
Metformin Modifies the Gut Microbiota of Mice Infected with Helicobacter pylori



Abstract
:1. Introduction
2. Results
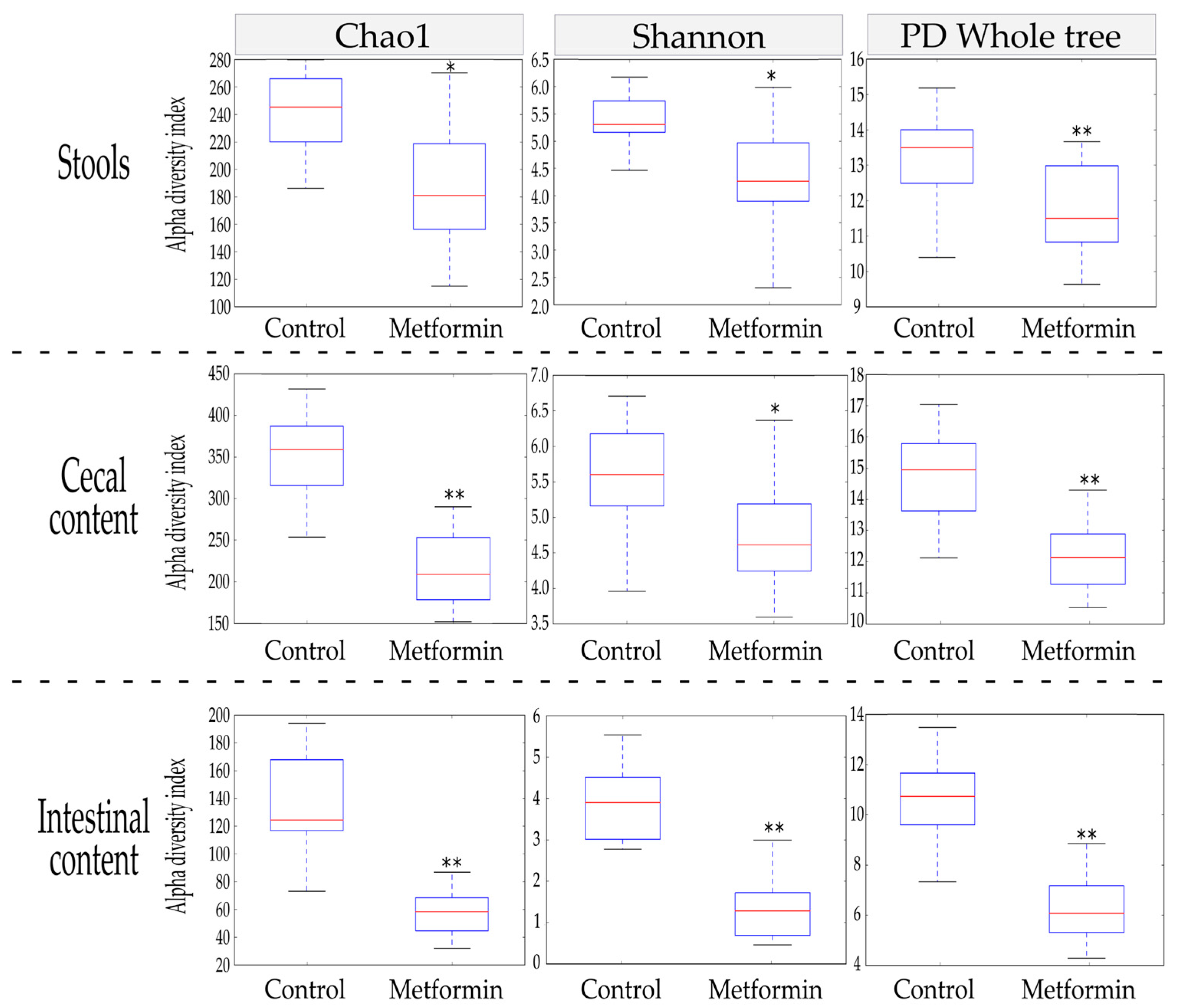
2.1. Alpha Diversity Indices Are Reduced in the Gut Microbiota of Mice Infected with H. pylori and Treated with Metformin
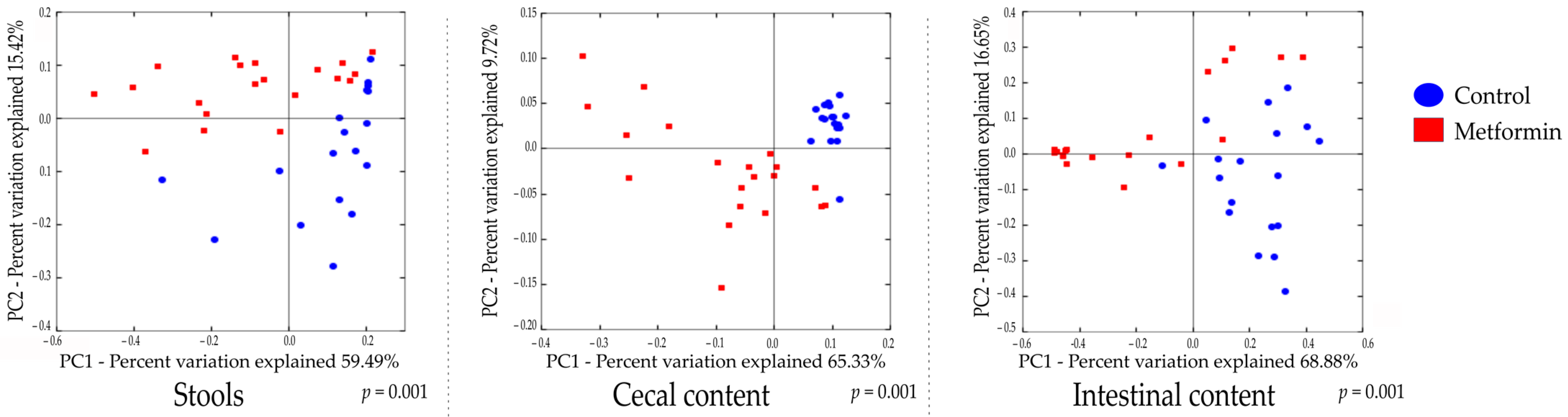
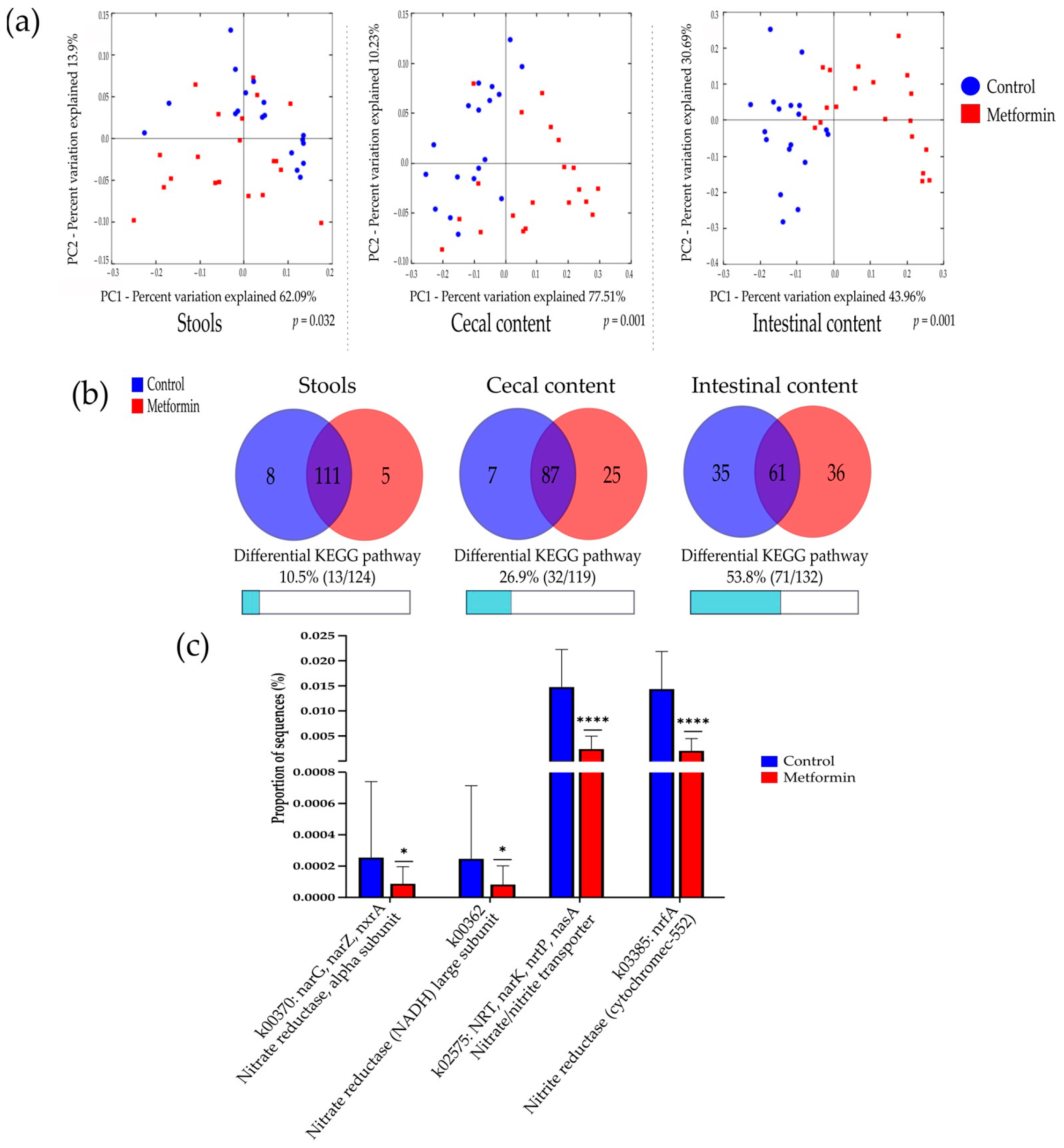
2.2. Beta Diversity Analysis Shows Changes in the Gut Microbiota of Mice Infected with H. pylori and Treated with Metformin
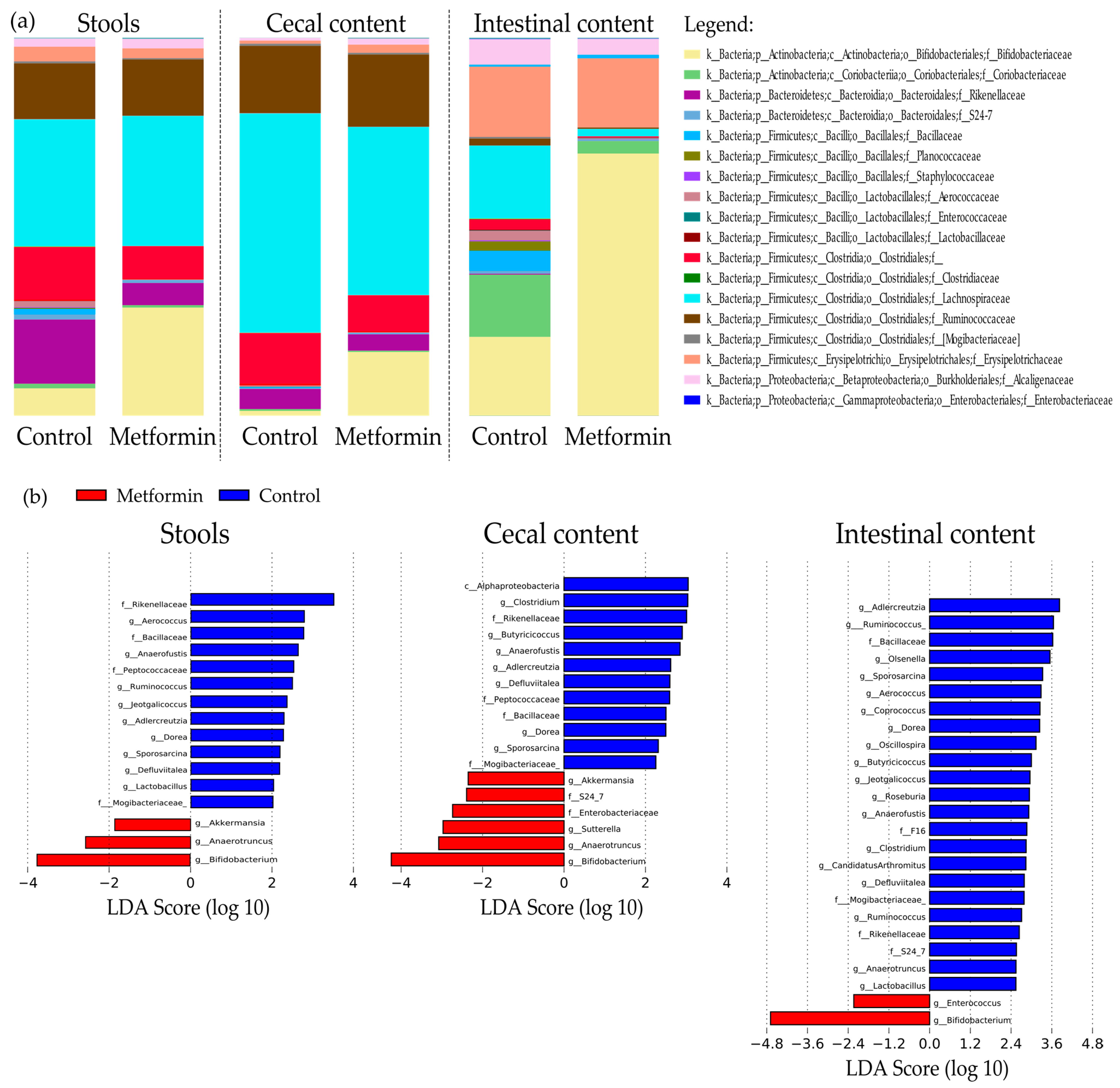
2.3. Metformin Treatment Changes Taxonomic Repartition in the Gut Microbiota of Mice Infected with H. pylori
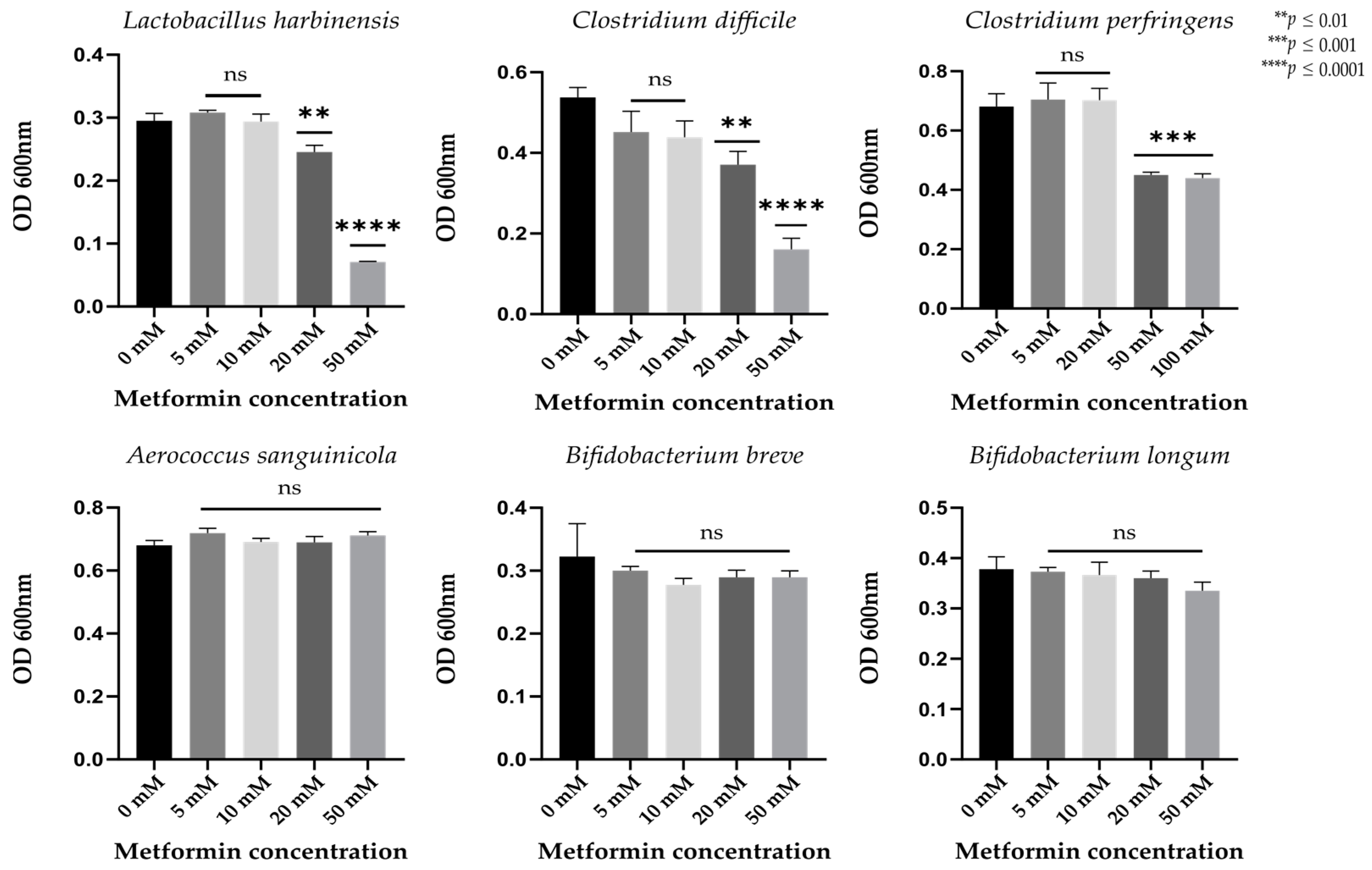
2.4. Metformin Directly Inhibits the Lactobacillus and Clostridium Gut Bacterial Strains In Vitro
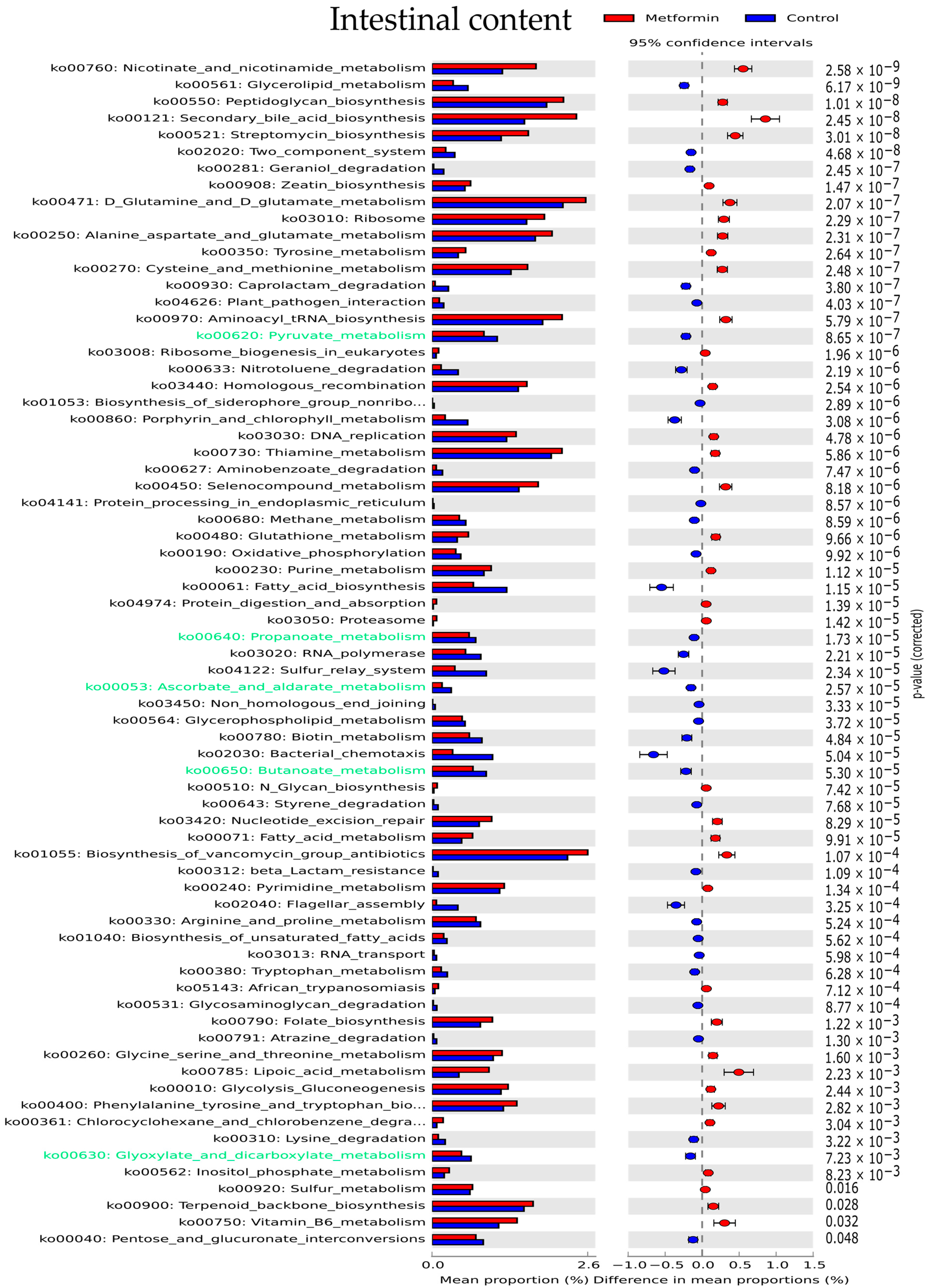
2.5. Potential Functional Pathways of Gut Microbiota Are Modified by Metformin Treatment in Mice Infected with H. pylori
3. Discussion
4. Materials and Methods
4.1. Animal Protocol and Sample Collection
4.2. DNA Extraction and rRNA Gene Sequencing
4.3. Functional Metagenome Predictions
4.4. Bioinformatics Analysis
4.5. In Vitro Bacterial Growth Experiments
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nathan, D.M.; Buse, J.B.; Davidson, M.B.; Ferrannini, E.; Holman, R.R.; Sherwin, R.; Zinman, B. Medical management of hyperglycemia in type diabetes: A consensus algorithm for the initiation and adjustment of therapy: A consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009, 32, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J.; Mynett, K.J.; Page, T. Importance of the intestine as a site of metformin-stimulated glucose utilization. Br. J. Pharm. 1994, 112, 671–675. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, C.; Bailey, C.J. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica 1994, 24, 49–57. [Google Scholar] [CrossRef]
