
Fully Automated GMP-Compliant Synthesis of [18F]FE-PE2I

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. General Considerations for the Production of [18F]FE-PE2I
2.2. Production of [18F]FE-PE2I Using a Scansys Module
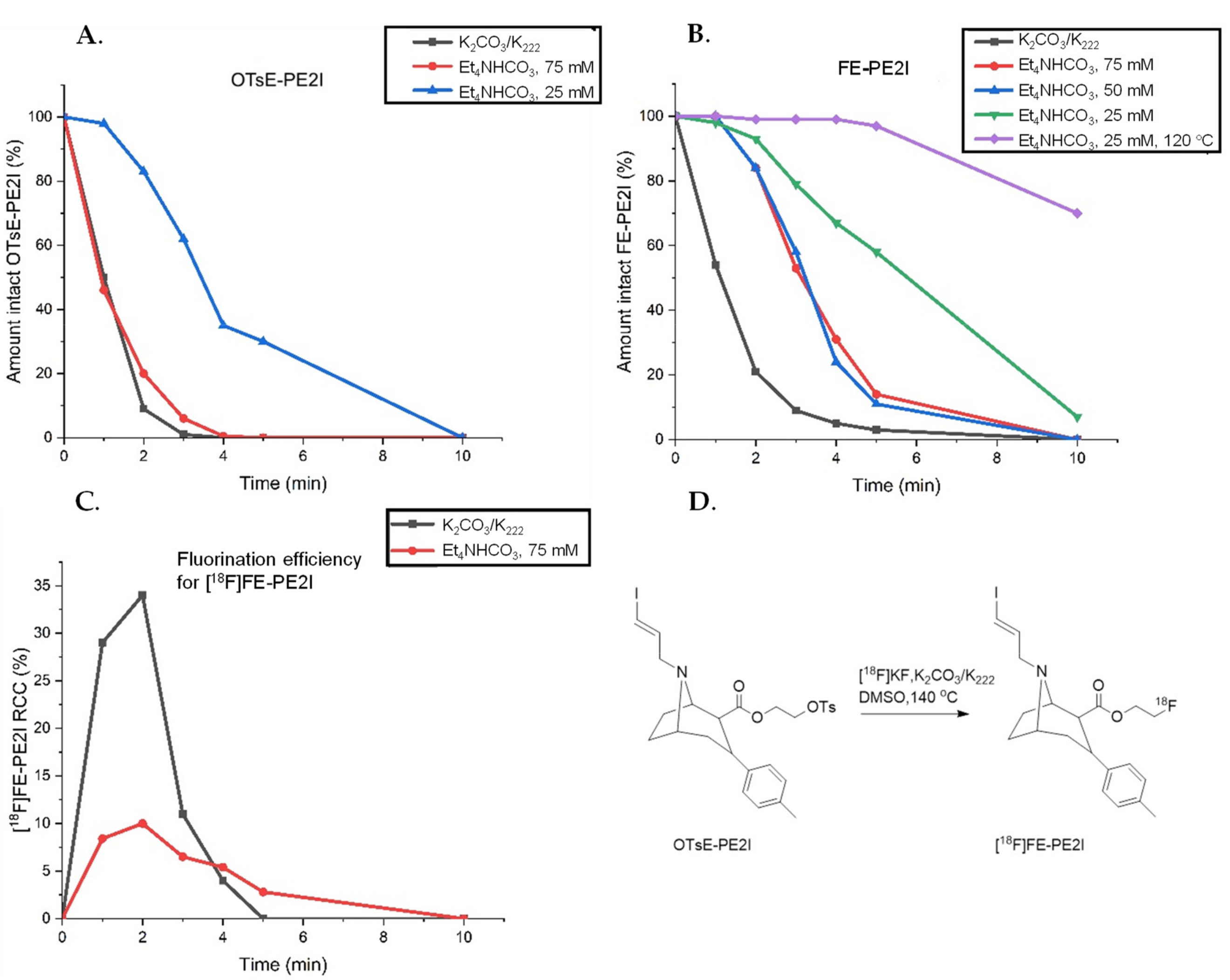
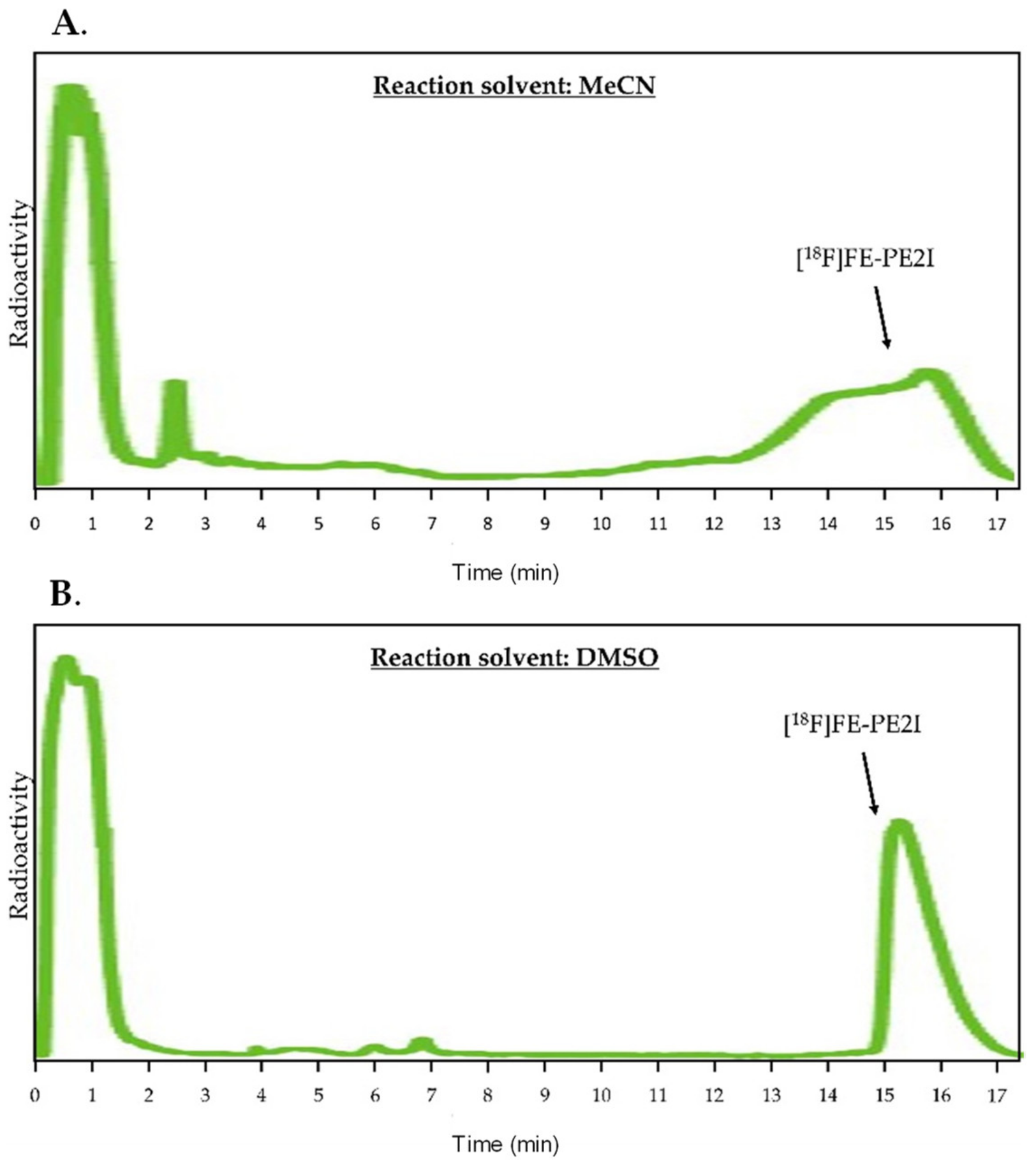
2.2.1. Manual Optimization of Reaction Parameters
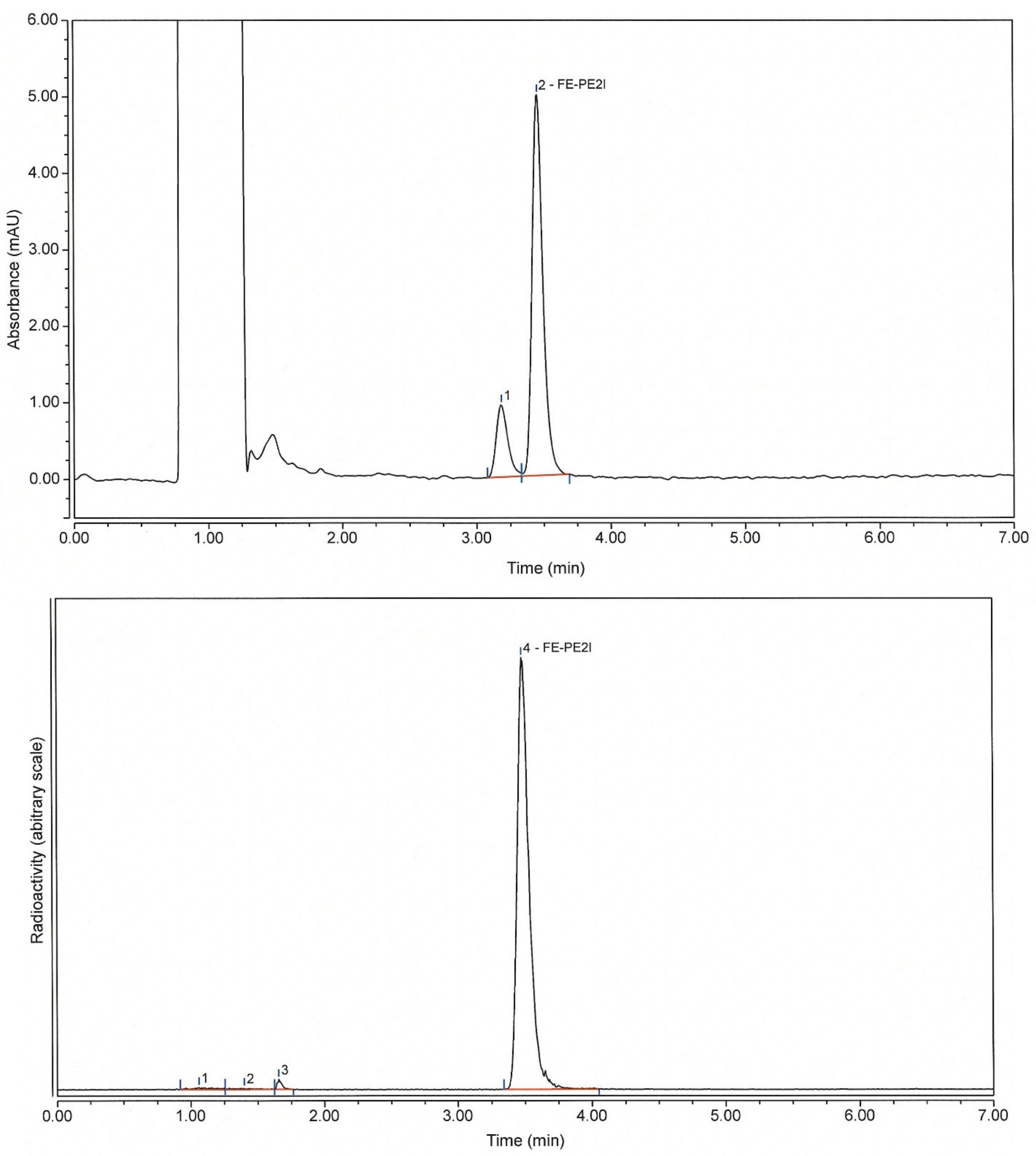
2.2.2. Preparative HPLC Method
2.2.3. Automated Production of [18F]FE-PE2I Using a Scansys Module
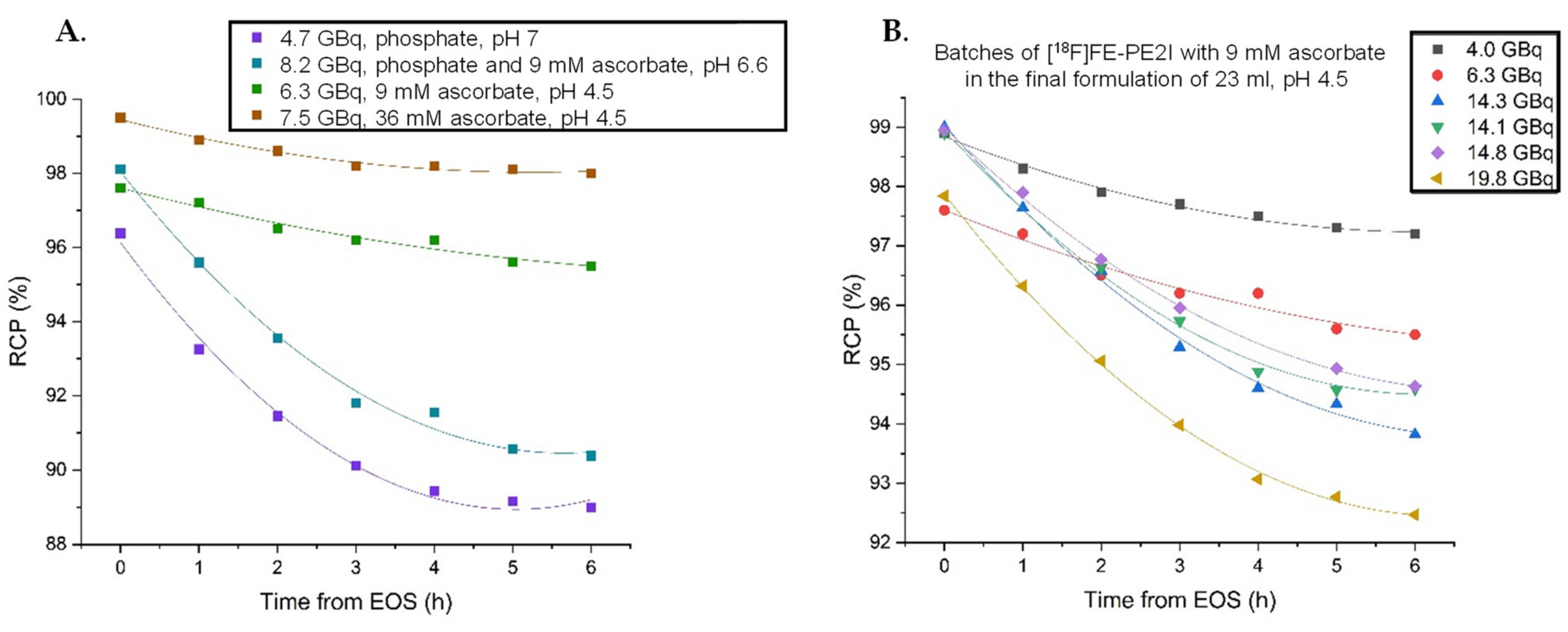
2.2.4. Stability Studies of the Formulated [18F]FE-PE2I
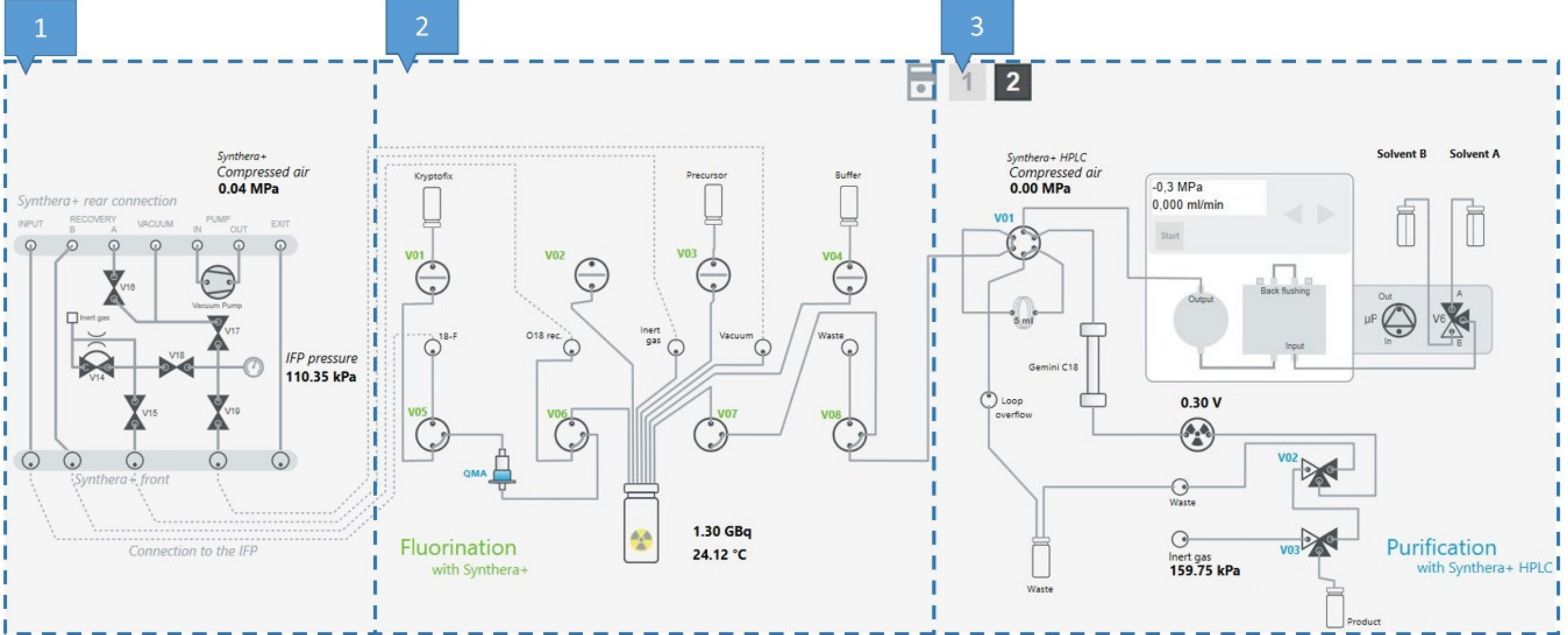
2.3. Production of [18F]FE-PE2I on Synthera®+
2.4. Implementing New Reaction Conditions for [18F]FE-PE2I
