Development of Potent, Selective, Orally Effective Cholecystokinin Antagonistst. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef]
- Bolz, S.N.; Adasme, M.F.; Schroeder, M. Toward an Understanding of Pan-Assay Interference Compounds and Promiscuity: A Structural Perspective on Binding Modes. J. Chem. Inf. Model. 2021, 61, 2248–2262. [Google Scholar] [CrossRef]
- Baell, J.B.; Nissink, J.W.M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations. ACS Chem. Biol. 2017, 13, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Lagorce, D.; Oliveira, N.; Miteva, M.A.; Villoutreix, B.O. Pan-assay interference compounds (PAINS) that may not be too painful for chemical biology projects. Drug Discov. Today 2017, 22, 1131–1133. [Google Scholar] [CrossRef]
- Jasial, S.; Hu, Y.; Bajorath, J. How Frequently Are Pan-Assay Interference Compounds Active? Large-Scale Analysis of Screening Data Reveals Diverse Activity Profiles, Low Global Hit Frequency, and Many Consistently Inactive Compounds. J. Med. Chem. 2017, 60, 3879–3886. [Google Scholar] [CrossRef]
- Pouliot, M.; Jeanmart, S. Pan Assay Interference Compounds (PAINS) and Other Promiscuous Compounds in Antifungal Research. J. Med. Chem. 2015, 59, 497–503. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.B. Observations on screening-based research and some concerning trends in the literature. Future Med. Chem. 2010, 2, 1529–1546. [Google Scholar] [CrossRef]
- Gupta, N.; Wish, J.B. Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors: A Potential New Treatment for Anemia in Patients With CKD. Am. J. Kidney Dis. 2017, 69, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.; Scheerens, H.; Li, S.-J.; Schultz, B.E.; Sprengeler, P.A.; Burrill, L.C.; Mendonca, R.V.; Sweeney, M.D.; Scott, K.C.K.; Grothaus, P.G.; et al. Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem 2007, 2, 58–61. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Bedos-Belval, F.; Rouch, A.; Vanucci-Bacqué, C.; Baltas, M. Diaryl ether derivatives as anticancer agents—A review. Medchemcomm 2012, 3, 1356–1372. [Google Scholar] [CrossRef]
- Chen, T.; Xiong, H.; Yang, J.F.; Zhu, X.L.; Qu, R.Y.; Yang, G.F. Diaryl Ether: A Privileged Scaffold for Drug and Agrochemical Discovery. J. Agric. Food Chem. 2020, 68, 9839–9877. [Google Scholar] [CrossRef]
- Kudalkar, S.N.; Ullah, I.; Bertoletti, N.; Mandl, H.K.; Cisneros, J.A.; Beloor, J.; Chan, A.H.; Quijano, E.; Saltzman, W.M.; Jorgensen, W.L.; et al. Structural and pharmacological evaluation of a novel non-nucleoside reverse transcriptase inhibitor as a promising long acting nanoformulation for treating HIV. Antivir. Res. 2019, 167, 110–116. [Google Scholar] [CrossRef]
- Adams, J.; Merluzzi, V.J. Discovery of Nevirapine, a Nonnucleoside Inhibitor of HIV-1 Reverse Transcriptase. In The Search for Antiviral Drugs; Birkhäuser: Boston, MA, USA, 1993; pp. 45–70. [Google Scholar]
- Wang, Y.; Xing, H.; Liao, L.; Wang, Z.; Su, B.; Zhao, Q.; Feng, Y.; Ma, P.; Liu, J.; Wu, J.; et al. The development of drug resistance mutations K103N Y181C and G190A in long term Nevirapine-containing antiviral therapy. AIDS Res. Ther. 2014, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Namasivayam, V.; Vanangamudi, M.; Kramer, V.G.; Kurup, S.; Zhan, P.; Liu, X.; Kongsted, J.; Byrareddy, S.N. The Journey of HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs) from Lab to Clinic. J. Med. Chem. 2019, 62, 4851–4883. [Google Scholar] [CrossRef]
- Bollini, M.; Domaoal, R.A.; Thakur, V.V.; Gallardo-Macias, R.; Spasov, K.A.; Anderson, K.S.; Jorgensen, W.L. Computationally-guided optimization of a docking hit to yield catechol diethers as potent anti-HIV agents. J. Med. Chem. 2011, 54, 8582–8591. [Google Scholar] [CrossRef] [Green Version]
- Frey, K.M.; Bollini, M.; Mislak, A.C.; Cisneros, J.A.; Gallardo-Macias, R.; Jorgensen, W.L.; Anderson, K.S. Crystal structures of HIV-1 reverse transcriptase with picomolar inhibitors reveal key interactions for drug design. J. Am. Chem. Soc. 2012, 134, 19501–19503. [Google Scholar] [CrossRef] [Green Version]
- Frey, K.M.; Puleo, D.E.; Spasov, K.A.; Bollini, M.; Jorgensen, W.L.; Anderson, K.S. Structure-Based Evaluation of Non-nucleoside Inhibitors with Improved Potency and Solubility That Target HIV Reverse Transcriptase Variants. J. Med. Chem. 2015, 58, 2737–2745. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Bollini, M.; Thakur, V.V.; Domaoal, R.A.; Spasov, K.A.; Anderson, K.S. Efficient discovery of potent anti-HIV agents targeting the Tyr181Cys variant of HIV reverse transcriptase. J. Am. Chem. Soc. 2011, 133, 15686–15696. [Google Scholar] [CrossRef] [Green Version]