- Morales, D.R.; Morris, A.D. Metformin in cancer treatment and prevention. Annu. Rev. Med. 2015, 66, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. Br. Med. Assoc. 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.-L.; Xue, W.-H.; Ding, X.-F.; Li, L.-F.; Dou, M.-M.; Zhang, W.-J.; Lv, Z.; Fan, Z.-R.; Zhao, J.; Wang, L.-X. Association between metformin and the risk of gastric cancer in patients with type 2 diabetes mellitus: A meta-analysis of cohort studies. Oncotarget 2017, 8, 55622–55631. [Google Scholar] [CrossRef] [Green Version]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process—First American cancer society award lecture on cancer epidemiology and prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar]
- González, C.A.; Megraud, F.; Buissonniere, A.; Barroso, L.L.; Agudo, A.; Duell, E.J.; Boutron-Ruault, M.C.; Clavel-Chapelon, F.; Palli, D.; Krogh, V.; et al. Helicobacter pylori infection assessed by ELISA and by immunoblot and noncardia gastric cancer risk in a prospective study: The Eurgast-EPIC project. Ann. Oncol. 2012, 23, 1320–1324. [Google Scholar] [CrossRef]
- Brown, L.M. Helicobacter pylori: Epidemiology and routes of transmission. Epidemiol. Rev. 2000, 22, 283–297. [Google Scholar] [CrossRef]
- Mégraud, F.; Bessède, E.; Varon, C. Helicobacter pylori infection and gastric carcinoma. Clin. Microbiol. Infect. 2015, 21, 984–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lofgren, J.L.; Whary, M.T.; Ge, Z.; Muthupalani, S.; Taylor, N.S.; Mobley, M.; Potter, A.; Varro, A.; Eibach, D.; Suerbaum, S.; et al. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology 2011, 140, 210–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Reddy, D.N. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef] [PubMed]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Cremonesi, E.; Governa, V.; Garzon, J.F.G.; Mele, V.; Amicarella, F.; Muraro, M.G.; Trella, E.; Galati-Fournier, V.; Oertli, D.; Däster, S.R.; et al. Gut microbiota modulate T cell trafficking into human colorectal cancer. Gut 2018, 67, 1984–1994. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Contreras, A.; Goldfarb, K.C.; Godoy-Vitorino, F.; Karaoz, U.; Contreras, M.; Blaser, M.J.; Brodie, E.L.; Dominguez-Bello, M.G. Structure of the human gastric bacterial community in relation to Helicobacter pylori status. ISME J. 2011, 5, 574–579. [Google Scholar] [CrossRef]
