
Cytostatic Action of Novel Histone Deacetylase Inhibitors in Androgen Receptor-Null Prostate Cancer Cells


 ,
,  , and
, and
Abstract
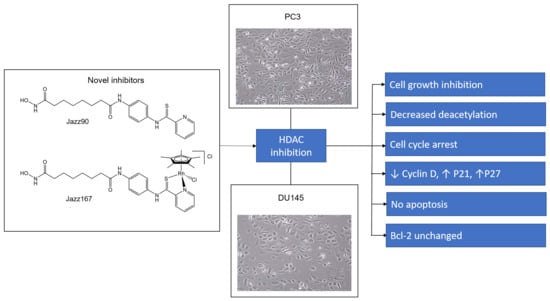
:
1. Introduction
2. Results
2.1. Cytotoxic Potency
2.2. HDAC Inhibition and Cellular Effects on Acetylation of Histone-3 Variant
2.3. Compound-Mediated Cytostatic or Cytotoxic Mechanisms
2.4. Compound Withdrawal Effects
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Maintenance
4.3. Cytotoxicity Assays and Time-Course Analysis
4.4. HDAC Inhibition
4.5. HDAC Docking
4.6. Preparation of Sample Lysates for Western Blotting
4.7. Western Blotting
4.8. Cell Cycle Analysis
4.9. Apoptosis Analysis
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Hancock, J. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017, 3, 524. [Google Scholar] [CrossRef] [PubMed]
- Saraon, P.; Drabovich, A.P.; Jarvi, K.A.; Diamandis, E.P. Mechanisms of Androgen-Independent Prostate Cancer. EJIFCC 2014, 25, 42–54. [Google Scholar] [PubMed]
- Huang, Y.; Jiang, X.; Liang, X.; Jiang, G. Molecular and cellular mechanisms of castration resistant prostate cancer. Oncol. Lett. 2018. [Google Scholar] [CrossRef] [Green Version]
- Sartor, A.O. Progression of metastatic castrate-resistant prostate cancer: Impact of therapeutic intervention in the post-docetaxel space. J. Hematol. Oncol. 2011, 4, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell. 2017, 32, 474–489.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Wang, Y.; Xiang, S.; Chan, F.L. Towards understanding androgen receptor-independent prostate cancer: An evolving paradigm. Transl. Cancer Res. 2020, 9, 415–417. [Google Scholar] [CrossRef]
- Shah, R.B.; Mehra, R.; Chinnaiyan, A.M.; Shen, R.; Ghosh, D.; Zhou, M. Androgen-Independent Prostate Cancer Is a Heterogeneous Group of Diseases: Lessons from a Rapid Autopsy Program. Cancer Res. 2004, 64, 9209–9216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 11428–11436. [Google Scholar] [CrossRef] [Green Version]
- Tucci, M.; Caffo, O.; Buttigliero, C.; Cavaliere, C.; D’aniello, C.; Di Maio, M.; Kinspergher, S.; Maines, F.; Rizzo, M.; Rossetti, S.; et al. Therapeutic options for first-line metastatic castration-resistant prostate cancer: Suggestions for clinical practise in the CHAARTED and LATITUDE era. Cancer Treat Rev. 2019, 74, 35–42. [Google Scholar] [CrossRef]
- Fléchon, A.; Pouessel, D.; Ferlay, C.; Perol, D.; Beuzeboc, P.; Gravis, G.; Joly, F.; Oudard, S.; Deplanque, G.; Zanetta, S.; et al. Phase II study of carboplatin and etoposide in patients with anaplastic progressive metastatic castration-resistant prostate cancer (mCRPC) with or without neuroendocrine differentiation: Results of the French Genito-Urinary Tumor Group (GETUG) P01 trial. Ann. Oncol. 2011, 22, 2476–2481. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009, 280, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.R.; Niesporek, S.; Denkert, C.; Dietel, M.; et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Wang, D.; Wang, Z. Yeh ETH. SENP1 Enhances Androgen Receptor-Dependent Transcription through Desumoylation of Histone Deacetylase 1. Mol. Cell Biol. 2004, 24, 6021–6028. [Google Scholar] [CrossRef] [Green Version]
- Chiao. Deficient histone acetylation and excessive deacetylase activity as epigenomic marks of prostate cancer cells. Int. J. Oncol. 2009, 35. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [Green Version]
- Raedler, L.A. Farydak (Panobinostat): First HDAC Inhibitor Approved for Patients with Relapsed Multiple Myeloma. Am. Health Drug Benefits 2016, 9, 84–87. [Google Scholar]