- Qin, B.; Jiang, X.; Lu, H.; Tian, X.; Barbault, F.; Huang, L.; Qian, K.; Chen, C.H.; Huang, R.; Jiang, S.; et al. Diarylaniline derivatives as a distinct class of HIV-1 non-nucleoside reverse transcriptase inhibitors. J. Med. Chem. 2010, 53, 4906–4916. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.Q.; Zhu, L.; Qian, K.; Qin, B.; Huang, L.; Chen, C.H.; Lee, K.H.; Xie, L. Design, synthesis, and preclinical evaluations of novel 4-substituted 1,5-diarylanilines as potent HIV-1 non-nucleoside reverse transcriptase inhibitor (NNRTI) drug candidates. J. Med. Chem. 2012, 55, 7219–7229. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Qin, B.; Sun, L.Q.; Yu, F.; Lu, L.; Jiang, S.; Lee, K.H.; Xie, L. Physicochemical property-driven optimization of diarylaniline compounds as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 3719–3723. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Wei, L.; Huang, L.; Yu, F.; Zheng, W.; Qin, B.; Zhu, D.Q.; Morris-Natschke, S.L.; Jiang, S.; Chen, C.H.; et al. Novel HIV-1 Non-nucleoside Reverse Transcriptase Inhibitor Agents: Optimization of Diarylanilines with High Potency against Wild-Type and Rilpivirine-Resistant E138K Mutant Virus. J. Med. Chem. 2016, 59, 3689–3704. [Google Scholar] [CrossRef] [Green Version]
- Chong, P.; Sebahar, P.; Youngman, M.; Garrido, D.; Zhang, H.; Stewart, E.L.; Nolte, R.T.; Wang, L.; Ferris, R.G.; Edelstein, M.; et al. Rational design of potent non-nucleoside inhibitors of HIV-1 reverse transcriptase. J. Med. Chem. 2012, 55, 10601–10609. [Google Scholar] [CrossRef]
- Ribone, S.R.; Leen, V.; Madrid, M.; Dehaen, W.; Daelemans, D.; Pannecouque, C.; Briñón, M.C. Synthesis, biological evaluation and molecular modeling of 4,6-diarylpyrimidines and diarylbenzenes as novel non-nucleosides HIV-1 reverse transcriptase inhibitors. Eur. J. Med. Chem. 2012, 58, 485–492. [Google Scholar] [CrossRef]
- Frączek, T.; Kamiński, R.; Krakowiak, A.; Naessens, E.; Verhasselt, B.; Paneth, P. Diaryl ethers with carboxymethoxyphenacyl motif as potent HIV-1 reverse transcriptase inhibitors with improved solubility. J. Enzyme Inhib. Med. Chem. 2018, 33, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.H.; Lee, W.G.; Spasov, K.A.; Cisneros, J.A.; Kudalkar, S.N.; Petrova, Z.O.; Buckingham, A.B.; Anderson, K.S.; Jorgensen, W.L. Covalent inhibitors for eradication of drug-resistant HIV-1 reverse transcriptase: From design to protein crystallography. Proc. Natl. Acad. Sci. USA 2017, 114, 9725–9730. [Google Scholar] [CrossRef] [Green Version]
- Talele, T.T.; Arora, P.; Kulkarni, S.S.; Patel, M.R.; Singh, S.; Chudayeu, M.; Kaushik-Basu, N. Structure-based virtual screening, synthesis and SAR of novel inhibitors of hepatitis C virus NS5B polymerase. Bioorganic Med. Chem. 2010, 18, 4630–4638. [Google Scholar] [CrossRef] [Green Version]
- Stammers, T.A.; Coulombe, R.; Duplessis, M.; Fazal, G.; Gagnon, A.; Garneau, M.; Goulet, S.; Jakalian, A.; Laplante, S.; Rancourt, J.; et al. Anthranilic acid-based Thumb Pocket 2 HCV NS5B polymerase inhibitors with sub-micromolar potency in the cell-based replicon assay. Bioorganic Med. Chem. Lett. 2013, 23, 6879–6885. [Google Scholar] [CrossRef]
- Hucke, O.; Coulombe, R.; Bonneau, P.; Bertrand-Laperle, M.; Brochu, C.; Gillard, J.; Joly, M.A.; Landry, S.; Lepage, O.; Llinàs-Brunet, M.; et al. Molecular dynamics simulations and structure-based rational design lead to allosteric HCV NS5B polymerase thumb pocket 2 inhibitor with picomolar cellular replicon potency. J. Med. Chem. 2014, 57, 1932–1943. [Google Scholar] [CrossRef]
- Aravapalli, S.; Lai, H.; Teramoto, T.; Alliston, K.R.; Lushington, G.H.; Ferguson, E.L.; Padmanabhan, R.; Groutas, W.C. Inhibitors of Dengue virus and West Nile virus proteases based on the aminobenzamide scaffold. Bioorganic Med. Chem. 2012, 20, 4140–4148. [Google Scholar] [CrossRef] [Green Version]
- Dou, D.; Mandadapu, S.R.; Alliston, K.R.; Kim, Y.; Chang, K.O.; Groutas, W.C. Design and synthesis of inhibitors of noroviruses by scaffold hopping. Bioorganic Med. Chem. 2011, 19, 5749–5755. [Google Scholar] [CrossRef] [Green Version]