- Coker, O.O.; Dai, Z.; Nie, Y.; Zhao, G.; Cao, L.; Nakatsu, G.; Wu, W.K.; Wong, S.H.; Chen, Z.; Sung, J.J.Y.; et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut 2018, 67, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Courtois, S.; Bénéjat, L.; Izotte, J.; Mégraud, F.; Varon, C.; Lehours, P.; Bessède, E. Metformin can inhibit Helicobacter pylori growth. Future Microbiol. 2018, 13, 1575–1583. [Google Scholar] [CrossRef]
- Derrien, M.; Belzer, C.; De Vos, W.M. Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog. 2017, 106, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Mannerås-Holm, L.; Ståhlman, M.; Olsson, L.M.; Serino, M.; Planas-Fèlix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, Y.; Kim, J.; An, J.; Lee, S.; Kong, H.; Song, Y.; Lee, C.-K.; Kim, K. Modulation of the gut microbiota by metformin improves metabolic profiles in aged obese mice. Gut Microbes 2018, 9, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Chen, J.; Meng, Y.; Yang, J.; Cui, Q.; Zhou, Y. Metformin alters gut microbiota of healthy mice: Implication for Its potential role in gut microbiota homeostasis. Front. Microbiol. 2018, 9, 1336. [Google Scholar] [CrossRef] [Green Version]
- Forslund, K.; Hildebrand, F.; Nielsen, T.R.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Sun, J.; Lim, G.E.; Fantus, I.G.; Brubaker, P.L.; Jin, T. Cross talk between the insulin and Wnt signaling pathways: Evidence from intestinal endocrine L cells. Endocrinology 2008, 149, 2341–2351. [Google Scholar] [CrossRef] [Green Version]
- McCreight, L.J.; Bailey, C.J.; Pearson, E.R. Metformin and the gastrointestinal tract. Diabetologia 2016, 59, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Morita, Y.; Nogami, M.; Sakaguchi, K.; Okada, Y.; Hirota, Y.; Sugawara, K.; Tamori, Y.; Zeng, F.; Murakami, T.; Ogawa, W. enhanced release of glucose into the intraluminal space of the intestine associated with metformin treatment as revealed by [18F]fluorodeoxyglucose PET-MRI. Diabetes Care 2020, 43, 1796–1802. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-H.; Guo, X.-L. Combinational strategies of metformin and chemotherapy in cancers. Cancer Chemother. Pharm. 2016, 78, 13–26. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a tool to target aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [Green Version]
