
Kinins and Kinin Receptors in Cardiovascular and Renal Diseases


Abstract
:1. Introduction
2. Physiological Role of Endogenously Produced Kinins
3. Pharmacological Activation of Kinin Receptors
3.1. Kinins as Therapeutic Agents in ACE/Kininase II Inhibitor or Angiotensin II AT1 Receptor Blocker Treatment
3.2. Direct Pharmacological Agonism of Kinin Receptors
4. Pharmacological and Genetic Inactivation of Kinin Receptors in Physiology and Therapeutic
4.1. Animal Studies
4.2. Clinical Studies in Angioedema
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Carey, R.M.; Wang, Z.Q.; Siragy, H.M. Role of the angiotensin type 2 receptor in the regulation of blood pressure and renal function. Hypertension 2000, 35, 155–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegbauer, J.; Coffman, T.M. New insights into angiotensin receptor actions: From blood pressure to aging. Curr. Opin. Neprol. Hypertens. 2011, 20, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regoli, D.; Marceau, F.; Barabé, J. De novo formation of vascular receptors for bradykinin. Can. J. Physiol. Pharmacol. 1978, 56, 674–677. [Google Scholar] [CrossRef]
- Regoli, D.; Barabé, J. Pharmacology of bradykinin and related kinins. Pharmacol. Rev. 1980, 32, 1–46. [Google Scholar] [PubMed]
- Pizard, A.; Marchetti, J.; Allegrini, J.; Alhenc-Gelas, F.; Rajerison, R.M. Negative cooperativity in the human bradykinin B2 receptor. J. Biol. Chem. 1998, 273, 1309–1315. [Google Scholar] [CrossRef] [Green Version]
- Leeb-Lundberg, L.M.F.; Marceau, F.; Müller-Esterl, W.; Pettibone, D.J.; Zuraw, B.L. International union of pharmacology. XLV. Classification of the kinin receptor family: From molecular mechanisms to pathophysiological consequences. Pharmacol. Rev. 2005, 57, 27–77. [Google Scholar] [CrossRef] [Green Version]
- Furchgott, R.F.; Vanhoutte, P.M. Endothelium-derived relaxing and contracting factors. FASEB J. 1989, 3, 2007–2018. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. The discovery of nitric oxide as the endogenous nitrovasodilator. Hypertension 1988, 12, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Alhenc-Gelas, F.; Tsai, S.J.; Callahan, K.S.; Campbell, W.B.; Johnson, A.R. Stimulation of prostaglandin formation by vasoactive mediators in cultured human endothelial cells. Prostaglandins 1982, 24, 723–742. [Google Scholar] [CrossRef]
- Campbell, W.B.; Fleming, I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers Arch. 2010, 459, 881–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, N.J.; Gainer, J.V.; Stein, C.M.; Vaughan, D.E. Bradykinin stimulates tissue plasminogen activator release in human vasculature. Hypertension 1999, 33, 1431–1435. [Google Scholar] [CrossRef]
- Margolius, H.S. Tissue kallikreins and kinins: Regulation and roles in hypertensive and diabetic diseases. Annu. Rev. Pharmacol. Toxicol. 1989, 29, 343–364. [Google Scholar] [CrossRef] [PubMed]
- Scicli, A.G.; Carbini, L.A.; Carretero, O.A. The molecular biology of the kallikrein-kinin system: II. The rat gene family. J. Hypertens. 1993, 11, 775–780. [Google Scholar] [CrossRef]