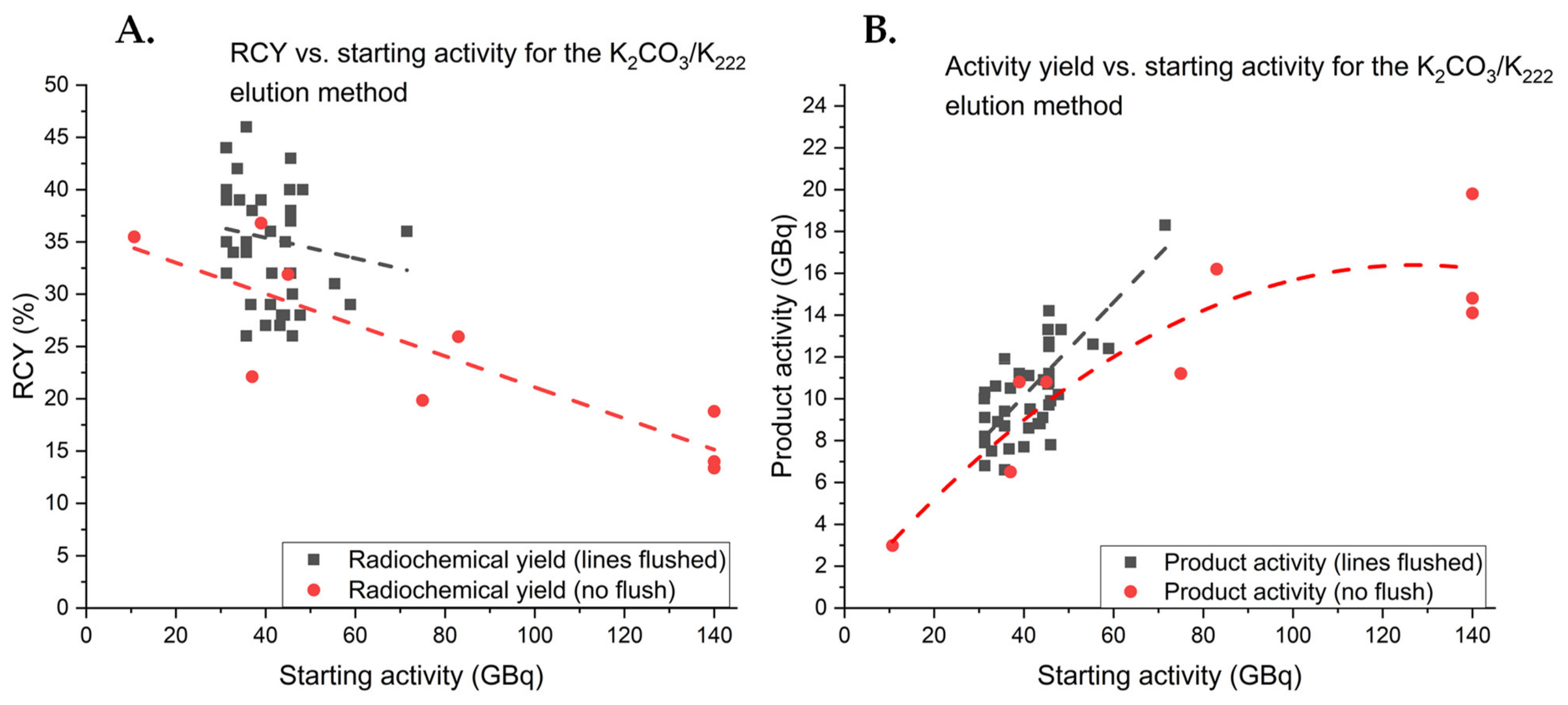
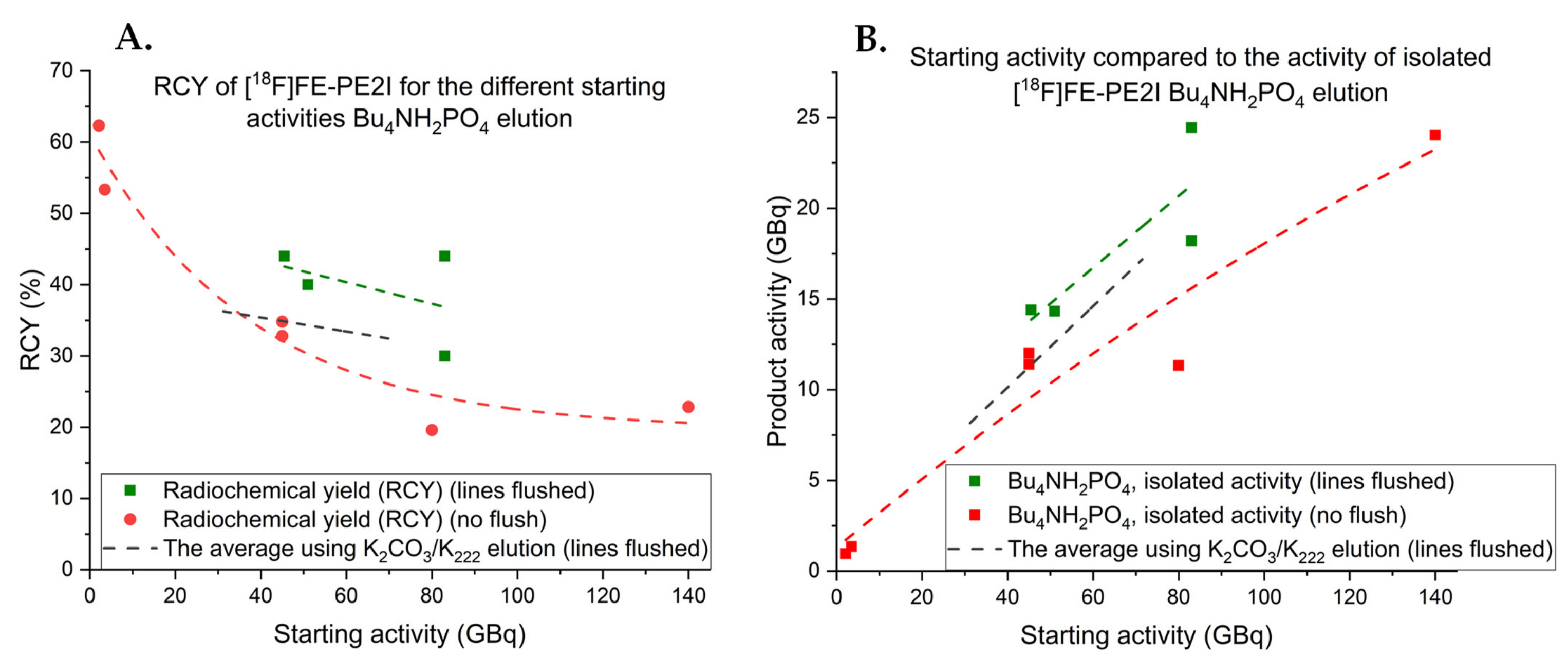
2.4.1. Elution Conditions for Improved Synthesis of [18F]FE-PE2I
2.4.2. Production of [18F]FE-PE2I with Improved Elution Conditions on Synthera®+
2.4.3. Limitations of the New Improved Synthesis of [18F]FE-PE2I on Synthera®+
3. Materials and Methods
3.1. General Considerations
3.1.1. Reagents and Consumables
3.1.2. [18F]Fluoride
3.2. General Description of the Manual Optimization of Reaction Conditions
3.2.1. General Method for Stability Studies of FE-PE2I and OTsE-PE2I
3.2.2. General Method for Manual Radiolabeling Reactions of [18F]FE-PE2I
3.3. Detailed Description of the Manual Optimization of Reaction Conditions; Details for Stability Studies of FE-PE2I and OTsE-PE2I and the Manual Fluorination Reaction of [18F]FE-PE2I