- Rajagopala, S.V.; Vashee, S.; Oldfield, L.M.; Suzuki, Y.; Venter, J.C.; Telenti, A.; Nelson, K.E. The human microbiome and cancer. Cancer Prev. Res. 2017, 10, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahmani, S.; Azarpira, N.; Moazamian, E. Anti-colon cancer activity of Bifidobacterium metabolites on colon cancer cell line SW742. Turk. J. Gastroenterol. 2019, 30, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Eslami, M.; Yousefi, B.; Kokhaei, P.; Hemati, M.; Nejad, Z.R.; Arabkari, V.; Namdar, A. Importance of probiotics in the prevention and treatment of colorectal cancer. J. Cell Physiol. 2019, 234, 17127–17143. [Google Scholar] [CrossRef] [PubMed]
- Al Kattar, S.; Jurjus, R.; Pinon, A.; Leger, D.Y.; Jurjus, A.; Boukarim, C.; Diab-Assaf, M.; Liagre, B. Metformin and probiotics in the crosstalk between colitis-associated colorectal cancer and diabetes in mice. Cancers 2020, 12, 1857. [Google Scholar] [CrossRef]
- Gao, J.-J.; Zhang, Y.; Gerhard, M.; Mejias-Luque, R.; Zhang, L.; Vieth, M.; Ma, J.-L.; Bajbouj, M.; Suchanek, S.; Liu, W.-D.; et al. Association between gut microbiota and Helicobacter pylori—Related gastric lesions in a high-risk population of gastric cancer. Front. Cell Infect. Microbiol. 2018, 8, 202. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; A Reyes, J.; Clemente, J.C.; E Burkepile, D.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Leach, S.A.; Thompson, M.; Hill, M. Bacterially catalysed N-nitrosation reactions and their relative importance in the human stomach. Carcinogenesis 1987, 8, 1907–1912. [Google Scholar] [CrossRef]
- Ambs, S.; Ogunfusika, M.O.; Merriam, W.G.; Bennett, W.P.; Billiar, T.R.; Harris, C.C. Up-regulation of inducible nitric oxide synthase expression in cancer-prone p53 knockout mice. Proc. Natl. Acad. Sci. USA 1998, 95, 8823–8828. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.W.; Wang, L.D.; Jiao, L.H.; Liu, B.; Zheng, S.; Xie, X.J. Expression of p53, inducible nitric oxide synthase and vascular endothelial growth factor in gastric precancerous and cancerous lesions: Correlation with clinical features. BMC Cancer 2002, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, R.M.; Pereira-Marques, J.; Pinto-Ribeiro, I.; Costa, J.L.; Carneiro, F.; Machado, J.C.; Figueiredo, C. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut 2018, 67, 226–236. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.M.W.; De Souza, R.; Kendall, C.W.C.; Emam, A.; Jenkins, D.J.A. Colonic health: Fermentation and short chain fatty acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef]
- Kaakoush, N.O.; Morris, M.J. The oesophageal microbiome: An unexplored link in obesity-associated oesophageal adenocarcinoma. FEMS Microbiol. Ecol. 2016, 92. [Google Scholar] [CrossRef] [Green Version]
- Belcheva, A.; Irrazabal, T.; Robertson, S.J.; Streutker, C.; Maughan, H.; Rubino, S.; Moriyama, E.H.; Copeland, J.K.; Surendra, A.; Kumar, S.; et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell 2014, 158, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Salani, B.; Del Rio, A.; Marini, C.; Sambuceti, G.; Cordera, R.; Maggi, D. Metformin, cancer and glucose metabolism. Endocr. Relat. Cancer 2014, 21, R461–R471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.H.; Ko, B.M.; Kim, S.H.; Myung, Y.S.; Choi, J.H.; Han, J.P.; Hong, S.J.; Jeon, S.R.; Kim, H.G.; Kim, J.O.; et al. Does metformin affect the incidence of colonic polyps and adenomas in patients with type 2 diabetes mellitus? Intest. Res. 2014, 12, 139–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Org, E.; Mehrabian, M.; Parks, B.W.; Shipkova, P.; Liu, X.; Drake, T.A.; Lusis, A.J. Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes 2016, 7, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Santos-Marcos, J.A.; Rangel-Zuñiga, O.A.; Jimenez-Lucena, R.; Quintana-Navarro, G.M.; Garcia-Carpintero, S.; Malagon, M.M.; Landa, B.B.; Tena-Sempere, M.; Perez-Martinez, P.; Lopez-Miranda, J.; et al. Influence of gender and menopausal status on gut microbiota. Maturitas 2018, 116, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; O’Rourke, J.; De Ungria, M.C.; Robertson, B.; Daskalopoulos, G.; Dixon, M.F. A standardized mouse model of Helicobacter pylori infection: Introducing the Sydney strain. Gastroenterology 1997, 112, 1386–1397. [Google Scholar] [CrossRef]
