
Development of a Rapid UHPLC-PDA Method for the Simultaneous Quantification of Flavonol Contents in Onions (Allium cepa L.)

 ,
,  ,
,  ,
,  ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. UHPLC Acquisition of the Responses
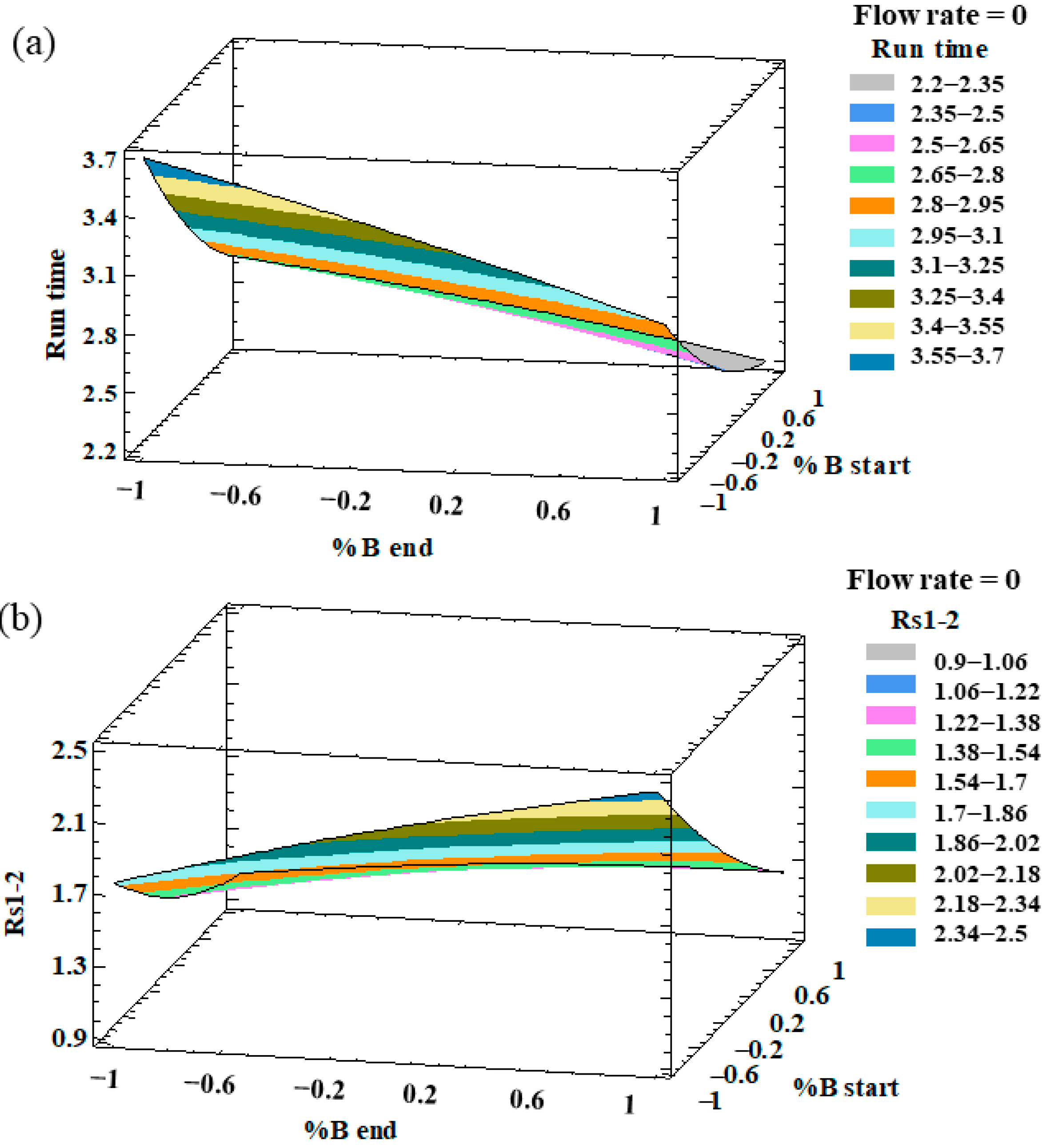
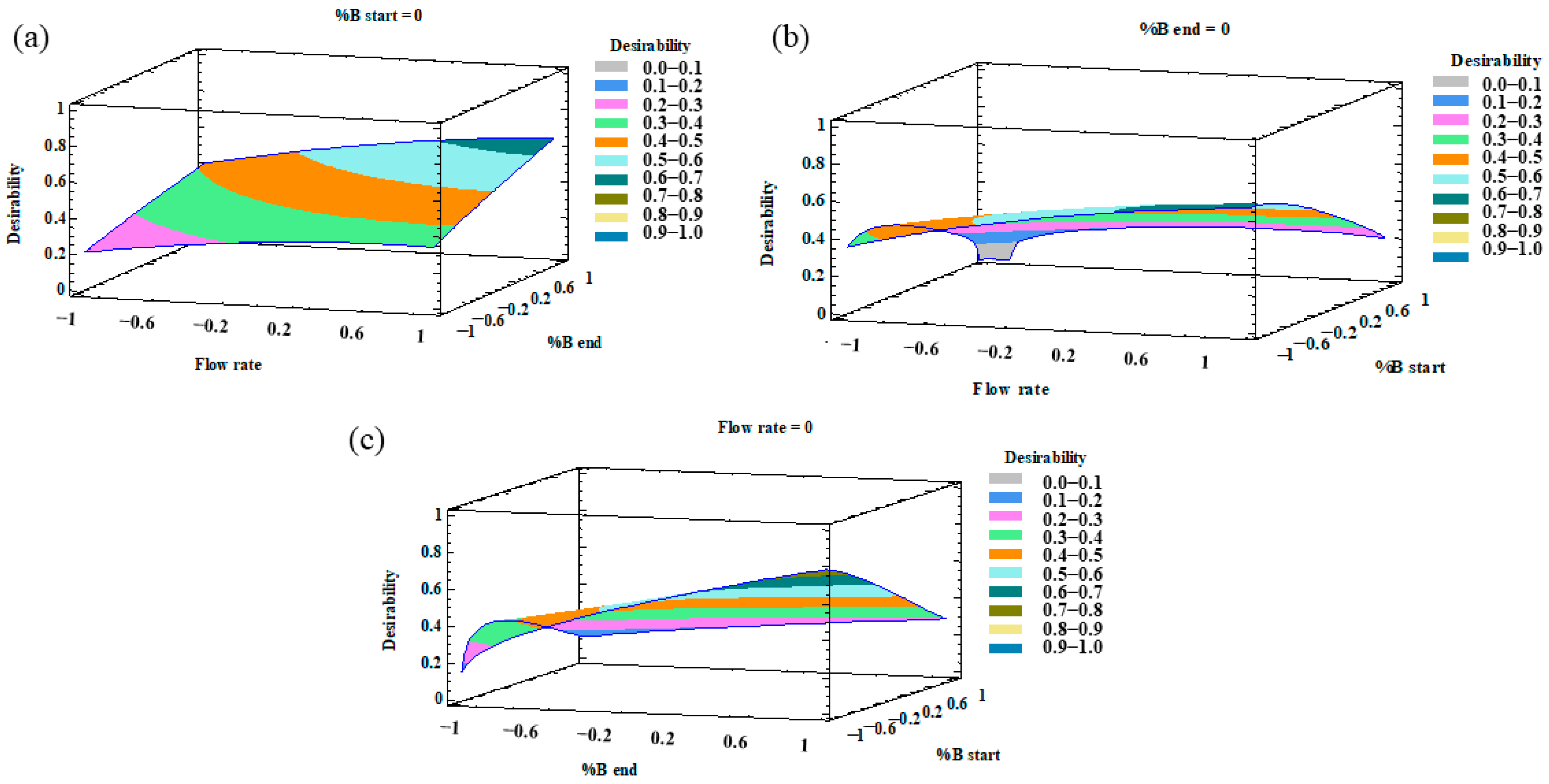
2.2. Optimization of the UHPLC Method
0.05X1X3 − 0.045833X22 − 0.125X2X3 + 0.104167X32
+ 0.0125X22 − 0.125X2X3 − 0.0875X32
− 0.325X1X3 − 0.0916667X22 + 0.05X2X3 + 0.158333 X32
+ 0.025X22 − 0.05X2X3 + 0.225X32
+ 0.0105X1X3 − 0.02375X22 + 0.08175X2X3 + 0.1315X32
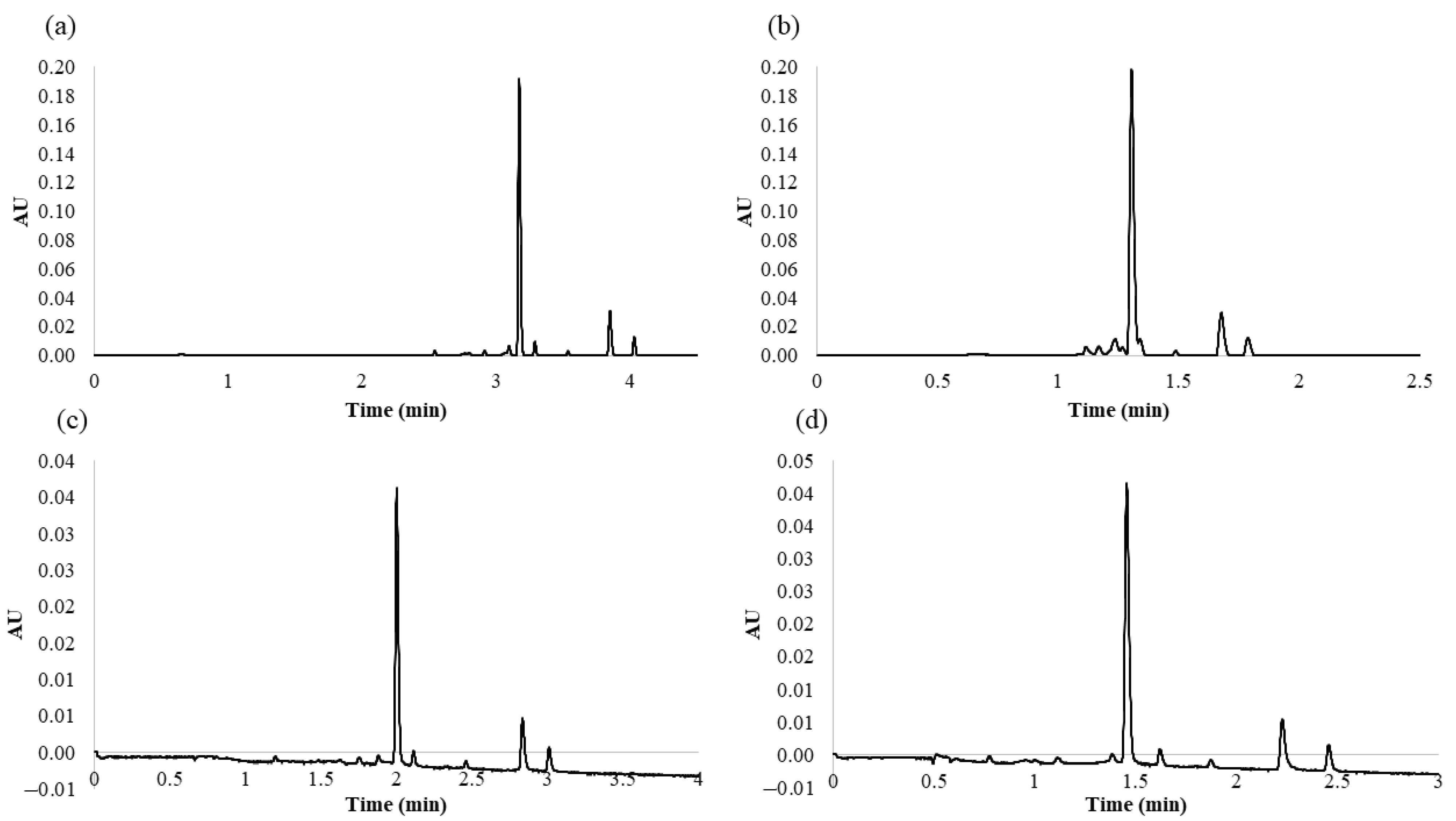
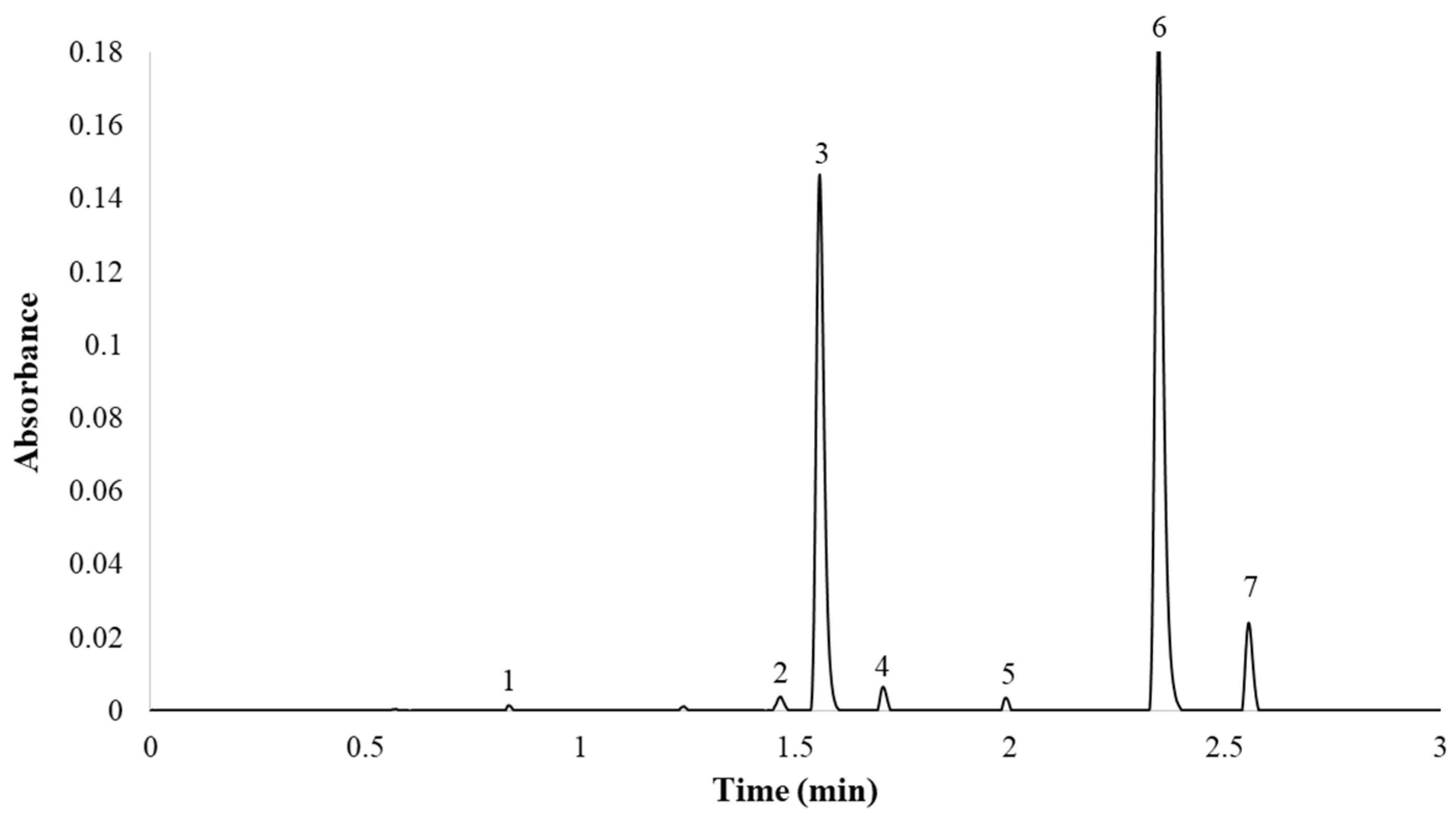
2.3. Characteristics of the Developed Method
2.4. Validation of the Developed Method
2.5. Application of the Developed Method to Different Onion Varieties
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Plant Material
3.3. Extraction Procedure
3.4. Identification of Flavonols by Liquid Chromatography Coupled to Mass Spectrometry
3.5. Separation and Quantification of Flavonols by UHPLC-PDA
3.6. Box–Behnken Design (BBD)
3.7. Validation Procedure and Chromatographic Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rodrigues, A.S.; Pérez-Gregorio, M.R.; García-Falcon, M.S.; Simal-Gándara, J.; Almeida, D.P.F. Effect of post-harvest practices on flavonoid content of red and white onion cultivars. Food Control 2010, 21, 878–884. [Google Scholar] [CrossRef]
- FAO. Global Forest Resources Assessment 2015; Food and Agriculture Organization of the United Nations: Rome, Italy, 2016. [Google Scholar]
- Block, E. The Organosulfur Chemistry of the Genus Allium—Implications for the Organic Chemistry of Sulfur. Angew. Chem. Int. Ed. Engl. 1992, 31, 1135–1178. [Google Scholar] [CrossRef]
- Özcan, M.M.; Süleyma, D.; Nurhan, U. Effect of Species on Total Phenol, Antioxidant Activity and Phenolic Compounds of Different Wild Onion Bulbs. J. Food Meas. Charact. 2018, 12, 902–905. [Google Scholar] [CrossRef]
- Ye, C.L.; Dai, D.H.; Hu, W.L. Antimicrobial and antioxidant activities of the essential oil from onion (Allium cepa L.). Food Control 2013, 30, 48–53. [Google Scholar] [CrossRef]
- Park, M.J.; Ryu, D.H.; Cho, J.Y.; Ha, I.J.; Moon, J.S.; Kang, Y.H. Comparison of the Antioxidant Properties and Flavonols in Various Parts of Korean Red Oni, ons by Multivariate Data Analysis. Hortic. Environ. Biotechnol. 2018, 59, 919–927. [Google Scholar] [CrossRef]