- Bonafoux, D.; Nanthakumar, S.; Bandarage, U.K.; Memmott, C.; Lowe, D.; Aronov, A.M.; Bhisetti, G.R.; Bonanno, K.C.; Coll, J.; Leeman, J.; et al. Fragment-Based Discovery of Dual JC Virus and BK Virus Helicase Inhibitors. J. Med. Chem. 2016, 59, 7138–7151. [Google Scholar] [CrossRef]
- Pevear, D.C.; Tull, T.M.; Seipel, M.E.; Groarke, J.M. Activity of pleconaril against enteroviruses. Antimicrob. Agents Chemother. 1999, 43, 2109–2115. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Jung, Y.K.; Kim, C.; Shin, J.S.; Scheers, E.; Lee, J.Y.; Han, S.B.; Lee, C.K.; Neyts, J.; Ha, J.D.; et al. A Novel Series of Highly Potent Small Molecule Inhibitors of Rhinovirus Replication. J. Med. Chem. 2017, 60, 5472–5492. [Google Scholar] [CrossRef]
- Xiong, R.; Zhang, L.; Li, S.; Sun, Y.; Ding, M.; Wang, Y.; Zhao, Y.; Wu, Y.; Shang, W.; Jiang, X.; et al. Novel and potent inhibitors targeting DHODH are broad-spectrum antivirals against RNA viruses including newly-emerged coronavirus SARS-CoV-2. Protein Cell 2020, 11, 723–739. [Google Scholar] [CrossRef]
- Das, P.; Deng, X.; Zhang, L.; Roth, M.G.; Fontoura, B.M.A.; Phillips, M.A.; De Brabander, J.K. SAR-based optimization of a 4-quinoline carboxylic acid analogue with potent antiviral activity. ACS Med. Chem. Lett. 2013, 4, 517–521. [Google Scholar] [CrossRef]
- Yang, Y.; Cao, L.; Gao, H.; Wu, Y.; Wang, Y.; Fang, F.; Lan, T.; Lou, Z.; Rao, Y. Discovery, Optimization, and Target Identification of Novel Potent Broad-Spectrum Antiviral Inhibitors. J. Med. Chem. 2019, 62, 4056–4073. [Google Scholar] [CrossRef]
- Zhang, M.Z.; Chen, Q.; Yang, G.F. A review on recent developments of indole-containing antiviral agents. Eur. J. Med. Chem. 2015, 89, 421–441. [Google Scholar] [CrossRef]
- Kumari, A.; Singh, R.K. Medicinal chemistry of indole derivatives: Current to future therapeutic prospectives. Bioorg. Chem. 2019, 89, 103021. [Google Scholar] [CrossRef]
- Dorababu, A. Indole-a promising pharmacophore in recent antiviral drug discovery. RSC Med. Chem. 2020, 11, 1335–1353. [Google Scholar] [CrossRef]
- Leneva, I.A.; Russell, R.J.; Boriskin, Y.S.; Hay, A.J. Characteristics of arbidol-resistant mutants of influenza virus: Implications for the mechanism of anti-influenza action of arbidol. Antivir. Res. 2009, 81, 132–140. [Google Scholar] [CrossRef]
- Paintsil, E.; Cheng, Y.C. Antiviral Agents. In Encyclopedia of Microbiology; Elsevier Inc.: Amsterdam, The Netherlands, 2009; pp. 223–257. [Google Scholar]
- Lawitz, E.; Gane, E.; Pearlman, B.; Tam, E.; Ghesquiere, W.; Guyader, D.; Alric, L.; Bronowicki, J.P.; Lester, L.; Sievert, W.; et al. Efficacy and safety of 12 weeks versus 18 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin for hepatitis C virus genotype 1 infection in previously untreated patients with cirrhosis and patients with previous null response with or without cirrhosis (C-WORTHY): A randomised, open-label phase 2 trial. Lancet 2015, 385, 1075–1086. [Google Scholar]
- Morse, G.D.; Reichman, R.C.; Fischl, M.A.; Para, M.; Leedom, J.; Powderly, W.; Demeter, L.M.; Resnick, L.; Bassiakos, Y.; Timpone, J.; et al. Concentration-targeted phase I trials of atevirdine mesylate in patients with HIV infection: Dosage requirements and pharmacokinetic studies. Antivir. Res. 2000, 45, 47–58. [Google Scholar] [CrossRef]
- Erhardt, A.; Deterding, K.; Benhamou, Y.; Reiser, M.; Forns, X.; Pol, S.; Calleja, J.L.; Ross, S.; Spangenberg, H.C.; Garcia-Samaniego, J.; et al. Safety, pharmacokinetics and antiviral effect of BILB 1941, a novel hepatitis C virus RNA poylmerase inhibitor, after 5 days oral treatment. Antivir. Ther. 2009, 14, 23–32. [Google Scholar]
- Ahmed, A.M.; Doheim, M.F.; Mattar, O.M.; Sherif, N.A.; Truong, D.H.; Hoa, P.T.L.; Hirayama, K.; Huy, N.T. Beclabuvir in combination with asunaprevir and daclatasvir for hepatitis C virus genotype 1 infection: A systematic review and meta-analysis. J. Med. Virol. 2018, 90, 907–918. [Google Scholar] [CrossRef]
- Shin, Y.H.; Park, C.M.; Kim, H.G.; Kim, D.E.; Choi, M.S.; Kim, J.A.; Choi, B.S.; Yoon, C.H. Identification of Aristolactam Derivatives That Act as Inhibitors of Human Immunodeficiency Virus Type 1 Infection and Replication by Targeting Tat-Mediated Viral Transcription. Virol. Sin. 2021, 36, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Massari, S.; Daelemans, D.; Barreca, M.L.; Knezevich, A.; Sabatini, S.; Cecchetti, V.; Marcello, A.; Pannecouque, C.; Tabarrini, O. A 1,8-Naphthyridone Derivative Targets the HIV-1 Tat-Mediated Transcription and Potently Inhibits the HIV-1 Replication. J. Med. Chem. 2009, 53, 641–648. [Google Scholar] [CrossRef]