- Lehours, P.; Ménard, A.; Dupouy, S.; Bergey, B.; Richy, F.; Zerbib, F.; Ruskoné-Fourmestraux, A.; Delchier, J.C.; Mégraud, F. Evaluation of the association of nine Helicobacter pylori virulence factors with strains involved in low-grade gastric mucosa-associated lymphoid tissue lymphoma. Infect. Immun. 2004, 72, 880–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Beiko, R.G. Identifying biologically relevant differences between metagenomic communities. Bioinformatics 2010, 26, 715–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stools | Caecal Content | Intestinal Content | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Control (%) | Metformin (%) | p | Control (%) | Metformin (%) | p | Control (%) | Metformin (%) | p | ||||
| Phylum | Firmicutes | 71.21 | 62.02 | NS | Firmicutes | 91.78 | 74.91 | 0.000004 | Firmicutes | 54.14 | 22.63 | 0.000042 |
| Bacteroidetes | 17.94 | 6.504 | 0.000068 | Bacteroidetes | 5.637 | 4.666 | NS | Actinobacteria | 37.08 | 70.97 | 0.000015 | |
| Actinobacteria | 8.519 | 28.76 | 0.000564 | Actinobacteria | 1.844 | 18.53 | 0.000003 | Proteobacteria | 7.367 | 5.667 | NS | |
| Proteobacteria | 2.157 | 2.455 | NS | Proteobacteria | 0.69 | 1.682 | 0.000717 | Bacteroidetes | 1.046 | 0.4659 | 0.0296 | |
| Verrucomicrobia | 0.1462 | 0.248 | NS | Verrucomicrobia | 0.04292 | 0.2031 | 0.000002 | Verrucomicrobia | 0.2619 | 0.1808 | NS | |
| Class | Clostridia | 64.08 | 59.39 | NS | Clostridia | 90.33 | 72.38 | <0.000001 | Clostridia | 25.2 | 2.885 | 0.000009 |
| Bacteroidia | 17.94 | 6.504 | 0.000068 | Bacteroidia | 5.637 | 4.666 | NS | Actinobacteria | 20.79 | 67.44 | <0.000001 | |
| Actinobacteria | 7.277 | 28.14 | 0.000283 | Actinobacteria | 1.393 | 18.15 | 0.000002 | Erysipelotrichi | 18.29 | 19.24 | NS | |
| Erysipelotrichi | 3.741 | 2.467 | NS | Erysipelotrichi | 0.8205 | 2.44 | NS | Coriobacteriia | 16.29 | 3.529 | 0.000095 | |
| Bacilli | 3.386 | 0.1616 | 0.00416 | Betaproteobacteria | 0.6795 | 1.667 | 0.000663 | Bacilli | 10.65 | 0.5026 | 0.018 | |
| Betaproteobacteria | 2.142 | 2.427 | NS | Bacilli | 0.6257 | 0.08574 | 0.0167 | Betaproteobacteria | 6.716 | 4.445 | NS | |