- Ferioli, F.; D’Antuono, L.F. Evaluation of Phenolics and Cysteine Sulfoxides in Local Onion and Shallot Germplasm from Italy and Ukraine. Genet. Resour. Crop Evol. 2016, 63, 601–614. [Google Scholar] [CrossRef]
- Larson, A.J.; Symons, J.D.; Jalili, T. Quercetin: A Treatment for Hypertension?—A Review of Efficacy and Mechanisms. Pharmaceuticals 2010, 3, 237–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abian, O.; Ortega-Alarcon, D.; Jimenez-alesanco, A.; Ceballos-laita, L.; Vega, S.; Reyburn, H.T.; Velazquez-campoy, A. Structural Stability of SARS-CoV-2 3CLpro and Identification of Quercetin as an Inhibitor by Experimental Screening. Int. J. Biol. Macromol. 2020, 164, 1693–1703. [Google Scholar] [CrossRef]
- Pérez-Gregorio, M.R.; Regueiro, J.; González-Barreiro, C.; Rial-Otero, R.; Simal-Gándara, J. Changes in Antioxidant Flavonoids during Freeze-Drying of Red Onions and Subsequent Storage. Food Control 2011, 22, 1108–1113. [Google Scholar] [CrossRef]
- Ko, E.Y.; Nile, S.H.; Jung, Y.S.; Keum, Y.S. Antioxidant and Antiplatelet Potential of Different Methanol Fractions and Flavonols Extracted from Onion (Allium cepa L.). 3 Biotech 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Jurinjak Tušek, A.; Benković, M.; Belščak Cvitanović, A.; Valinger, D.; Jurina, T.; Gajdoš Kljusurić, J. Kinetics and Thermodynamics of the Solid-Liquid Extraction Process of Total Polyphenols, Antioxidants and Extraction Yield from Asteraceae Plants. Ind. Crop. Prod. 2016, 91, 205–214. [Google Scholar] [CrossRef]
- Tistaert, C.; Dejaegher, B.; Heyden, Y.V. Chromatographic Separation Techniques and Data Handling Methods for Herbal Fingerprints: A Review. Anal. Chim. Acta 2011, 690, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Stipcovich, T.; Barbero, G.F.; Ferreiro-González, M.; Palma, M.; Barroso, C.G. Fast Analysis of Capsaicinoids in Naga Jolokia Extracts (Capsicum Chinense) by High-Performance Liquid Chromatography Using Fused Core Columns. Food Chem. 2018, 239, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Rojas, J.M.; Moreno-Ortega, A.; Ordóñez, J.L.; Moreno-Rojas, R.; Pérez-Aparicio, J.; Pereira-Caro, G. Development and Validation of UHPLC-HRMS Methodology for the Determination of Flavonoids, Amino Acids and Organosulfur Compounds in Black Onion, a Novel Derived Product from Fresh Shallot Onions (Allium Cepa Var. Aggregatum). LWT-Food Sci. Technol. 2018, 97, 376–383. [Google Scholar] [CrossRef]