- Barbarotta, L.; Hurley, K. Romidepsin for the Treatment of Peripheral T-Cell Lymphoma. J. Adv. Pract. Oncol. 2015, 6, 22–36. [Google Scholar]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.Z.; et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef]
- Hanif, M.; Arshad, J.; Astin, J.W.; Rana, Z.; Zafar, A.; Movassaghi, S.; Leung, E.; Patel, K.; Söhnel, T.; Reynisson, J.; et al. A Multitargeted Approach: Organorhodium Anticancer Agent Based on Vorinostat as a Potent Histone Deacetylase Inhibitor. Angew. Chem. Int. Ed. 2020, 132, 14717–14722. [Google Scholar] [CrossRef]
- Ritchie, T.J.; Macdonald, S.J.F. The impact of aromatic ring count on compound developability-are too many aromatic rings a liability in drug design? Drug Discov. Today 2009, 14, 1011–1020. [Google Scholar] [CrossRef]
- Meier, S.M.; Hanif, M.; Adhireksan, Z.; Pichler, V.; Novak, M.; Jirkovsky, E.; Hartinger, C.G. Novel metal(ii) arene 2-pyridinecarbothioamides: A rationale to orally active organometallic anticancer agents. Chem. Sci. 2013, 4, 1837. [Google Scholar] [CrossRef]
- Lauffer, B.E.L.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone Deacetylase (HDAC) Inhibitor Kinetic Rate Constants Correlate with Cellular Histone Acetylation but Not Transcription and Cell Viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef] [Green Version]
- Mahanta, N.; Szantai-Kis, D.M.; Petersson, E.J.; Mitchell, D.A. Biosynthesis and Chemical Applications of Thioamides. ACS Chem. Biol. 2019, 14, 142–163. [Google Scholar] [CrossRef]
- Laurenzana, A.; Balliu, M.; Cellai, C.; Romanelli, M.N.; Paoletti, F. Effectiveness of the Histone Deacetylase Inhibitor (S)-2 against LNCaP and PC3 Human Prostate Cancer Cells. PLoS ONE 2013, 8, e58267. [Google Scholar] [CrossRef]
- Patra, N.; De, U.; Kim, T.H.; Lee, Y.J.; Ahn, M.Y.; Kim, N.D.; Yoon, J.H.; Choi, W.S.; Moon, H.R.; Lee, B.M.; et al. A novel histone deacetylase (HDAC) inhibitor MHY219 induces apoptosis via up-regulation of androgen receptor expression in human prostate cancer cells. Biomed. Pharm. 2013, 67, 407–415. [Google Scholar] [CrossRef]
- Ni, X.; Li, L.; Pan, G. HDAC inhibitor-induced drug resistance involving ATP-binding cassette transporters (Review). Oncol. Lett. 2015, 9, 515–521. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Huang, C.; Zhao, L.; Zhang, H.; Yang, J.M.; Luo, P.; Zhan, B.-X.; Pan, Q.; Li, J.; Wang, B.-L. Histone deacetylase inhibitors regulate P-gp expression in colorectal cancer via transcriptional activation and mRNA stabilization. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.M.; Kuay, K.T.; Koh, P.F.; Asad, M.; Tan, T.Z.; Chung, V.Y.; Lee, S.C.; Thiery, J.P.; Huang, R.Y.-J. An epithelial marker promoter induction screen identifies histone deacetylase inhibitors to restore epithelial differentiation and abolishes anchorage independence growth in cancers. Cell Death Discov. 2016, 2, 16041. [Google Scholar] [CrossRef] [PubMed]
- Kortenhorst, M.S.; Wissing, M.D.; Rodriguez, R.; Kachhap, S.K.; Jans, J.J.; Van der Groep, P.; Verheul, H.M.W.; Gupta, A.; Aiyetan, P.O.; Van der Wall, E.; et al. Analysis of the genomic response of human prostate cancer cells to histone deacetylase inhibitors. Epigenetics 2013, 8, 907–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.-Y.; Ding, W.; Li, T.-Q.; Zhang, Y.-X.; Zhao, S.-C. Histone Deacetylase (HDAC) Inhibitor, Suberoylanilide Hydroxamic Acid (SAHA), Induces Apoptosis in Prostate Cancer Cell Lines via the Akt/FOXO3a Signaling Pathway. Med. Sci. Monit. 2017, 23, 5793–5802. [Google Scholar] [CrossRef] [PubMed]