| Coriobacteriia | 1.241 | 0.6206 | 0.0301 | Coriobacteriia | 0.451 | 0.3785 | NS | Bacteroidia | 1.044 | 0.4628 | 0.0294 | |
| Verrucomicrobiae | 0.1462 | 0.248 | NS | Verrucomicrobiae | 0.04292 | 0.2031 | 0.000002 | Alphaproteobacteria | 0.5376 | 1.076 | NS | |
| Verrucomicrobiae | 0.2619 | 0.1808 | NS | |||||||||
| Order | Clostridiales | 64.08 | 59.39 | NS | Clostridiales | 90.33 | 72.38 | <0.000001 | Clostridiales | 25.2 | 2.885 | 0.000009 |
| Bacteroidales | 17.94 | 6.504 | 0.000068 | Bacteroidales | 5.637 | 4.666 | NS | Bifidobacteriales | 20.79 | 67.44 | <0.000001 | |
| Bifidobacteriales | 7.277 | 28.14 | 0.000283 | Bifidobacteriales | 1.392 | 18.15 | 0.000002 | Erysipelotrichales | 18.29 | 19.24 | NS | |
| Erysipelotrichales | 3.741 | 2.467 | NS | Erysipelotrichales | 0.8205 | 2.44 | NS | Coriobacteriales | 16.29 | 3.529 | 0.000095 | |
| Burkholderiales | 2.142 | 2.427 | NS | Burkholderiales | 0.6795 | 1.667 | 0.000663 | Bacillales | 7.818 | 0.05888 | 0.0213 | |
| Bacillales | 1.779 | 0.02667 | 0.0147 | Coriobacteriales | 0.451 | 0.3785 | NS | Burkholderiales | 6.716 | 4.445 | NS | |
| Lactobacillales | 1.606 | 0.135 | 0.00112 | Bacillales | 0.3566 | 0.01136 | 0.0275 | Lactobacillales | 2.833 | 0.4437 | 0.0178 | |
| Coriobacteriales | 1.241 | 0.6206 | 0.0301 | Lactobacillales | 0.2691 | 0.07438 | 0.014 | Bacteroidales | 1.044 | 0.4628 | 0.0294 | |
| Verrucomicrobiales | 0.1462 | 0.248 | NS | Verrucomicrobiales | 0.04292 | 0.2031 | 0.000002 | Rickettsiales | 0.5069 | 1.056 | NS | |
| Verrucomicrobiales | 0.2619 | 0.1808 | NS | |||||||||
| Family | Lachnospiraceae | 34.13 | 34.93 | NS | Lachnospiraceae | 57.86 | 43.3 | 0.00035 | Bifidobacteriaceae | 20.79 | 67.44 | <0.000001 |
| Rikenellaceae | 16.58 | 5.729 | 0.000128 | Ruminococcaceae | 18.02 | 18.98 | NS | Lachnospiraceae | 19.8 | 2.11 | 0.000009 | |
| Ruminococcaceae | 14.88 | 15.05 | NS | Rikenellaceae | 5.371 | 4.218 | NS | Erysipelotrichaceae | 18.29 | 19.24 | NS | |
| Bifidobacteriaceae | 7.277 | 28.14 | 0.000283 | Bifidobacteriaceae | 1.392 | 18.15 | 0.000002 | Coriobacteriaceae | 16.29 | 3.529 | 0.000095 | |