- Alhenc Gelas, F.; Bouby, N.; Girolami, J. Kallikrein/K1, Kinins, and ACE/Kininase II in Homeostasis and in Disease Insight From Human and Experimental Genetic Studies, Therapeutic Implication. Front. Med. 2019, 6, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meneton, P.; Bloch-Faure, M.; Hagege, A.A.; Ruetten, H.; Huang, W.; Bergaya, S.; Ceiler, D.; Gehring, D.; Martins, I.; Salmon, G.; et al. Cardiovascular abnormalities with normal blood pressure in tissue kallikrein-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 2634–2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slim, R.; Torremocha, F.; Moreau, T.; Pizard, A.; Hunt, S.C.; Vuagnat, A.; Williams, G.H.; Gauthier, F.; Jeunemaitre, X.; Alhenc-Gelas, F. Loss-of-function polymorphism of the human kallikrein gene with reduced urinary kallikrein activity. J. Am. Soc. Nephrol. 2002, 13, 968–976. [Google Scholar]
- Borkowski, J.A.; Ransom, R.W.; Seabrook, G.R.; Trumbauer, M.; Chen, H.; Hill, R.G.; Strader, C.D.; Hess, J.F. Targeted disruption of a B2 bradykinin receptor gene in mice eliminates bradykinin action in smooth muscle and neurons. J. Biol. Chem. 1995, 270, 13706–13710. [Google Scholar] [CrossRef] [Green Version]
- Kakoki, M.; Takahashi, N.; Jennette, J.C.; Smithies, O. Diabetic nephropathy is markedly enhanced in mice lacking the bradykinin B2 receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 13302–13305. [Google Scholar] [CrossRef] [Green Version]
- Waeckel, L.; Potier, L.; Richer, C.; Roussel, R.; Bouby, N.; Alhenc-Gelas, F. Pathophysiology of genetic deficiency in tissue kallikrein activity in mouse and man. Thromb. Haemost. 2013, 110, 476–483. [Google Scholar] [CrossRef]
- Bergaya, S.; Meneton, P.; Bloch-Faure, M.; Mathieu, E.; Alhenc-Gelas, F.; Lévy, B.I.; Boulanger, C.M. Decreased Flow-Dependent Dilation in Carotid Arteries of Tissue Kallikrein–Knockout Mice. Circ. Res. 2001, 88, 593–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilgers, R.H.; Bergaya, S.; Schiffers, P.M.; Meneton, P.; Boulanger, C.M.; Henrion, D.; Lévy, B.I.; De Mey, J.G. Uterine artery structural and functional changes during pregnancy in tissue kallikrein-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1826–1832. [Google Scholar] [CrossRef]
- Azizi, M.; Boutouyrie, P.; Bissery, A.; Agharazii, M.; Verbeke, F.; Stern, N.; Bura-Rivière, A.; Laurent, S.; Alhenc-Gelas, F.; Jeunemaitre, X. Arterial and renal consequences of partial genetic deficiency in tissue kallikrein activity in humans. J. Clin. Investig. 2005, 115, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Picard, N.; Van Abel, M.; Campone, C.; Seiler, M.; Bloch-Faure, M.; Hoenderop, J.G.; Loffing, J.; Meneton, P.; Bindels, R.J.; Paillard, M.; et al. Tissue Kallikrein–Deficient Mice Display a Defect in Renal Tubular Calcium Absorption. J. Am. Soc. Nephrol. 2005, 16, 3602–3610. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, A.; Azizi, M.; Peyrard, S.; Stern, N.; Alhenc-Gelas, F.; Houillier, P.; Jeunemaitre, X. Partial human genetic deficiency in tissue kallikrein activity and renal calcium handling. Clin. J. Am. Soc. Nephrol. 2007, 2, 320–325. [Google Scholar] [CrossRef] [Green Version]
- El Moghrabi, S.; Houillier, P.; Picard, N.; Sohet, F.; Wootla, B.; Bloch-Faure, M.; Leviel, F.; Cheval, L.; Frische, S.; Meneton, P.; et al. Tissue kallikrein permits early renal adaptation to potassium load. Proc. Natl. Acad. Sci. USA 2010, 107, 13526–13531. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, J.S.; Blanchard, A.; Curis, E.; Chambrey, R.; Jeunemaitre, X.; Azizi, M. Partial genetic deficiency in tissue kallikrein impairs adaptation to high potassium intake in humans. Kidney Int. 2013, 84, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Trabold, F.; Pons, S.; Hagege, A.A.; Bloch-Faure, M.; Alhenc-Gelas, F.; Giudicelli, J.F.; Richer-Giudicelli, C.; Meneton, P. Cardiovascular phenotypes of kinin B2 receptor- and tissue kallikrein-deficient mice. Hypertension 2002, 40, 90–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griol-Charhbili, V.; Messadi-Laribi, E.; Bascands, J.L.; Heudes, D.; Meneton, P.; Giudicelli, J.F.; Alhenc-Gelas, F.; Richer, C. Role of tissue kallikrein in the cardioprotective effects of ischemic and pharmacological preconditioning in myocardial ischemia. FASEB J. 2005, 19, 1172–1174. [Google Scholar] [CrossRef]
- Bodin, S.; Chollet, C.; Goncalves-Mendes, N.; Gardes, J.; Pean, F.; Heudes, D.; Bruneval, P.; Marre, M.; Alhenc-Gelas, F.; Bouby, N. Kallikrein protects against microalbuminuria in experimental type I diabetes. Kidney Int. 2009, 76, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Kayashima, Y.; Smithies, O.; Kakoki, M. The kallikrein–kinin system and oxidative stress. Curr. Opin. Neprol. Hypertens. 2012, 21, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Silvestre, J.S.; Bergaya, S.; Tamarat, R.; Duriez, M.; Boulanger, C.M.; Levy, B.I. Proangiogenic effect of angiotensin-converting enzyme inhibition is mediated by the bradykinin B(2) receptor pathway. Circ. Res. 2001, 89, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Kränkel, N.; Katare, R.G.; Siragusa, M.; Barcelos, L.S.; Campagnolo, P.; Mangialardi, G.; Fortunato, O.; Spinetti, G.; Tran, N.; Zacharowski, K.; et al. Role of kinin B2 receptor signaling in the recruitment of circulating progenitor cells with neovascularization potential. Circ. Res. 2008, 103, 1335–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinetti, G.; Fortunato, O.; Cordella, D.; Portararo, P.; Kränkel, N.; Katare, R.; Sala-Newby, G.B.; Richer, C.; Vincent, M.P.; Alhenc-Gelas, F.; et al. Tissue kallikrein is essential for invasive capacity of circulating proangiogenic cells. Circ. Res. 2011, 108, 284–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pons, S.; Griol-Charhbili, V.; Heymes, C.; Fornes, P.; Heudes, D.; Hagege, A.; Loyer, X.; Meneton, P.; Giudicelli, J.F.; Samuel, J.L.; et al. Tissue kallikrein deficiency aggravates cardiac remodelling and decreases survival after myocardial infarction in mice. Eur. J. Heart Fail. 2008, 10, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Kakoki, M.; McGarrah, R.W.; Kim, H.-S.; Smithies, O. Bradykinin B1 and B2 receptors both have protective roles in renal ischemia/reperfusion injury. Proc. Natl. Acad. Sci. USA 2007, 104, 7576–7581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, O.A.; Richer, C.; Emanueli, C.; van Weel, V.; Quax, P.H.; Katare, R.; Kraenkel, N.; Campagnolo, P.; Barcelos, L.S.; Siragusa, M.; et al. Critical role of tissue kallikrein in vessel formation and maturation: Implications for therapeutic revascularization. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Carretero, O.A.; Sun, Y.; Shesely, E.G.; Rhaleb, N.E.; Liu, Y.H.; Liao, T.D.; Yang, J.J.; Bader, M.; Yang, X.P. Role of the B1 kinin receptor in the regulation of cardiac function and remodeling after myocardial infarction. Hypertension 2005, 45, 747–753. [Google Scholar] [CrossRef] [Green Version]