- Seim, I.; Jeffery, P.L.; Thomas, P.B.; Nelson, C.C.; Chopin, L.K. Whole-Genome Sequence of the Metastatic PC3 and LNCaP Human Prostate Cancer Cell Lines. G3 Genes Genomes Genetics 2017, 7, 1731–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlietstra, R.J.; van Alewijk, D.C.; Hermans, K.G.; van Steenbrugge, G.J.; Trapman, J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res. 1998, 58, 2720–2723. [Google Scholar] [PubMed]
- Chappell, W.H.; Lehmann, B.D.; Terrian, D.M.; Abrams, S.L.; Steelman, L.S.; McCubrey, J.A. p53 expression controls prostate cancer sensitivity to chemotherapy and the MDM2 inhibitor Nutlin-3. Cell Cycle 2012, 11, 4579–4588. [Google Scholar] [CrossRef] [Green Version]
- Chuang, M.-J.; Wu, S.-T.; Tang, S.-H.; Lai, X.-M.; Lai, H.-C.; Hsu, K.-H.; Sun, G.-H.; Chang, S.-Y.; Yu, D.-S.; Hsiao, P.-W.; et al. The HDAC Inhibitor LBH589 Induces ERK-Dependent Prometaphase Arrest in Prostate Cancer via HDAC6 Inactivation and Down-Regulation. PLoS ONE 2013, 8, e73401. [Google Scholar] [CrossRef]
- Inks, E.S.; Josey, B.J.; Jesinkey, S.R.; Chou, C.J. A Novel Class of Small Molecule Inhibitors of HDAC6. ACS Chem. Biol. 2012, 7, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.A.; No, M.; Lee, J.M.; Shin, J.H.; Oh, J.S.; Choi, E.J.; Kim, H., II; Atadja, P.; Bernhard, E.J. Epigenetic modulation of radiation response in human cancer cells with activated EGFR or HER-2 signaling: Potential role of histone deacetylase 6. Radiother. Oncol. 2009, 92, 125–132. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruscetti, M.; Leibold, J.; Bott, M.J.; Fennell, M.; Kulick, A.; Salgado, N.R.; Chen, C.C.; Ho, Y.J.; Sanchez-Rivera, F.J.; Feucht, J.; et al. NK cell–mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 2018, 362, 1416–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. JNCI J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C.; et al. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cancer/Preneoplastic Cell Lines | EC50 Values (μM) (Cancer Cells) | EC50 Values (μM) (PNT1A Cells) | Selectivity Index 1 |
|---|---|---|---|---|
| Jazz90 | PC3 | 2.09 ± 0.33 | 6.13 ± 0.74 | 2.93 |
| DU145 | 2.50 ± 0.65 | 2.45 | ||
| LnCaP | 3.04 ± 0.93 | 2.02 | ||
| NIH 3T3 | 3.93 ± 0.65 | 0.64 | ||
| Jazz167 | PC3 | 2.39 ± 0.16 * | 8.36 ± 0.10 | 3.50 |
| DU145 | 6.44 ± 1.26 * | 1.30 | ||
| LnCaP | 3.36 ± 0.71 | 2.53 | ||
| NIH 3T3 | 16.79 ± 1.77 * | 0.50 | ||
| SAHA | PC3 | 1.13 ± 0.11 | 1.87 ± 0.21 | 1.65 |
| DU145 | 0.69 ± 0.08 | 2.71 | ||
| LnCaP | 0.61 ± 0.08 | 3.07 | ||
| NIH 3T3 | 1.41 ± 0.21 | 1.33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rana, Z.; Tyndall, J.D.A.; Hanif, M.; Hartinger, C.G.; Rosengren, R.J. Cytostatic Action of Novel Histone Deacetylase Inhibitors in Androgen Receptor-Null Prostate Cancer Cells. Pharmaceuticals 2021, 14, 103. https://doi.org/10.3390/ph14020103
Rana Z, Tyndall JDA, Hanif M, Hartinger CG, Rosengren RJ. Cytostatic Action of Novel Histone Deacetylase Inhibitors in Androgen Receptor-Null Prostate Cancer Cells. Pharmaceuticals. 2021; 14(2):103. https://doi.org/10.3390/ph14020103
Chicago/Turabian StyleRana, Zohaib, Joel D. A. Tyndall, Muhammad Hanif, Christian G. Hartinger, and Rhonda J. Rosengren. 2021. "Cytostatic Action of Novel Histone Deacetylase Inhibitors in Androgen Receptor-Null Prostate Cancer Cells" Pharmaceuticals 14, no. 2: 103. https://doi.org/10.3390/ph14020103
APA StyleRana, Z., Tyndall, J. D. A., Hanif, M., Hartinger, C. G., & Rosengren, R. J. (2021). Cytostatic Action of Novel Histone Deacetylase Inhibitors in Androgen Receptor-Null Prostate Cancer Cells. Pharmaceuticals, 14(2), 103. https://doi.org/10.3390/ph14020103