| Erysipelotrichaceae | 3.741 | 2.467 | NS | Erysipelotrichaceae | 0.8205 | 2.44 | NS | Alcaligenaceae | 6.716 | 4.445 | NS | |
| Alcaligenaceae | 2.138 | 2.427 | NS | Alcaligenaceae | 0.6795 | 1.667 | 0.000663 | Bacillaceae | 5.125 | 0.002976 | 0.0411 | |
| Aerococcaceae | 1.521 | 0.1177 | 0.00143 | [Mogibacteriaceae] | 0.579 | 0.473 | 0.0326 | Aerococcaceae | 2.499 | 0.2731 | 0.0228 | |
| Bacillaceae | 1.361 | 0 | 0.019 | Coriobacteriaceae | 0.451 | 0.3785 | NS | Planococcaceae | 2.439 | 0.001683 | 0.00703 | |
| S24-7 | 1.358 | 0.7749 | 0.0403 | S24-7 | 0.266 | 0.4476 | 0.0237 | Ruminococcaceae | 1.95 | 0.3866 | 0.00399 | |
| Coriobacteriaceae | 1.241 | 0.6206 | 0.0301 | Bacillaceae | 0.2608 | 0 | 0.0373 | S24-7 | 0.7812 | 0.4473 | NS | |
| [Mogibacteriaceae] | 0.548 | 0.3557 | 0.00734 | Aerococcaceae | 0.2524 | 0.0623 | 0.013 | [Mogibacteriaceae] | 0.4774 | 0.03272 | 0.0223 | |
| Planococcaceae | 0.2407 | 0 | 0.0147 | Verrucomicrobiaceae | 0.04292 | 0.2031 | 0.000002 | Rikenellaceae | 0.2633 | 0.01555 | 0.0232 | |
| Verrucomicrobiaceae | 0.1462 | 0.248 | NS | Verrucomicrobiaceae | 0.2619 | 0.1808 | NS | |||||
| Staphylococcaceae | 0.2544 | 0.05422 | NS | |||||||||
| Lactobacillaceae | 0.2505 | 0.0467 | 0.00019 | |||||||||
| Enterococcaceae | 0.08181 | 0.1239 | NS | |||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jauvain, M.; Courtois, S.; Lehours, P.; Bessède, E. Metformin Modifies the Gut Microbiota of Mice Infected with Helicobacter pylori. Pharmaceuticals 2021, 14, 329. https://doi.org/10.3390/ph14040329
Jauvain M, Courtois S, Lehours P, Bessède E. Metformin Modifies the Gut Microbiota of Mice Infected with Helicobacter pylori. Pharmaceuticals. 2021; 14(4):329. https://doi.org/10.3390/ph14040329
Chicago/Turabian StyleJauvain, Marine, Sarah Courtois, Philippe Lehours, and Emilie Bessède. 2021. "Metformin Modifies the Gut Microbiota of Mice Infected with Helicobacter pylori" Pharmaceuticals 14, no. 4: 329. https://doi.org/10.3390/ph14040329
APA StyleJauvain, M., Courtois, S., Lehours, P., & Bessède, E. (2021). Metformin Modifies the Gut Microbiota of Mice Infected with Helicobacter pylori. Pharmaceuticals, 14(4), 329. https://doi.org/10.3390/ph14040329