- Souza, D.G.; Lomez, E.S.; Pinho, V.; Pesquero, J.B.; Bader, M.; Pesquero, J.L.; Teixeira, M.M. Role of bradykinin B2 and B1 receptors in the local, remote, and systemic inflammatory responses that follow intestinal ischemia and reperfusion injury. J. Immunol. 2004, 172, 2542–2548. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.F.; Smith, R.S., Jr.; Shen, B.; Yang, Z.R.; Borlongan, C.V.; Chao, L.; Chao, J. Postischemic brain injury is exacerbated in mice lacking the kinin B2 receptor. Hypertension 2006, 47, 752–761. [Google Scholar] [CrossRef] [Green Version]
- Austinat, M.; Braeuninger, S.; Pesquero, J.B.; Brede, M.; Bader, M.; Stoll, G.; Renné, T.; Kleinschnitz, C. Blockade of bradykinin receptor B1 but not bradykinin receptor B2 provides protection from cerebral infarction and brain edema. Stroke 2009, 40, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Kakoki, M. Senescence-associated phenotypes in Akita diabetic mice are enhanced by absence of bradykinin B2 receptors. J. Clin. Investig. 2006, 116, 1302–1309. [Google Scholar] [CrossRef] [Green Version]
- Erdös, E.G. Angiotensin I converting enzyme and the changes in our concepts through the years. Lewis, K. Dahl memorial lecture. Hypertension 1990, 16, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Cushman, D.W.; Ondetti, M.A. Design of angiotensin converting enzyme inhibitors. Nat. Med. 1999, 5, 1110–1113. [Google Scholar] [CrossRef]
- Heart Outcomes Prevention Evaluation Study Investigators; Yusuf, S.; Sleight, P.; Pogue, J.; Bosch, J.; Davies, J.; Dagenais, G. Effects of an Angiotensin-Converting–Enzyme Inhibitor, Ramipril, on Cardiovascular Events in High-Risk Patients. N. Engl. J. Med. 2000, 342, 145–153. [Google Scholar]
- Campbell, D.J.; Kladis, A.; Duncan, A.M. Effects of converting enzyme inhibitors on angiotensin and bradykinin peptides. Hypertension 1994, 23, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Pellacani, A.; Brunner, H.R.; Nussberger, J. Plasma Kinins Increase after Angiotensin-Converting Enzyme Inhibition in Human Subjects. Clin. Sci. 1994, 87, 567–574. [Google Scholar] [CrossRef]
- Alhenc-Gelas, F.; Bouby, N.; Richer, C.; Potier, L.; Roussel, R.; Marre, M. Kinins as Therapeutic Agents in Cardiovascular and Renal Diseases. Curr. Pharm. Des. 2011, 17, 2654–2662. [Google Scholar] [CrossRef] [PubMed]
- Rhaleb, N.-E.; Yang, X.-P.; Carretero, O.A. The Kallikrein-Kinin System as a Regulator of Cardiovascular and Renal Function. In Comprehensive Physiology; John Wiley & Sons: Hoboken, NJ, USA, 2011; p. c100053. [Google Scholar]
- Komers, R.; Cooper, M.E. Acute renal hemodynamic effects of ACE inhibition in diabetic hyperfiltration: Role of kinins. Am. J. Physiol.-Ren. Physiol. 1995, 268, F588–F594. [Google Scholar] [CrossRef]
- Allen, T.J.; Cao, Z.; Youssef, S.; Hulthen, U.L.; Cooper, M.E. Role of angiotensin II and bradykinin in experimental diabetic nephropathy. Functional and structural studies. Diabetes 1997, 46, 1612–1618. [Google Scholar] [CrossRef] [PubMed]
- Tschöpe, C.; Seidl, U.; Reinecke, A.; Riester, U.; Graf, K.; Schultheiss, H.P.; Hilgenfeldt, U.; Unger, T. Kinins are involved in the antiproteinuric effect of angiotensin-converting enzyme inhibition in experimental diabetic nephropathy. Int. Immunopharmacol. 2003, 3, 335–344. [Google Scholar] [CrossRef]
- Allard, J.; Buléon, M.; Cellier, E.; Renaud, I.; Pecher, C.; Praddaude, F.; Conti, M.; Tack, I.; Girolami, J.P. ACE inhibitor reduces growth factor receptor expression and signaling but also albuminuria through B2-kinin glomerular receptor activation in diabetic rats. Am. J. Physiol.-Renal Physiol. 2007, 293, F1083–F1092. [Google Scholar] [CrossRef] [PubMed]