- Park, S.K.; Jin, D.E.; Park, C.H.; Seung, T.W.; Guo, T.J.; Song, J.W.; Kim, J.H.; Kim, D.O.; Heo, H.J. Ameliorating effects of ethyl acetate fraction from onion (Allium cepa L.) flesh and peel in mice following trimethyltin-induced learning and memory impairment. Food Res. Int. 2015, 75, 53–60. [Google Scholar] [CrossRef]
- Lee, S.U.; Lee, J.H.; Choi, S.H.; Lee, J.S.; Ohnisi-Kameyama, M.; Kozukue, N.; Levin, C.E.; Friedman, M. Flavonoid content in fresh, home-processed, and light-exposed onions and in dehydrated commercial onion products. J. Agric. Food Chem. 2008, 56, 8541–8547. [Google Scholar] [CrossRef] [PubMed]
- Majid, I.; Vikas, N. Instrumental Texture and FLavonoid Profile of Paste Developed from Sprouted Onion Varieties of Indian Origin. Int. J. Food Prop. 2017, 20, 2511–2526. [Google Scholar] [CrossRef] [Green Version]
- González de Peredo, A.V.; Vázquez-Espinosa, M.; Piñeiro, Z.; Espada-Bellido, E.; Ferreiro-González, M.; Barbero, G.F.; Palma, M. Development of a Rapid and Accurate UHPLC-PDA-FL Method for the Quantification of Phenolic Compounds in Grapes. Food Chem. 2020, 334, 127569. [Google Scholar] [CrossRef]
- Setyaningsih, W.; Saputro, I.E.; Carrera, C.A.; Palma, M.; Barroso, C.G. Multiresponse optimization of a UPLC method for the simultaneous determination of tryptophan and 15 tryptophan-derived compounds using a Box-Behnken design with a desirability function. Food Chem. 2017, 225, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wani, T.A.; Ahmad, A.; Zargar, S.; Khalil, N.Y.; Darwish, I.A. Use of response surface methodology for development of new microwell-based spectrophotometric method for determination of atrovastatin calcium in tablets. Chem. Cent. J. 2012, 6, 134. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Cai, M.; Liang, H.H. Desirability Function Approach for the Optimization of Microwave-Assisted Extraction of Saikosaponins from Radix Bupleuri. Sep. Purif. Technol. 2008, 61, 266–275. [Google Scholar] [CrossRef]
- Zheng, J.; Polyakova, Y.; Row, K.H. Retention Factors and Resolutions of Amino Benzoic Acid Isomers with Some Lonic Liquids. Biotechnol. Bioproc. E 2006, 11, 477–483. [Google Scholar] [CrossRef]
- Sharma, K.; Assefa, A.D.; Kim, S.; Ko, E.Y.; Lee, E.T.; Park, S.W. Evaluation of Total Phenolics, Flavonoids and Antioxidant Activity of 18 Korean Onion Cultivars: A Comparative Study. J. Sci. Food Agric. 2014, 94, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.; Turner, P.; Jacobson, G.; Almgren, K.; Waldebäck, M.; Sjöberg, P.; Karlsson, E.N.; Markides, K.E. Subcritical Water Extraction and β-Glucosidase-Catalyzed Hydrolysis of Quercetin Glycosides in Onion Waste. Green Chem. 2006, 8, 949–959. [Google Scholar] [CrossRef]