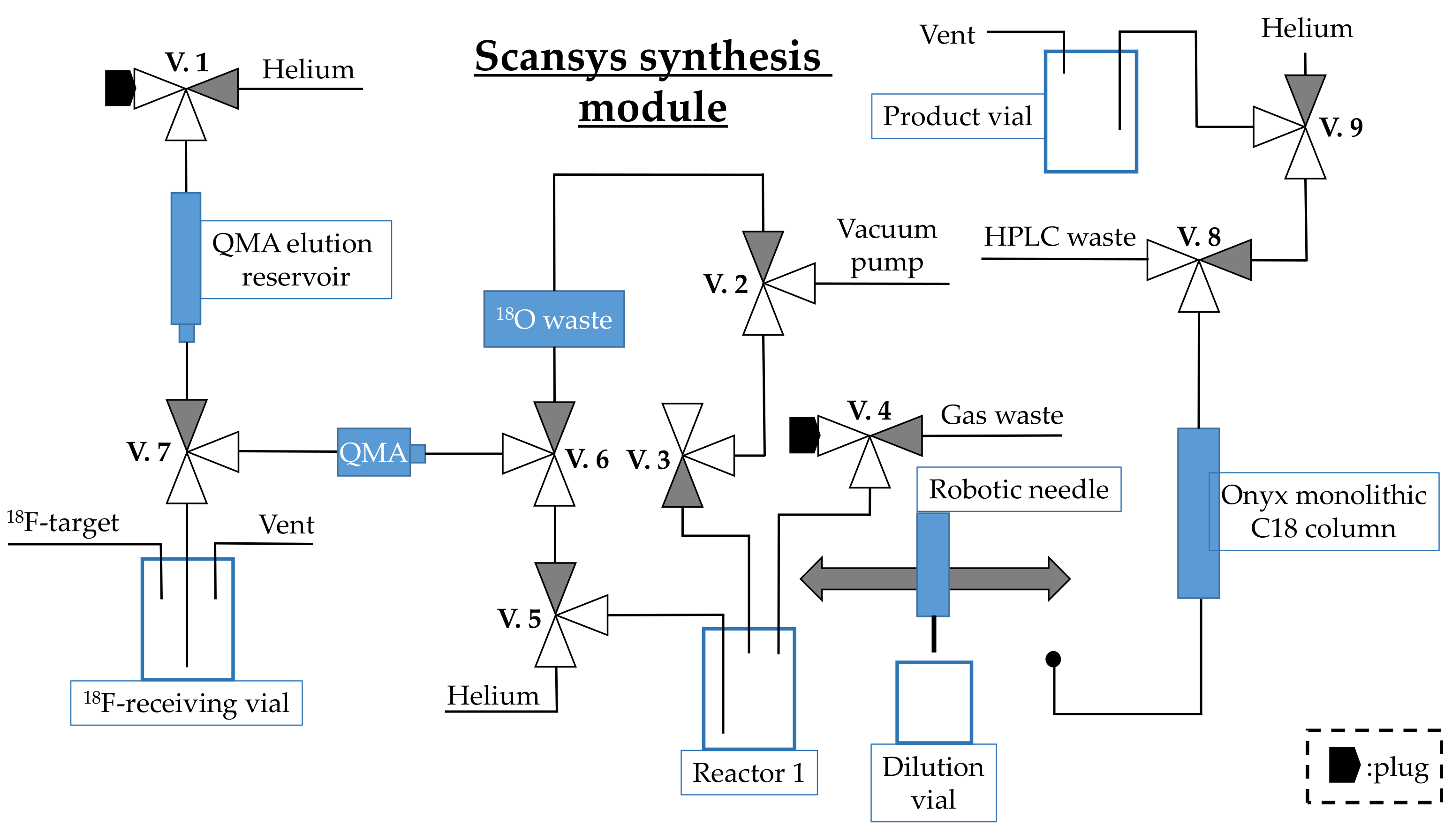
3.4. Process Description of the Production of [18F]FE-PE2I on a Scansys Synthesis Module
3.5. Process Description of the Production of [18F]FE-PE2I on a Synthera®+ Synthesis Module
3.6. Quality Control
3.6.1. HPLC
3.6.2. GC Analysis
3.6.3. TLC Analysis
3.6.4. Other Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kristensen, J.L.; Herth, M.M. In vivo Imaging in Drug Discovery. In Textbook of Drug Design and Discovery; Strømgaard, K., Krogsgaard-Larsen, P., Madsen, U., Eds.; CRC Press: Copenhagen, Denmark, 2017; ISBN 9781498702782. [Google Scholar]