- Buléon, M.; Allard, J.; Jaafar, A.; Praddaude, F.; Dickson, Z.; Ranera, M.T.; Pecher, C.; Girolami, J.P.; Tack, I. Pharmacological blockade of B2-kinin receptor reduces renal protective effect of angiotensin-converting enzyme inhibition in db/db mice model. Am. J. Physiol.-Ren. Physiol. 2008, 294, F1249–F1256. [Google Scholar] [CrossRef]
- Timmermans, P.B.; Wong, P.C.; Chiu, A.T.; Herblin, W.F.; Benfield, P.; Carini, D.J.; Lee, R.J.; Wexler, R.R.; Saye, J.A.; Smith, R.D. Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol. Rev. 1993, 45, 205–251. [Google Scholar]
- Elliott, W.J. Therapeutic trials comparing angiotensin converting enzyme inhibitors and angiotensin II receptor blockers. Curr. Hypertens. Rep. 2000, 2, 402–411. [Google Scholar] [CrossRef]
- Bergaya, S.; Hilgers, R.H.; Meneton, P.; Dong, Y.; Bloch-Faure, M.; Inagami, T.; Alhenc-Gelas, F.; Lévy, B.I.; Boulanger, C.M. Flow-Dependent Dilation Mediated by Endogenous Kinins Requires Angiotensin AT2 Receptors. Circ. Res. 2004, 94, 1623–1629. [Google Scholar] [CrossRef] [Green Version]
- Messadi-Laribi, E.; Griol-Charhbili, V.; Pizard, A.; Vincent, M.P.; Heudes, D.; Meneton, P.; Alhenc-Gelas, F.; Richer, C. Tissue Kallikrein Is Involved in the Cardioprotective Effect of AT1-Receptor Blockade in Acute Myocardial Ischemia. J. Pharmacol. Exp. Ther. 2007, 323, 210–216. [Google Scholar] [CrossRef]
- Liu, Y.H.; Yang, X.P.; Sharov, V.G.; Nass, O.; Sabbah, H.N.; Peterson, E.; Carretero, O.A. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure. Role of kinins and angiotensin II type 2 receptors. J. Clin. Investig. 1997, 99, 1926–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, R.M.; Wang, Z.Q.; Siragy, H.M. Update: Role of the angiotensin type-2 (AT(2)) receptor in blood pressure regulation. Curr. Hypertens. Rep. 2000, 2, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Abadir, P.M.; Periasamy, A.; Carey, R.M.; Siragy, H.M. Angiotensin II type 2 receptor-bradykinin B2 receptor functional heterodimerization. Hypertension 2006, 48, 316–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Castaño, M.E.; Schanstra, J.P.; Neau, E.; Praddaude, F.; Pecher, C.; Ader, J.L.; Girolami, J.P.; Bascands, J.L. Induction of Functional Bradykinin B1 -Receptors in Normotensive Rats and Mice Under Chronic Angiotensin-Converting Enzyme Inhibitor Treatment. Circulation 2002, 105, 627–632. [Google Scholar] [CrossRef] [Green Version]
- Israili, Z.H. Cough and Angioneurotic Edema Associated with Angiotensin-Converting Enzyme Inhibitor Therapy: A Review of the Literature and Pathophysiology. Ann. Intern. Med. 1992, 117, 234. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.J.; Lalloo, U.G.; Belvisi, M.G.; Bernareggi, M.; Chung, K.F.; Barnes, P.J. Bradykinin–evoked sensitization of airway sensory nerves: A mechanism for ACE–inhibitor cough. Nat. Med. 1996, 2, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.J. Neprilysin Inhibitors and Bradykinin. Front. Med. 2018, 5, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanchi, A.; Maillard, M.; Burnier, M. Recent clinical trials with omapatrilat: New developments. Curr. Hypertens. Rep. 2003, 5, 346–352. [Google Scholar] [CrossRef]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [Green Version]