- Guendel, I.; Iordanskiy, S.; Van Duyne, R.; Kehn-Hall, K.; Saifuddin, M.; Das, R.; Jaworski, E.; Sampey, G.C.; Senina, S.; Shultz, L.; et al. Novel Neuroprotective GSK-3 Inhibitor Restricts Tat-Mediated HIV-1 Replication. J. Virol. 2014, 88, 1189–1208. [Google Scholar] [CrossRef] [Green Version]
- Tabarrini, O.; Massari, S.; Sancineto, L.; Daelemans, D.; Sabatini, S.; Manfroni, G.; Cecchetti, V.; Pannecouque, C. Structural Investigation of the Naphthyridone Scaffold: Identification of a 1,6-Naphthyridone Derivative with Potent and Selective Anti-HIV Activity. ChemMedChem 2011, 6, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Cihan-Üstündağ, G.; Zopun, M.; Vanderlinden, E.; Ozkirimli, E.; Persoons, L.; Çapan, G.; Naesens, L. Superior inhibition of influenza virus hemagglutinin-mediated fusion by indole-substituted spirothiazolidinones. Bioorganic Med. Chem. 2020, 28, 115130. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Rankovic, Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. The Physicochemical Challenges of Designing Multiple Ligands. J. Med. Chem. 2006, 49, 4961–4970. [Google Scholar] [CrossRef]
- Zhan, P.; Liu, X. Designed Multiple Ligands: An Emerging Anti-HIV Drug Discovery Paradigm. Curr. Pharm. Des. 2009, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Liu, X. Rationally Designed Multitarget Anti-HIV Agents. Curr. Med. Chem. 2013, 20, 1743–1758. [Google Scholar] [CrossRef] [PubMed]
- Sancineto, L.; Iraci, N.; Barreca, M.L.; Massari, S.; Manfroni, G.; Corazza, G.; Cecchetti, V.; Marcello, A.; Daelemans, D.; Pannecouque, C.; et al. Exploiting the anti-HIV 6-desfluoroquinolones to design multiple ligands. Bioorg. Med. Chem. 2014, 22, 4658–4666. [Google Scholar] [CrossRef]
- Wang, Z.; Bennett, E.M.; Wilson, D.J.; Salomon, C.; Vince, R. Rationally Designed Dual Inhibitors of HIV Reverse Transcriptase and Integrase. J. Med. Chem. 2007, 50, 3416–3419. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Yu, G.; Yu, Y.; Yang, S.; Duan, Z.; Wang, W.; Liu, Y.; Yu, R.; Li, J.; Zhu, T.; et al. Chemoreactive-Inspired Discovery of Influenza A Virus Dual Inhibitor to Block Hemagglutinin-Mediated Adsorption and Membrane Fusion. J. Med. Chem. 2020, 63, 6924–6940. [Google Scholar] [CrossRef]
- Fatma, B.; Kumar, R.; Singh, V.A.; Nehul, S.; Sharma, R.; Kesari, P.; Kuhn, R.J.; Tomar, S. Alphavirus capsid protease inhibitors as potential antiviral agents for Chikungunya infection. Antivir. Res. 2020, 179, 104808. [Google Scholar] [CrossRef]
- Xu, L.; Liu, H.; Hong, A.; Vivian, R.; Murray, B.P.; Callebaut, C.; Choi, Y.C.; Lee, M.S.; Chau, J.; Tsai, L.K.; et al. Structure–activity relationships of diamine inhibitors of cytochrome P450 (CYP) 3A as novel pharmacoenhancers. Part II: P2/P3 region and discovery of cobicistat (GS-9350). Bioorg. Med. Chem. Lett. 2014, 24, 995–999. [Google Scholar] [CrossRef]
- Yang, J.; Chen, W.; Kang, D.; Lu, X.; Li, X.; Liu, Z.; Huang, B.; Daelemans, D.; Pannecouque, C.; De Clercq, E.; et al. Design, synthesis and anti-HIV evaluation of novel diarylpyridine derivatives targeting the entrance channel of NNRTI binding pocket. Eur. J. Med. Chem. 2016, 109, 294–304. [Google Scholar] [CrossRef]
- Han, Z.; Liang, X.; Wang, Y.; Qing, J.; Cao, L.; Shang, L.; Yin, Z. The discovery of indole derivatives as novel hepatitis C virus inhibitors. Eur. J. Med. Chem. 2016, 116, 147–155. [Google Scholar] [CrossRef]
- Leal, E.S.; Adler, N.S.; Fernández, G.A.; Gebhard, L.G.; Battini, L.; Aucar, M.G.; Videla, M.; Monge, M.E.; Hernández de los Ríos, A.; Acosta Dávila, J.A.; et al. De novo design approaches targeting an envelope protein pocket to identify small molecules against dengue virus. Eur. J. Med. Chem. 2019, 182, 111628. [Google Scholar] [CrossRef] [PubMed]
- Dou, D.; Tiew, K.C.; Mandadapu, S.R.; Gunnam, M.R.; Alliston, K.R.; Kim, Y.; Chang, K.O.; Groutas, W.C. Potent norovirus inhibitors based on the acyclic sulfamide scaffold. Bioorganic Med. Chem. 2012, 20, 2111–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Wang, W.; Jiang, L.; Wan, S.; Zhang, L.; Yu, R.; Jiang, T. 2-Pyridinyl-4(3H)-Quinazolinone: A Scaffold for Anti-influenza A Virus Compounds. Chem. Biol. Drug Des. 2015, 86, 1221–1225. [Google Scholar] [CrossRef]