- Snyder, L.R.; Dolan, J.W. High-Performance Gradient Elution; Wiley-Interscience: Hoboken, NJ, USA, 2006. [Google Scholar]
- ICH. ICH Topic Q2 (R1) Validation of Analytical Procedures: Text and Methodology; ICH: Geneva, Switzerland, 2005; Volume 1994. [Google Scholar]
- AOAC. AOAC Peer Verified Methods Program, Manual on Policies and Procedures; AOAC International: Rockville, MD, USA, 2012. [Google Scholar]
- Pérez-Gregorio, M.R.; García-Falcón, M.S.; Simal-Gándara, J.; Rodrigues, A.S.; Almeida, D.P.F. Identification and Quantification of Flavonoids in Traditional Cultivars of Red and White Onions at Harvest. J. Food Compos. Anal. 2010, 23, 592–598. [Google Scholar] [CrossRef]
- Vallverdú-Queralt, A.; Medina-Remón, A.; Andres-Lacueva, C.; Lamuela-Raventos, R.M. Changes in Phenolic Profile and Antioxidant Activity during Production of Diced Tomatoes. Food Chem. 2011, 126, 1700–1707. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Espinosa, M.; Espada-Bellido, E.; González de Peredo, A.V.; Ferreiro-González, M.; Carrera, C.; Palma, M.; Barroso, C.G.; Barbero, G.F. Optimization of Microwave-Assisted Extraction for the Recovery of Bioactive Compounds from the Chilean Superfruit (Aristotelia Chilensis (Mol.) Stuntz). Agronomy 2018, 8, 240. [Google Scholar] [CrossRef] [Green Version]
- González de Peredo, A.V.; Vázquez-Espinosa, M.; Espada-Bellido, E.; Ferreiro-González, M.; Amores-Arrocha, A.; Palma, M.; Barbero, G.F.; Jiménez-Cantizano, A. Alternative Ultrasound-Assisted Method for the Extraction of the Bioactive Compounds Present in Myrtle (Myrtus communis L.). Molecules 2019, 24, 882. [Google Scholar] [CrossRef] [Green Version]
- Bonaccorsi, P.; Caristi, C.; Gargiulli, C.; Leuzzi, U. Flavonol glucosides in Allium species: A comparative study by means of HPLC-DAD-ESI-MS-MS. Food Chem. 2008, 107, 1668–1673. [Google Scholar] [CrossRef]
- Katsampa, P.; Valsamedou, E.; Grigorakis, S.; Makris, D.P. A green ultrasound-assisted extraction process for the recovery of antioxidant polyphenols and pigments from onion solid wastes using Box–Behnken experimental design and kinetics. Ind. Crop. Prod. 2015, 77, 535–543. [Google Scholar] [CrossRef]
- Tedesco, I.; Carbone, V.; Spagnuolo, C.; Minasi, P.; Russo, G.L. Identification and Quantification of Flavonoids from Two Southern Italian Cultivars of Allium cepa L., Tropea (Red Onion) and Montoro (Copper Onion), and Their Capacity to Protect Human Erythrocytes from Oxidative Stress. J. Agric. Food Chem. 2015, 63, 5229–5238. [Google Scholar] [CrossRef] [PubMed]