- Kolodka, T.; Charles, M.L.; Raghavan, A.; Radichev, I.A.; Amatya, C.; Ellefson, J.; Savinov, A.Y.; Nag, A.; Williams, M.S.; Robbins, M.S. Preclinical Characterization of Recombinant Human Tissue Kallikrein-1 as a Novel Treatment for Type 2 Diabetes Mellitus. PLoS ONE 2014, 9, e103981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koid, S.S.; Ziogas, J.; Campbell, D.J. Aliskiren reduces myocardial ischemia-reperfusion injury by a bradykinin B2 receptor- and angiotensin AT2 receptor-mediated mechanism. Hypertension 2014, 63, 768–773. [Google Scholar] [CrossRef] [Green Version]
- Bélanger, S.; Bovenzi, V.; Côté, J.; Neugebauer, W.; Amblard, M.; Martinez, J.; Lammek, B.; Savard, M.; Gobeil, F., Jr. Structure-activity relationships of novel peptide agonists of the human bradykinin B2 receptor. Peptides 2009, 30, 777–787. [Google Scholar]
- Côté, J.; Savard, M.; Bovenzi, V.; Bélanger, S.; Morin, J.; Neugebauer, W.; Larouche, A.; Dubuc, C.; Gobeil, F., Jr. Novel kinin B1 receptor agonists with improved pharmacological profiles. Peptides 2009, 30, 788–795. [Google Scholar]
- Potier, L.; Waeckel, L.; Vincent, M.P.; Chollet, C.; Gobeil FJr Marre, M.; Bruneval, P.; Richer, C.; Roussel, R.; Alhenc-Gelas, F.; Bouby, N. Selective kinin receptor agonists as cardioprotective agents in myocardial ischemia and diabetes. J. Pharmacol. Exp. Ther. 2013, 346, 23–30. [Google Scholar] [CrossRef]
- Côté, J.; Savard, M.; Bovenzi, V.; Dubuc, C.; Tremblay, L.; Tsanaclis, A.M.; Fortin, D.; Lepage, M.; Gobeil, F., Jr. Selective tumor blood-brain barrier opening with the kinin B2 receptor agonist [Phe(8)psi(CH(2)NH)Arg(9)]-BK in a F98 glioma rat model: An MRI study. Neuropeptides 2010, 44, 177–185. [Google Scholar]
- Sikpa, D.; Whittingstall, L.; Savard, M.; Lebel, R.; Côté, J.; McManus, S.; Chemtob, S.; Fortin, D.; Lepage, M.; Gobeil, F. Pharmacological Modulation of Blood–Brain Barrier Permeability by Kinin Analogs in Normal and Pathologic Conditions. Pharmaceuticals 2020, 13, 279. [Google Scholar] [CrossRef]
- Manolis, A.J.; Marketou, M.E.; Gavras, I.; Gavras, H. Cardioprotective properties of bradykinin: Role of the B2 receptor. Hypertens. Res. 2010, 33, 772–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duka, I.; Kintsurashvili, E.; Gavras, I.; Johns, C.; Bresnahan, M.; Gavras, H. Vasoactive potential of the b(1) bradykinin receptor in normotension and hypertension. Circ. Res. 2001, 88, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desposito, D.; Zadigue, G.; Taveau, C.; Adam, C.; Alhenc-Gelas, F.; Bouby, N.; Roussel, R. Neuroprotective effect of kinin B1 receptor activation in acute cerebral ischemia in diabetic mice. Sci. Rep. 2017, 7, 9410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desposito, D.; Potier, L.; Chollet, C.; Gobeil FJr Roussel, R.; Alhenc-Gelas, F.; Bouby, N.; Waeckel, L. Kinin receptor agonism restores hindlimb postischemic neovascularization capacity in diabetic mice. J. Pharmacol. Exp. Ther. 2015, 352, 218–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desposito, D.; Chollet, C.; Taveau, C.; Descamps, V.; Alhenc-Gelas, F.; Roussel, R.; Bouby, N.; Waeckel, L. Improvement of skin wound healing in diabetic mice by kinin B2 receptor blockade. Clin. Sci. 2016, 130, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Regoli, D.; Nsa Allogho, S.; Rizzi, A.; Gobeil, F.J. Bradykinin receptors and their antagonists. Eur. J. Pharmacol. 1998, 348, 1–10. [Google Scholar] [CrossRef]