- Li, H.; Chen, L.; Li, S.; Liao, Y.; Wang, L.; Liu, Z.; Liu, S.; Song, G. Incorporation of privileged structures into 3-O-β-chacotriosyl ursolic acid can enhance inhibiting the entry of the H5N1 virus. Bioorganic Med. Chem. Lett. 2019, 29, 2675–2680. [Google Scholar] [CrossRef] [PubMed]
- Opsenica, I.; Burnett, J.C.; Gussio, R.; Opsenica, D.; Todorović, N.; Lanteri, C.A.; Sciotti, R.J.; Gettayacamin, M.; Basilico, N.; Taramelli, D.; et al. A chemotype that inhibits three unrelated pathogenic targets: The botulinum neurotoxin serotype a light chain, P. falciparum malaria, and the Ebola filovirus. J. Med. Chem. 2011, 54, 1157–1169. [Google Scholar] [CrossRef] [Green Version]
- Selaković, Ž.; Tran, J.P.; Kota, K.P.; Lazić, M.; Retterer, C.; Besh, R.; Panchal, R.G.; Soloveva, V.; Sean, V.A.; Jay, W.B.; et al. Second generation of diazachrysenes: Protection of Ebola virus infected mice and mechanism of action. Eur. J. Med. Chem. 2019, 162, 32–50. [Google Scholar] [CrossRef]
- Klimenko, K.; Lyakhov, S.; Shibinskaya, M.; Karpenko, A.; Marcou, G.; Horvath, D.; Zenkova, M.; Goncharova, E.; Amirkhanov, R.; Krysko, A.; et al. Virtual screening, synthesis and biological evaluation of DNA intercalating antiviral agents. Bioorganic Med. Chem. Lett. 2017, 27, 3915–3919. [Google Scholar] [CrossRef]
- Su, X.Z.; Miller, L.H. The discovery of artemisinin and the Nobel Prize in Physiology or Medicine. Sci. China Life Sci. 2015, 58, 1175–1179. [Google Scholar] [CrossRef] [Green Version]
- Efferth, T.; Romero, M.R.; Wolf, D.G.; Stamminger, T.; Marin, J.J.G.; Marschall, M. The antiviral activities of artemisinin and artesunate. Clin. Infect. Dis. 2008, 47, 804–811. [Google Scholar] [CrossRef] [Green Version]
- Efferth, T. Beyond malaria: The inhibition of viruses by artemisinin-type compounds. Biotechnol. Adv. 2018, 36, 1730–1737. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Iram, S.; Thomas, J.; Hayat, M.Q.; Pannecouque, C.; Dehaen, W. Application of the triazolization reaction to afford dihydroartemisinin derivatives with anti-HIV activity. Molecules 2017, 22, 303. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, B.; Ashraf, U.; Zhang, H.; Cao, C.; Li, Q.; Chen, Z.; Imran, M.; Chen, H.; Cao, S.; et al. Artemisinin inhibits the replication of flaviviruses by promoting the type I interferon production. Antivir. Res. 2020, 179, 104810. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Marschall, M.; Wang, X.; Huong, S.M.; Hauber, I.; Olbrich, A.; Kronschnabl, M.; Stamminger, T.; Huang, E.S. Antiviral activity of artesunate towards wild-type, recombinant, and ganciclovir-resistant human cytomegaloviruses. J. Mol. Med. 2002, 80, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Kaptein, S.J.F.; Efferth, T.; Leis, M.; Rechter, S.; Auerochs, S.; Kalmer, M.; Bruggeman, C.A.; Vink, C.; Stamminger, T.; Marschall, M. The anti-malaria drug artesunate inhibits replication of cytomegalovirus in vitro and in vivo. Antivir. Res. 2006, 69, 60–69. [Google Scholar] [CrossRef]
- Chou, S.; Marousek, G.; Auerochs, S.; Stamminger, T.; Milbradt, J.; Marschall, M. The unique antiviral activity of artesunate is broadly effective against human cytomegaloviruses including therapy-resistant mutants. Antivir. Res. 2011, 92, 364–368. [Google Scholar] [CrossRef]
- He, R.; Mott, B.T.; Rosenthal, A.S.; Genna, D.T.; Posner, G.H.; Arav-Boger, R. An artemisinin-derived dimer has highly potent anti-cytomegalovirus (CMV) and anti-cancer activities. PLoS ONE 2011, 6, e24334. [Google Scholar] [CrossRef] [Green Version]
- Mott, B.T.; He, R.; Chen, X.; Fox, J.M.; Civin, C.I.; Arav-Boger, R.; Posner, G.H. Artemisinin-derived dimer phosphate esters as potent anti-cytomegalovirus (anti-CMV) and anti-cancer agents: A structure-activity study. Bioorganic Med. Chem. 2013, 21, 3702–3707. [Google Scholar] [CrossRef] [Green Version]
- He, R.; Forman, M.; Mott, B.T.; Venkatadri, R.; Posner, G.H.; Arav-Boger, R. Unique and highly selective anticytomegalovirus activities of artemisinin-derived dimer diphenyl phosphate stem from combination of dimer unit and a diphenyl phosphate moiety. Antimicrob. Agents Chemother. 2013, 57, 4208–4214. [Google Scholar] [CrossRef] [Green Version]