- de Souza Dias, F.; Lovillo, M.P.; Barroso, C.G.; David, J.M. Optimization and validation of a method for the direct determination of catechin and epicatechin in red wines by HPLC/fluorescence. Microchem. J. 2010, 96, 17–20. [Google Scholar] [CrossRef]
- Piñeiro, Z.; Palma, M.; Barroso, C.G. Determination of Catechins by Means of Extraction with Pressurized Liquids. J. Chromatogr. A 2004, 1026, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Setyaningsih, W.; Saputro, I.E.; Carrera, C.A.; Palma, M.; García-Barroso, C. Fast Determination of Phenolic Compounds in Rice Grains by Ultraperformance Liquid Chromatography Coupled to Photodiode Array Detection: Method Development and Validation. J. Agric. Food Chem. 2019, 67, 3018–3027. [Google Scholar] [CrossRef]
- Maran, J.; Manikandan, S.; Thirugnanasambandham, K.; Vigna Nivetha, C.; Dinesh, R. Box-Behnken Design Based Statistical Modeling for Ultrasound-Assisted Extraction of Corn Silk Polysaccharide. Carbohydr. Polym. 2013, 92, 604–611. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
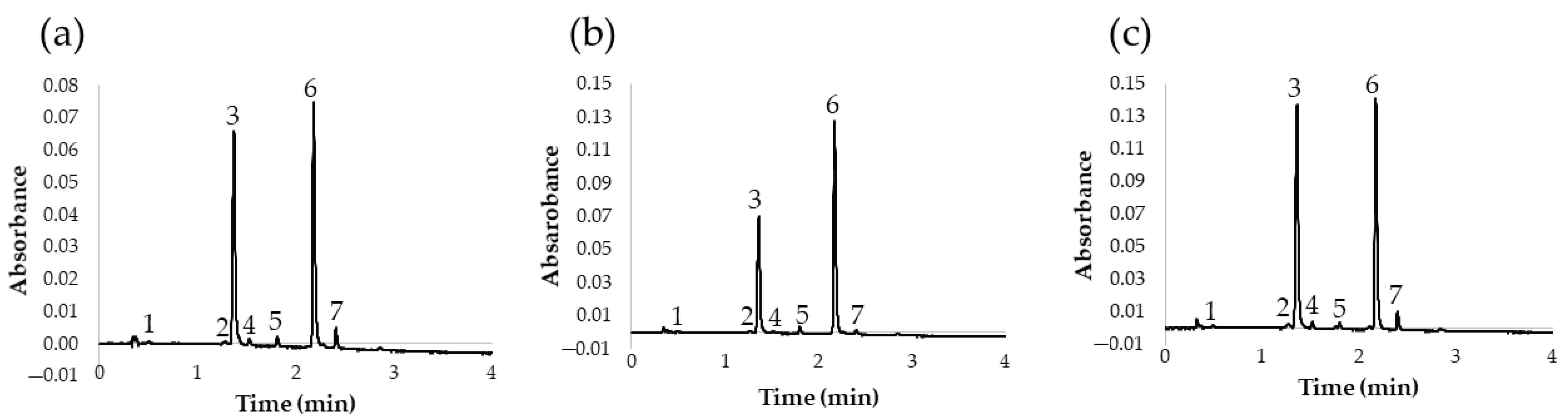{kind=link}
| Run | Factors | Responses | ||||||
|---|---|---|---|---|---|---|---|---|
| X1 | X2 | X3 | Rs3–4 | Rs4–5 | Rs5–6 | Rs6–7 | Run Time (min) | |
| 1 | 0 | 0 | 0 | 1.50 | 3.62 | 3.34 | 1.74 | 2.761 |
| 2 | 1 | 0 | 1 | 1.22 | 3.68 | 2.72 | 1.31 | 2.326 |
| 3 | 0 | 0 | 0 | 1.77 | 4.53 | 4.10 | 2.02 | 2.775 |
| 4 | 0 | −1 | 1 | 1.08 | 2.46 | 2.12 | 1.22 | 2.658 |
| 5 | −1 | 0 | −1 | 2.01 | 3.93 | 3.68 | 2.20 | 3.574 |
| 6 | −1 | 0 | 1 | 1.00 | 2.83 | 2.79 | 1.27 | 2.659 |
| 7 | 0 | 1 | 1 | 1.18 | 3.53 | 3.08 | 1.47 | 2.210 |
| 8 | 0 | 0 | 0 | 1.36 | 3.62 | 2.89 | 1.41 | 2.759 |
| 9 | 0 | 1 | −1 | 2.35 | 5.40 | 4.81 | 2.83 | 2.924 |
| 10 | 1 | −1 | 0 | 1.25 | 3.60 | 3.49 | 1.75 | 2.892 |
| 11 | 0 | −1 | −1 | 1.80 | 3.88 | 4.01 | 2.35 | 3.699 |
| 12 | −1 | −1 | 0 | 1.13 | 3.38 | 3.05 | 1.61 | 3.242 |
| 13 | −1 | 1 | 0 | 1.49 | 3.76 | 3.01 | 1.58 | 2.705 |
| 14 | 1 | 1 | 0 | 1.82 | 4.30 | 3.63 | 1.88 | 2.298 |
| 15 | 1 | 0 | −1 | 2.02 | 4.33 | 4.91 | 2.92 | 3.199 |
| Peak | Compounds | tR (min) | Width (Wb) | t’R | Retention Factor (k) | Selectivity (α) | Resolution (Rs) |
|---|---|---|---|---|---|---|---|
| 1 | Quercetin 7,3,4′-O-triglucoside | 0.945 | 2.85 | 0.448 | 1.36 | - | - |
| 2 | Quercetin 7,4′-O-diglucoside | 1.478 | 3.45 | 1.084 | 2.69 | 1.98 | 7.69 |