- Linz, W.; Wiemer, G.; Gohlke, P.; Unger, T.; Schölkens, B.A. Contribution of kinins to the cardiovascular actions of angiotensin-converting enzyme inhibitors. Pharmacol. Rev. 1995, 47, 25–49. [Google Scholar] [PubMed]
- Pesquero, J.B.; Araujo, R.C.; Heppenstall, P.A.; Stucky, C.L.; Silva JAJr Walther, T.; Oliveira, S.M.; Pesquero, J.L.; Paiva, A.C.; Calixto, J.B.; Lewin, G.R.; et al. Hypoalgesia and altered inflammatory responses in mice lacking kinin B1 receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 8140–8145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cayla, C.; Todiras, M.; Iliescu, R.; Saul, V.V.; Gross, V.; Pilz, B.; Chai, G.; Merino, V.F.; Pesquero, J.B.; Baltatu, O.C.; et al. Mice deficient for both kinin receptors are normotensive and protected from endotoxin-induced hypotension. FASEB J. 2007, 21, 1689–1698. [Google Scholar] [CrossRef]
- Huang, W.; Gallois, Y.; Bouby, N.; Bruneval, P.; Heudes, D.; Belair, M.F.; Krege, J.H.; Meneton, P.; Marre, M.; Smithies, O.; et al. Genetically increased angiotensin I-converting enzyme level and renal complications in the diabetic mouse. Proc. Natl. Acad. Sci. USA 2001, 98, 13330–13334. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Keum, J.-S.; Wang, B.; McHenry, M.B.; Lipsitz, S.R.; Jaffa, A.A. Targeted deletion of B2-kinin receptors protects against the development of diabetic nephropathy. Am. J. Physiol.-Ren. Physiol. 2007, 293, F1026–F1035. [Google Scholar] [CrossRef] [PubMed]
- Brosius, F.C.; Alpers, C.E.; Bottinger, E.P.; Breyer, M.D.; Coffman, T.M.; Gurley, S.B.; Harris, R.C.; Kakoki, M.; Kretzler, M.; Leiter, E.H.; et al. Mouse Models of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2009, 20, 2503–2512. [Google Scholar] [CrossRef] [Green Version]
- Mage, M.; Pécher, C.; Neau, E.; Cellier, E.; Dos Reiss, M.L.; Schanstra, J.P.; Couture, R.; Bascands, J.L.; Girolami, J.P. Induction of B1 receptors in streptozotocin diabetic rats: Possible involvement in the control of hyperglycemia-induced glomerular Erk1 and 2 phosphorylation. Can. J. Physiol. Pharmacol. 2002, 80, 328–333. [Google Scholar] [CrossRef]
- Phipps, J.A.; Feener, E.P. The kallikrein-kinin system in diabetic retinopathy: Lessons for the kidney. Kidney Int. 2008, 73, 1114–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, M.; Pouliot, M.; Couture, R.; Vaucher, E. The kallikrein-kinin system in diabetic retinopathy. Prog. Drug Res. 2014, 69, 111–143. [Google Scholar]
- Pouliot, M.; Talbot, S.; Sénécal, J.; Dotigny, F.; Vaucher, E.; Couture, R. Ocular application of the kinin B1 receptor antagonist LF22–0542 inhibits retinal inflammation and oxidative stress in streptozotocin-diabetic rats. PLoS ONE 2012, 7, e33864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmaier, A.H. The hereditary angioedema syndromes. J. Clin. Investig. 2018, 129, 66–68. [Google Scholar] [CrossRef] [Green Version]
- Nussberger, J.; Cugno, M.; Amstutz, C.; Cicardi, M.; Pellacani, A.; Agostoni, A. Plasma bradykinin in angio-oedema. Lancet 1998, 351, 1693–1697. [Google Scholar] [CrossRef]
- Cicardi, M.; Banerji, A.; Bracho, F.; Malbrán, A.; Rosenkranz, B.; Riedl, M.; Bork, K.; Lumry, W.; Aberer, W.; Bier, H.; et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N. Engl. J. Med. 2010, 363, 532–541. [Google Scholar] [CrossRef] [Green Version]