- Reiter, C.; Fröhlich, T.; Gruber, L.; Hutterer, C.; Marschall, M.; Voigtländer, C.; Friedrich, O.; Kappes, B.; Efferth, T.; Tsogoeva, S.B. Highly potent artemisinin-derived dimers and trimers: Synthesis and evaluation of their antimalarial, antileukemia and antiviral activities. Bioorganic Med. Chem. 2015, 23, 5452–5458. [Google Scholar] [CrossRef]
- Wild, M.; Hahn, F.; Grau, B.; Herrmann, L.; Niesar, A.; Schütz, M.; Lorion, M.M.; Ackermann, L.; Tsogoeva, S.B.; Marschall, M. The artemisinin-derived autofluorescent compound bg95 exerts strong anticytomegaloviral activity based on a mitochondrial targeting mechanism. Int. J. Mol. Sci. 2020, 21, 5578. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, A.G.; Fernandez-Dolon, M.; Sanchez-Vicente, L.; Maestre, A.D.; Gomez-San Miguel, A.B.; Alvarez, M.; Serrano, M.A.; Jansen, H.; Efferth, T.; Marin, J.J.G.; et al. Novel artemisinin derivatives with potential usefulness against liver/colon cancer and viral hepatitis. Bioorganic Med. Chem. 2013, 21, 4432–4441. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.K.; Villanueva, R.A.; Thomas, D.L.; Wakita, T.; Lemon, S.M. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc. Natl. Acad. Sci. USA 2006, 103, 2310–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obeid, S.; Alen, J.; Nguyen, V.H.; Pham, V.C.; Meuleman, P.; Pannecouque, C.; Le, T.N.; Neyts, J.; Dehaen, W.; Paeshuyse, J. Artemisinin analogues as potent inhibitors of in vitro hepatitis C virus replication. PLoS ONE 2013, 8, e81783. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhan, P.; Liu, X. 1,3,4-Oxadiazole: A Privileged Structure in Antiviral Agents. Mini-Rev. Med. Chem. 2011, 11, 1130–1142. [Google Scholar] [CrossRef]
- Salahuddin; Mazumder, A.; Yar, M.S.; Mazumder, R.; Chakraborthy, G.S.; Ahsan, M.J.; Rahman, M.U. Updates on synthesis and biological activities of 1,3,4-oxadiazole: A review. Synth. Commun. 2017, 47, 1805–1847. [Google Scholar] [CrossRef]
- Shah, P.; Naik, D.; Jariwala, N.; Bhadane, D.; Kumar, S.; Kulkarni, S.; Bhutani, K.K.; Singh, I.P. Synthesis of C-2 and C-3 substituted quinolines and their evaluation as anti-HIV-1 agents. Bioorg. Chem. 2018, 80, 591–601. [Google Scholar] [CrossRef]
- Benmansour, F.; Eydoux, C.; Querat, G.; De Lamballerie, X.; Canard, B.; Alvarez, K.; Guillemot, J.C.; Barral, K. Novel 2-phenyl-5-[(E)-2-(thiophen-2-yl)ethenyl]-1,3,4-oxadiazole and 3-phenyl-5-[(E)-2-(thiophen-2-yl)ethenyl]-1,2,4-oxadiazole derivatives as dengue virus inhibitors targeting NS5 polymerase. Eur. J. Med. Chem. 2016, 109, 146–156. [Google Scholar] [CrossRef]
- El Mansouri, A.E.; Maatallah, M.; Ait Benhassou, H.; Moumen, A.; Mehdi, A.; Snoeck, R.; Andrei, G.; Zahouily, M.; Lazrek, H.B. Design, synthesis, chemical characterization, biological evaluation, and docking study of new 1,3,4-oxadiazole homonucleoside analogs. Nucleosides Nucleotides Nucleic Acids 2020, 39, 1088–1107. [Google Scholar] [CrossRef] [PubMed]
- Tomašić, T.; Peterlin Mašič, L. Rhodanine as a scaffold in drug discovery: A critical review of its biological activities and mechanisms of target modulation. Expert Opin. Drug Discov. 2012, 7, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Rajamaki, S.; Innitzer, A.; Falciani, C.; Tintori, C.; Christ, F.; Witvrouw, M.; Debyser, Z.; Massa, S.; Botta, M. Exploration of novel thiobarbituric acid-, rhodanine- and thiohydantoin-based HIV-1 integrase inhibitors. Bioorganic Med. Chem. Lett. 2009, 19, 3615–3618. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, M.; Tintori, C.; Franchi, L.; Vignaroli, G.; Innitzer, A.; Massa, S.; Esté, J.A.; Gonzalo, E.; Christ, F.; Debyser, Z.; et al. A Versatile and Practical Synthesis toward the Development of Novel HIV-1 Integrase Inhibitors. ChemMedChem 2011, 6, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Sancineto, L.; Iraci, N.; Tabarrini, O.; Santi, C. NCp7: Targeting a multitasking protein for next-generation anti-HIV drug development part 1: Covalent inhibitors. Drug Discov. Today 2018, 23, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Kovalenko, L.; Lyonnais, S.; Antaki, D.; Torbett, B.E.; Botta, M.; Mirambeau, G.; Mély, Y. Nucleocapsid Protein: A Desirable Target for Future Therapies Against HIV-1. Curr. Top. Microbiol. Immunol. 2015, 389, 53–92. [Google Scholar]
- Goudreau, N.; Hucke, O.; Faucher, A.M.; Grand-Maître, C.; Lepage, O.; Bonneau, P.R.; Mason, S.W.; Titolo, S. Discovery and Structural Characterization of a New Inhibitor Series of HIV-1 Nucleocapsid Function: NMR Solution Structure Determination of a Ternary Complex Involving a 2:1 Inhibitor/NC Stoichiometry. J. Mol. Biol. 2013, 425, 1982–1998. [Google Scholar] [CrossRef]