| 3 | Quercetin 3,4′-O-diglucoside | 1.688 | 4.10 | 1.174 | 3.22 | 1.19 | 2.15 |
| 4 | Isorhamnetin 3,4′-O-diglucoside | 1.835 | 2.40 | 1.315 | 3.59 | 1.11 | 1.93 |
| 5 | Quercetin 3′-O-glucoside | 2.119 | 2.55 | 1.599 | 4.30 | 1.20 | 4.56 |
| 6 | Quercetin 4′-O-glucoside | 2.478 | 5.65 | 1.951 | 5.19 | 1.21 | 3.68 |
| 7 | Isorhamnetin 4′-O-glucoside | 2.693 | 3.35 | 2.157 | 5.73 | 1.10 | 2.23 |
| Peak | Compounds | Linear Equation | R2 | LOD (mg L−1) | LOQ (mg L−1) | CV 1 for Repeatability (%) | CV 1 for Intermediate Precision (%) | ||
|---|---|---|---|---|---|---|---|---|---|
| tR | Rs | tR | Rs | ||||||
| 1 | Quercetin 7,3,4′-O-triglucoside | y = 5069.90x + 8282.83 | 0.9997 | 0.0437 | 0.1454 | 2.33 | - | 2.80 | - |
| 2 | Quercetin 7,4′-O-diglucoside | y = 6382.50x + 8282.83 | 0.9997 | 0.0347 | 0.1155 | 1.20 | 1.88 | 1.93 | 4.13 |
| 3 | Quercetin 3,4′-O-diglucoside | y = 6382.52x + 8282.83 | 0.9997 | 0.0347 | 0.1155 | 1.17 | 3.89 | 1.91 | 4.59 |
| 4 | Isorhamnetin 3,4′-O-diglucoside | y = 6247.89x + 8282.83 | 0.9997 | 0.0354 | 0.1181 | 0.95 | 2.52 | 1.76 | 2.72 |
| 5 | Quercetin 3′-O-glucoside | y = 8610.35x + 8282.83 | 0.9997 | 0.0257 | 0.0857 | 0.96 | 4.88 | 1.70 | 4.90 |
| 6 | Quercetin 4′-O-glucoside | y = 8610.35x + 8282.83 | 0.9997 | 0.0257 | 0.0857 | 0.88 | 1.53 | 3.17 | 3.65 |
| 7 | Isorhamnetin 4′-O-glucoside | y = 8358.38x + 8282.83 | 0.9997 | 0.0265 | 0.0883 | 0.78 | 4.14 | 1.39 | 4.03 |
| Column Temperatures (°C) | Flow Rates (mL min−1) | Injection Volumes (µL) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 52 | 55 | 58 | 0.57 | 0.60 | 0.63 | 2.8 | 3.0 | 3.2 | ||
| Retention time (min) | Quercetin 7,3,4′-O-triglucoside | 1.026 a | 0.945 b | 0.882 c | 1.026 a | 0.945 b | 0.912 c | 0.955 a | 0.945 a | 0.929 a |
| Quercetin 7,4′-O-diglucoside | 1.501 a | 1.477 a | 1.497 a | 1.590 a | 1.477 b | 1.540 b | 1.530 a | 1.477 a | 1.559 a | |
| Quercetin 3,4′-O-diglucoside | 1.797 a | 1.687 b | 1.679 b | 1.748 a | 1.687 b | 1.627 c | 1.694 a | 1.687 a | 1.674 a | |
| Isorhamnetin 3,4′-O-diglucoside | 1.936 a | 1.835 b | 1.804 b | 1.887 a | 1.835 b | 1.775 c | 1.839 a | 1.835 a | 1.823 a | |
| Quercetin 3′-O-glucoside | 2.231 a | 2.119 b | 2.002 c | 2.161 a | 2.119 b | 2.056 c | 2.126 a | 2.119 a | 2.111 a | |
| Quercetin 4′-O-glucoside | 2.591 a | 2.478 b | 2.357 c | 2.512 a | 2.478 a | 2.417 b | 2.484 a | 2.478 a,b | 2.471 b | |
| Isorhamnetin 4′-O-glucoside | 2.797 a | 2.693 a | 2.574 b | 2.721 a | 2.692 b | 2.633 c | 2.697 a | 2.693 a,b | 2.683 b | |