- Lumry, W.R.; Li, H.H.; Levy, R.J.; Potter, P.C.; Farkas, H.; Moldovan, D.; Riedl, M.; Li, H.; Craig, T.; Bloom, B.J.; et al. Randomized placebo-controlled trial of the bradykinin B2 receptor antagonist icatibant for the treatment of acute attacks of hereditary angioedema: The FAST-3 trial. Ann. Allergy Asthma Immunol. 2011, 107, 529–537. [Google Scholar] [CrossRef]
- Baş, M.; Greve, J.; Stelter, K.; Havel, M.; Strassen, U.; Rotter, N.; Veit, J.; Schossow, B.; Hapfelmeier, A.; Kehl, V.; et al. A randomized trial of icatibant in ACE-inhibitor-induced angioedema. N. Engl. J. Med. 2015, 372, 418–425. [Google Scholar] [CrossRef] [Green Version]
- Straka, B.T.; Ramirez, C.E.; Byrd, J.B.; Stone, E.; Woodard-Grice, A.; Nian, H.; Yu, C.; Banerji, A.; Brown, N.J. Effect of bradykinin receptor antagonism on ACE inhibitor-associated angioedema. J. Allergy Clin. Immunol. 2017, 140, 242–248.e2. [Google Scholar] [CrossRef] [Green Version]
- Sinert, R.; Levy, P.; Bernstein, J.A.; Body, R.; Sivilotti, M.L.A.; Moellman, J.; Schranz, J.; Baptista, J.; Kimura, A.; Nothaft, W.; et al. Randomized Trial of Icatibant for Angiotensin-Converting Enzyme Inhibitor-Induced Upper Airway Angioedema. J. Allergy Clin. Immunol Pract. 2017, 5, 1402–1409.e3. [Google Scholar] [CrossRef]
- Jeon, J.; Lee, Y.J.; Lee, S.-Y. Effect of icatibant on angiotensin-converting enzyme inhibitor-induced angioedema: A meta-analysis of randomized controlled trials. J. Clin. Pharm. Ther. 2019, 44, 685–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AbdAlla, S.; Lother, H.; Quitterer, U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature 2000, 407, 94–98. [Google Scholar] [CrossRef]


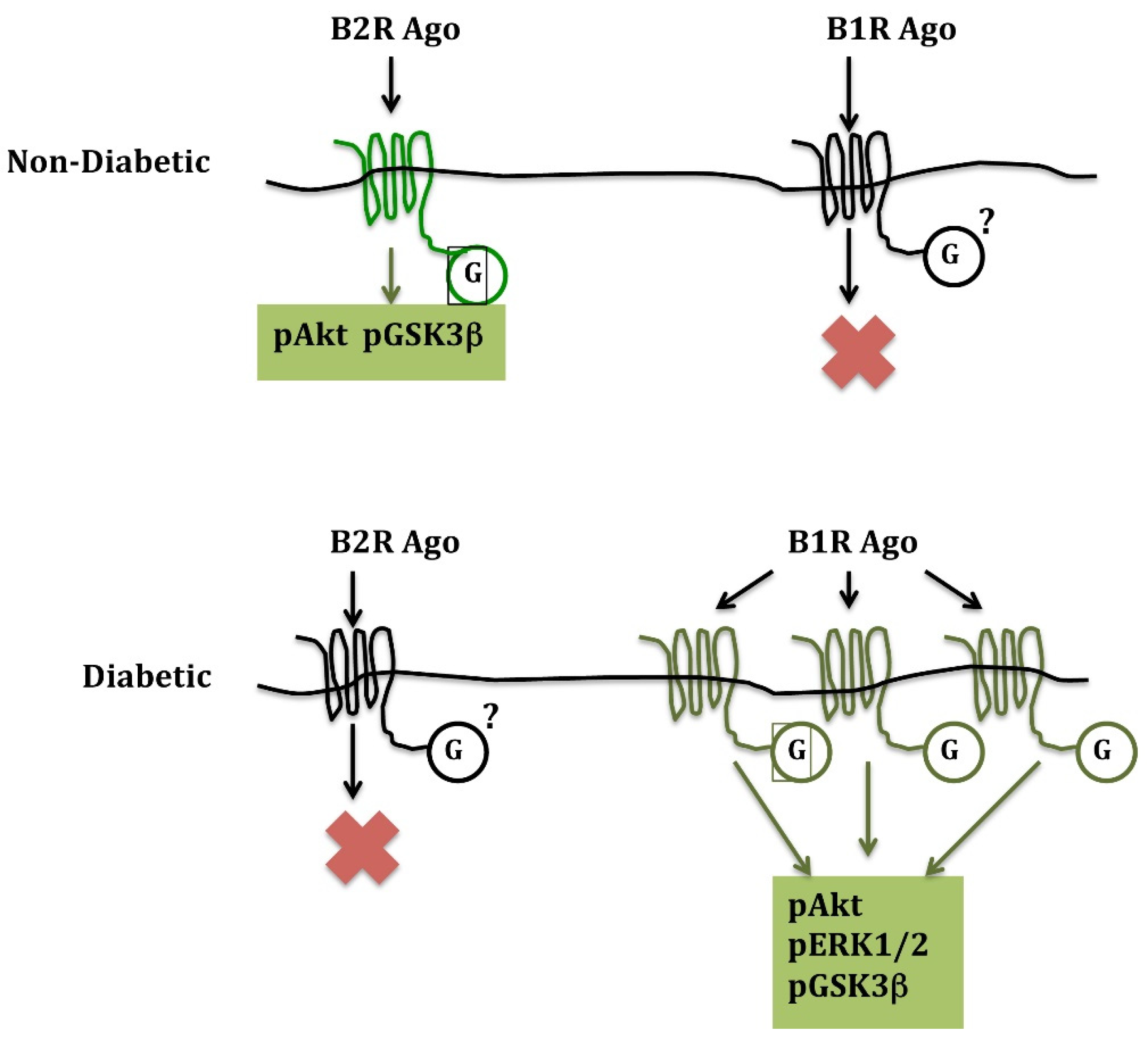{kind=link}
| B1R Effects | B2R Effects | |
|---|---|---|
| Agonists | ||
| Experimental | B1R agonist reduces heart infarct size in diabetic mice [71] | B2R agonist acutely but not chronically reduces blood pressure [71] |
| B1R agonist enhances peripheral post-ischemic angiogenesis in diabetic mice [77] | B2R agonist reduces heart infarct size in non-diabetic mice [71,74] | |
| B1R agonist increases blood–brain barrier permeability in mice [73] | B2R agonist enhances peripheral post-ischemic angiogenesis in diabetic mice [77] | |
| B1R agonist reduces brain infarct size in diabetic mice [76] | B2R agonist opens blood brain barrier in mice [72] | |
| Antagonists | ||
| Experimental | B1R antagonist inhibits retinal inflammation in diabetic rats [89] | B2R antagonist improves skin wound healing in diabetic mice [78] |
| Clinical | B2R antagonist accelerates clinical recovery in attacks of hereditary angioedema [92,93] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girolami, J.-P.; Bouby, N.; Richer-Giudicelli, C.; Alhenc-Gelas, F. Kinins and Kinin Receptors in Cardiovascular and Renal Diseases. Pharmaceuticals 2021, 14, 240. https://doi.org/10.3390/ph14030240
Girolami J-P, Bouby N, Richer-Giudicelli C, Alhenc-Gelas F. Kinins and Kinin Receptors in Cardiovascular and Renal Diseases. Pharmaceuticals. 2021; 14(3):240. https://doi.org/10.3390/ph14030240
Chicago/Turabian StyleGirolami, Jean-Pierre, Nadine Bouby, Christine Richer-Giudicelli, and Francois Alhenc-Gelas. 2021. "Kinins and Kinin Receptors in Cardiovascular and Renal Diseases" Pharmaceuticals 14, no. 3: 240. https://doi.org/10.3390/ph14030240
APA StyleGirolami, J. -P., Bouby, N., Richer-Giudicelli, C., & Alhenc-Gelas, F. (2021). Kinins and Kinin Receptors in Cardiovascular and Renal Diseases. Pharmaceuticals, 14(3), 240. https://doi.org/10.3390/ph14030240