- Nitsche, C.; Schreier, V.N.; Behnam, M.A.M.; Kumar, A.; Bartenschlager, R.; Klein, C.D. Thiazolidinone-peptide hybrids as dengue virus protease inhibitors with antiviral activity in cell culture. J. Med. Chem. 2013, 56, 8389–8403. [Google Scholar] [CrossRef]
- Stahla-Beek, H.J.; April, D.G.; Saeedi, B.J.; Hannah, A.M.; Keenan, S.M.; Geiss, B.J. Identification of a Novel Antiviral Inhibitor of the Flavivirus Guanylyltransferase Enzyme. J. Virol. 2012, 86, 8730–8739. [Google Scholar] [CrossRef] [Green Version]
- Quek, J.P.; Liu, S.; Zhang, Z.; Li, Y.; Ng, E.Y.; Loh, Y.R.; Hung, A.W.; Luo, D.; Kang, C.B. Identification and structural characterization of small molecule fragments targeting Zika virus NS2B-NS3 protease. Antivir. Res. 2020, 175, 104707. [Google Scholar] [CrossRef]
- Murray, C.W.; Rees, D.C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; McDowell, R.S.; O’Brien, T. Fragment-Based Drug Discovery. J. Med. Chem. 2004, 47, 3463–3482. [Google Scholar] [CrossRef] [PubMed]
- Jadav, S.S.; Sinha, B.N.; Hilgenfeld, R.; Pastorino, B.; De Lamballerie, X.; Jayaprakash, V. Thiazolidone derivatives as inhibitors of chikungunya virus. Eur. J. Med. Chem. 2015, 89, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.C.; Freiberg, A.N.; Zhang, T.; Akyol-Ataman, Z.; Grock, A.; Hong, P.W.; Li, J.; Watson, N.F.; Fang, A.Q.; Aguilar, H.C.; et al. A broad-spectrum antiviral targeting entry of enveloped viruses. Proc. Natl. Acad. Sci. USA 2010, 107, 3157–3162. [Google Scholar] [CrossRef] [Green Version]
- Vigant, F.; Lee, J.; Hollmann, A.; Tanner, L.B.; Akyol Ataman, Z.; Yun, T.; Shui, G.; Aguilar, H.C.; Zhang, D.; Meriwether, D.; et al. A Mechanistic Paradigm for Broad-Spectrum Antivirals that Target Virus-Cell Fusion. PLoS Pathog. 2013, 9, e1003297. [Google Scholar] [CrossRef]
- Hollmann, A.; Castanho, M.A.R.B.; Lee, B.; Santos, N.C. Singlet oxygen effects on lipid membranes: Implications for the mechanism of action of broad-spectrum viral fusion inhibitors. Biochem. J. 2014, 459, 161–170. [Google Scholar] [CrossRef]
- Cagno, V.; Tintori, C.; Civra, A.; Cavalli, R.; Tiberi, M.; Botta, L.; Brai, A.; Poli, G.; Tapparel, C.; Lembo, D.; et al. Novel broad spectrum virucidal molecules against enveloped viruses. PLoS ONE 2018, 13, e0208333. [Google Scholar] [CrossRef]
- Tintori, C.; Iovenitti, G.; Ceresola, E.R.; Ferrarese, R.; Zamperini, C.; Brai, A.; Poli, G.; Dreassi, E.; Cagno, V.; Lembo, D.; et al. Rhodanine derivatives as potent anti-HIV and anti-HSV microbicides. PLoS ONE 2018, 13, e0198478. [Google Scholar] [CrossRef]
- Maga, G.; Falchi, F.; Radi, M.; Botta, L.; Casaluce, G.; Bernardini, M.; Irannejad, H.; Manetti, F.; Garbelli, A.; Samuele, A.; et al. Toward the Discovery of Novel Anti-HIV Drugs. Second-Generation Inhibitors of the Cellular ATPase DDX3 with Improved Anti-HIV Activity: Synthesis, Structure-Activity Relationship Analysis, Cytotoxicity Studies, and Target Validation. ChemMedChem 2011, 6, 1371–1389. [Google Scholar] [CrossRef]
- Perlíková, P.; Hocek, M. Pyrrolo[2,3-d]pyrimidine (7-deazapurine) as a privileged scaffold in design of antitumor and antiviral nucleosides. Med. Res. Rev. 2017, 37, 1429–1460. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Yu, J.; Hussain, M.; Zhou, Y.; Duan, A.; Pan, W.; Yuan, J.; Zhang, J. Design, synthesis, and biological evaluation of novel 7-deazapurine nucleoside derivatives as potential anti-dengue virus agents. Antivir. Res. 2018, 149, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Alagarsamy, V.; Chitra, K.; Saravanan, G.; Solomon, V.R.; Sulthana, M.T.; Narendhar, B. An overview of quinazolines: Pharmacological significance and recent developments. Eur. J. Med. Chem. 2018, 151, 628–685. [Google Scholar] [CrossRef] [PubMed]
- Modh, R.P.; De Clercq, E.; Pannecouque, C.; Chikhalia, K.H. Design, synthesis, antimicrobial activity and anti-HIV activity evaluation of novel hybrid quinazoline–triazine derivatives. J. Enzym. Inhib. Med. Chem. 2014, 29, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancineto, L.; Iraci, N.; Massari, S.; Attanasio, V.; Corazza, G.; Barreca, M.L.; Sabatini, S.; Manfroni, G.; Avanzi, N.R.; Cecchetti, V.; et al. Computer-aided design, synthesis and validation of 2-phenylquinazolinone fragments as CDK9 inhibitors with anti-HIV-1 tat-mediated transcription activity. ChemMedChem 2013, 8, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Rothan, H.A.; Faraj, F.L.; Teoh, T.C.; Yusof, R. Novel Quinazoline derivatives inhibited HCV Serine protease and viral replication in Huh-7 cells. bioRxiv 2019, 671313. [Google Scholar]