| Peak area | Quercetin 7,3,4′-O-triglucoside | 1584 a | 1749 a | 1992 a | 1815 a | 1749 a | 1846 a | 1493 a | 1749 a | 2270 b |
| Quercetin 7,4′-O-diglucoside | 4073 a | 4459 a | 5070 a | 5733 a | 4459 a | 5004 a | 3246 b | 4459 a,b | 5251 b | |
| Quercetin 3,4′-O-diglucoside | 104,048 a | 112,705 a | 123,675 a | 123,884 a | 112,705 a | 122,463 a | 88,187 a | 112,705 a | 140,119 b | |
| Isorhamnetin 3,4′-O-diglucoside | 5073 a | 5245 a | 5931 a | 5892 a | 5245 a | 5800 a | 4233 a | 5245 a | 6905 b | |
| Quercetin 3′-O-glucoside | 5045 a | 4899 a | 5396 a | 5330 a | 4899 a | 5448 a | 4015 a | 4899 a,b | 5811 b | |
| Quercetin 4′-O-glucoside | 151,175 a | 162,031 a | 178,379 a | 175,285 a | 159,754 a | 173,719 a | 130,086 a | 165,384 a | 198,383 b | |
| Isorhamnetin 4′-O-glucoside | 18,933 a | 20,539 a | 22,383 a | 22,187 a | 20,539 a | 21,882 a | 16,308 a | 20,539 a | 25,220 b | |
| Peak resolution | Quercetin 7,3,4′-O-triglucoside | - | - | - | - | - | - | - | - | - |
| Quercetin 7,4′-O-diglucoside | 6.22 a | 7.69 a | 6.94 a | 6.95 a | 7.69 a | 7.68 a | 7.62 a | 7.69 a | 8.15 a | |
| Quercetin 3,4′-O-diglucoside | 2.89 a | 2.15 a,b | 1.6 b | 1.62 a | 2.15 a | 0.810 b | 1.80 a | 2.15 b | 1.20 a | |
| Isorhamnetin 3,4′-O-diglucoside | 1.92 a | 1.93 a | 1.59 a | 2.09 a | 1.93 a | 1.95 a | 2.10 a | 1.93 a | 1.94 a | |
| Quercetin 3′-O-glucoside | 4.54 a | 4.56 a | 2.9 b | 4.60 a | 4.72 a | 4.56 a | 4.48 a | 4.56 a | 4.44 a | |
| Quercetin 4′-O-glucoside | 3.30 a | 3.68 a | 3.47 a | 3.70 a | 3.67 a | 3.68 a | 3.43 a | 3.68 a | 3.82 a | |
| Isorhamnetin 4′-O-glucoside | 1.87 a | 2.23 b | 2.11 a,b | 2.14 a | 2.23 a | 2.15 a | 2.10 a | 2.23 a | 2.16 a | |
| Flavonol Composition (mg/10 g DW) 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Quercetin 7,3,4′-O-triglucoside | Quercetin 7,4′-O-diglucoside | Quercetin 3,4′-O-diglucoside | Isorhamnetin 3,4′-O-diglucoside | Quercetin 3′-O-glucoside | Quercetin 4′-O-glucoside | Isorhamnetin 4′-O-glucoside | Total | |
| Spring white onion 1 | 0.09 ± 0.00 | 0.62 ± 0.00 | 14.31 ± 0.19 | 0.56 ± 0.00 | 0.56 ± 0.00 | 11.74 ± 0.14 | 1.11 ± 0.02 | 28.98 |
| Sweet white onion 2 | 0.08 ± 0.01 | 0.12 ± 0.00 | 5.61 ± 0.06 | 0.23 ± 0.00 | 0.43 ± 0.02 | 2.50 ± 0.07 | 0.20 ± 0.00 | 9.17 |
| Spring white onion 3 | 0.15 ± 0.00 | 0.51 ± 0.00 | 9.17 ± 0.06 | 0.48 ± 0.01 | 0.47 ± 0.00 | 7.36 ± 0.04 | 0.73 ± 0.01 | 18.88 |
| Sweet white onion 4 | 0.11 ± 0.00 | 0.11 ± 0.03 | 5.58 ± 0.22 | 0.25 ± 0.00 | 3.77 ± 0.00 | 1.03 ± 0.10 | 0.40 ± 0.02 | 11.23 |