- Caruso, A.; Ceramella, J.; Iacopetta, D.; Saturnino, C.; Mauro, M.V.; Bruno, R.; Aquaro, S.; Sinicropi, M.S. Carbazole derivatives as antiviral agents: An overview. Molecules 2019, 24, 1912. [Google Scholar] [CrossRef] [Green Version]
- Spizzichino, S.; Mattedi, G.; Lauder, K.; Valle, C.; Aouadi, W.; Canard, B.; Decroly, E.; Kaptein, S.J.F.; Neyts, J.; Graham, C.; et al. Design, Synthesis and Discovery of N,N’-Carbazoyl-aryl-urea Inhibitors of Zika NS5 Methyltransferase and Virus Replication. ChemMedChem 2020, 15, 385–390. [Google Scholar] [CrossRef]
- Baell, J.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nat. News 2014, 513, 481–483. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Structure | Activity | Molecular Target | Status | References |
|---|---|---|---|---|---|
| Umifenovir |  | Anti-Influenza | Fusion inhibitor | Approved * | [44] |
| Delavirdine |  | Anti-HIV | Reverse transcriptase | Approved | [45] |
| Enfuvirtide | AcFTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-CONH2 (28) | Anti-HIV | Fusion inhibitor | Approved | [45] |
| Elbasvir |  | Anti-HCV | NS5A inhibitor | Approved | [46] |
| Atevirdine |  | Anti-HIV | Reverse transcriptase | Phase I | [47] |
| BILB 1941 |  | Anti-HCV | NS5B polymerase | Phase I | [48] |
| Beclabuvir |  | Anti-HCV | NS5B polymerase | Phase II | [49] |
| Diaryl Ether | Indole | 2-(1,3-Oxazinan-3-yl)ethan-1-amine | Artemisinin | 1,3,4-Oxadiazole | Rhodanine | 7-Deazapurine | Quinazolin | Carbazole | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Virus and Molecular Target | HIV | Reverse transcriptase | [17,18,19,20,21,22,23,24,25,26,27,28,29] | [42,43] | [64] | [76] | [89,90] | [114] | [118] | ||
| Integrase | [95,96] | [104] | |||||||||
| Fusion | [42,43] | ||||||||||
| Ncp7 | [99] | ||||||||||
| Tat | [50] | [116] | |||||||||
| Unknown | [42] | [91] | [114] | [118] | |||||||
| HCV | NS3-4A protease | [117] | |||||||||
| NS5B polymerase | [30,31,32] | [42,43] | [87] | [30] | [112] | [118] | |||||
| NS5A protein | [41] | ||||||||||
| Virus entry | [65] | ||||||||||
| Unknown | [70,71] | [118] | |||||||||
| HBV | Unknown | [86] | [112] | ||||||||
| FLV | NS2B-NS3 protease | [33] | [100,102] | ||||||||
| RNA polymerase | [92] | [112] | |||||||||
| E protein | [66] | ||||||||||
| NS5 RNA capping enzyme | [101] | ||||||||||
| NS5 methyltransferase | [119] | ||||||||||
| Interferon production enhancement | [77] | ||||||||||
| Unknown | [113] | [114] | |||||||||
| IAV/IBV | Fusion | [54,61] | [68] | ||||||||
| Neuraminidase | [61] | [69] | |||||||||
| Herpesviruses | Viral kinase | [114] | |||||||||
| Unknown | [78,79,81,82,83,84,85] | [93] | [112] | [114] | [118] | ||||||
| Polyomaviruses | Helicase | [35] | |||||||||
| Rhinoviruses | Viral coat protein (VP1) | [36,37] | |||||||||
| Unknown | [112] | ||||||||||
| Alphaviruses | Capsid protease | [62] | |||||||||
| NSP2 protease | [105] | ||||||||||
| Adenoviruses | Unknown | [114] | |||||||||
| Noroviruses | Unknown | [34] | [67] | ||||||||
| HPV | Unknown | [118] | |||||||||
| Ebola virus | Unknown | [70,71] | |||||||||
| Vaccinia virus | Nucleic acid intercalation | [72] | |||||||||
| Host target | dihydroorotate dehydrogenase | [38,39,40] | |||||||||
| cytochrome P450 | [63] | ||||||||||
| DEAD-box RNA helicase/ATPase DDX3 | [111] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skoreński, M.; Sieńczyk, M. The Fellowship of Privileged Scaffolds—One Structure to Inhibit Them All. Pharmaceuticals 2021, 14, 1164. https://doi.org/10.3390/ph14111164
Skoreński M, Sieńczyk M. The Fellowship of Privileged Scaffolds—One Structure to Inhibit Them All. Pharmaceuticals. 2021; 14(11):1164. https://doi.org/10.3390/ph14111164
Chicago/Turabian StyleSkoreński, Marcin, and Marcin Sieńczyk. 2021. "The Fellowship of Privileged Scaffolds—One Structure to Inhibit Them All" Pharmaceuticals 14, no. 11: 1164. https://doi.org/10.3390/ph14111164
APA StyleSkoreński, M., & Sieńczyk, M. (2021). The Fellowship of Privileged Scaffolds—One Structure to Inhibit Them All. Pharmaceuticals, 14(11), 1164. https://doi.org/10.3390/ph14111164