| Sweet white onion 5 | 0.06 ± 0.03 | 0.12 ± 0.02 | 8.41 ± 0.15 | 0.12 ± 0.01 | 0.45 ± 0.00 | 9.42 ± 0.09 | 0.84 ± 0.00 | 19.42 |
| Sweet white onion 6 | 0.07 ± 0.00 | 0.10 ± 0.01 | 7.28 ± 0.07 | 0.21 ± 0.00 | 0.12 ± 0.00 | 6.61 ± 0.05 | 0.98 ± 0.00 | 15.36 |
| Yellow onion 1 | 0.21 ± 0.02 | 0.53 ± 0.03 | 12.32 ± 0.02 | 0.46 ± 0.02 | 0.52 ± 0.00 | 15.47 ± 0.00 | 0.65 ± 0.01 | 30.17 |
| Yellow onion 2 | 0.29 ± 0.01 | 0.10 ± 0.02 | 7.13 ± 0.13 | 0.52 ± 0.03 | 0.40 ± 0.12 | 5.18 ± 0.01 | 0.91 ± 0.02 | 14.53 |
| Yellow onion 3 | 0.18 ± 0.00 | 0.22 ± 0.01 | 8.13 ± 0.02 | 0.32 ± 0.00 | 5.00 ± 0.01 | 1.37 ± 0.05 | 0.13 ± 0.00 | 15.35 |
| Red onion 1 | 0.36 ± 0.02 | 0.88 ± 0.06 | 25.58 ± 0.16 | 0.72 ± 0.01 | 0.57 ± 0.02 | 18.42 ± 0.15 | 1.47 ± 0.00 | 48.00 |
| Red onion 2 | 0.34 ± 0.03 | 0.69 ± 0.04 | 16.36 ± 0.04 | 0.83 ± 0.01 | 0.57 ± 0.00 | 18.69 ± 0.03 | 2.44 ± 0.00 | 39.92 |
| Red onion 3 | 0.28 ± 0.00 | 0.56 ± 0.00 | 17.60 ± 0.05 | 0.52 ± 0.03 | 0.92 ± 0.01 | 21.94 ± 1.03 | 1.03 ± 0.01 | 42.86 |
| Red onion 4 | 0.29 ± 0.02 | 0.86 ± 0.02 | 24.97 ± 0.32 | 0.83 ± 0.02 | 0.56 ± 0.00 | 16.73 ± 0.91 | 1.99 ± 0.06 | 46.23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-de-Peredo, A.V.; Vázquez-Espinosa, M.; Carrera, C.; Espada-Bellido, E.; Ferreiro-González, M.; F. Barbero, G.; Palma, M. Development of a Rapid UHPLC-PDA Method for the Simultaneous Quantification of Flavonol Contents in Onions (Allium cepa L.). Pharmaceuticals 2021, 14, 310. https://doi.org/10.3390/ph14040310
González-de-Peredo AV, Vázquez-Espinosa M, Carrera C, Espada-Bellido E, Ferreiro-González M, F. Barbero G, Palma M. Development of a Rapid UHPLC-PDA Method for the Simultaneous Quantification of Flavonol Contents in Onions (Allium cepa L.). Pharmaceuticals. 2021; 14(4):310. https://doi.org/10.3390/ph14040310
Chicago/Turabian StyleGonzález-de-Peredo, Ana V., Mercedes Vázquez-Espinosa, Ceferino Carrera, Estrella Espada-Bellido, Marta Ferreiro-González, Gerardo F. Barbero, and Miguel Palma. 2021. "Development of a Rapid UHPLC-PDA Method for the Simultaneous Quantification of Flavonol Contents in Onions (Allium cepa L.)" Pharmaceuticals 14, no. 4: 310. https://doi.org/10.3390/ph14040310
APA StyleGonzález-de-Peredo, A. V., Vázquez-Espinosa, M., Carrera, C., Espada-Bellido, E., Ferreiro-González, M., F. Barbero, G., & Palma, M. (2021). Development of a Rapid UHPLC-PDA Method for the Simultaneous Quantification of Flavonol Contents in Onions (Allium cepa L.). Pharmaceuticals, 14(4), 310. https://doi.org/10.3390/ph14040310