- Edem, P.E.; Steen, E.J.L.; Kjær, A.; Herth, M.M. Chapter 2—Fluorine-18 Radiolabeling Strategies—Advantages and Disadvantages of Currently Applied Labeling Methods. In Late-Stage Fluorination of Bioactive Molecules and Biologically-Relevant Substrates; Postigo, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 29–103. ISBN 978-0-12-812958-6. [Google Scholar]
- Delva, A.; Van Weehaeghe, D.; Koole, M.; Van Laere, K.; Vandenberghe, W. Loss of Presynaptic Terminal Integrity in the Substantia Nigra in Early Parkinson’s Disease. Mov. Disord. 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Moriya, H.; Tiger, M.; Tateno, A.; Sakayori, T.; Masuoka, T.; Kim, W.C.; Arakawa, R.; Okubo, Y. Low dopamine transporter binding in the nucleus accumbens in geriatric patients with severe depression. Psychiatry Clin. Neurosci. 2020, 74, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Ikoma, Y.; Sasaki, T.; Kimura, Y.; Seki, C.; Okubo, Y.; Suhara, T.; Ito, H. Evaluation of semi-quantitative method for quantification of dopamine transporter in human PET study with [18F]FE-PE2I. Ann. Nucl. Med. 2015, 29, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Booij, J.; Tissingh, G.; Boer, G.J.; Speelman, J.D.; Stoof, J.C.; Janssen, A.G.M.; Wolters, E.C.; Van Royen, E.A. [123I]FP-CIT SPECT shows a pronounced decline of striatal dopamine transporter labelling in early and advanced Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1997, 62, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobson Mo, S.; Axelsson, J.; Jonasson, L.; Larsson, A.; Ögren, M.J.; Ögren, M.; Varrone, A.; Eriksson, L.; Bäckström, D.; af Bjerkén, S.; et al. Dopamine transporter imaging with [18F]FE-PE2I PET and [123I]FP-CIT SPECT—A clinical comparison. EJNMMI Res. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delva, A.; Van Weehaeghe, D.; van Aalst, J.; Ceccarini, J.; Koole, M.; Baete, K.; Nuyts, J.; Vandenberghe, W.; Van Laere, K. Quantification and discriminative power of [18F]FE-PE2I PET in patients with Parkinson’s disease. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1913–1926. [Google Scholar] [CrossRef] [PubMed]
- Schou, M.; Steiger, C.; Varrone, A.; Guilloteau, D.; Halldin, C. Synthesis, radiolabeling and preliminary in vivo evaluation of [18F]FE-PE2I, a new probe for the dopamine transporter. Bioorganic Med. Chem. Lett. 2009, 19, 4843–4845. [Google Scholar] [CrossRef] [PubMed]
- Stepanov, V.; Krasikova, R.; Raus, L.; Loog, O.; Hiltunen, J.; Halldin, C. An efficient one-step radiosynthesis of [ 18F]FE-PE2I, a PET radioligand for imaging of dopamine transporters. J. Label. Compd. Radiopharm. 2012, 55, 206–210. [Google Scholar] [CrossRef]
- Bratteby, K.; Shalgunov, V.; Battisti, U.; Petersen, I.; Broek, S.; Ohlsson, T.; Gillings, N.; Erlandsson, M.; Herth, M. Insights into Elution of Anion Exchange Cartridges: Opening the Path towards Aliphatic 18F-Radiolabeling of Base-Sensitive Tracers. ChemRxiv. Prepr. 2021. [Google Scholar] [CrossRef]
- European Medicines Agency. ICH Topic Q 2 (R1) Validation of Analytical Procedures: Text and Methodology; European Medicines Agency: Amsterdam, The Netherlands, 1995. [Google Scholar]
- Herth, M.M.; Ametamey, S.; Antuganov, D.; Bauman, A.; Berndt, M.; Brooks, A.F.; Bormans, G.; Seong, Y.; Gillings, N.; Häfeli, U.O.; et al. On the consensus nomenclature rules for radiopharmaceutical chemistry—Reconsideration of radiochemical conversion. Nucl. Med. Biol. 2021, 93, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Murali, D.; Zammit, M.; DiFilippo, A.; Tullis, T.; Higgins, A.; Barnhart, T.; Engle, J.; Christian, B. Improving radiochemical purity of [11C]PIB. J. Nucl. Med. 2019, 60, 197. [Google Scholar]
- Berridge, M.S.; Apana, S.M.; Hersha, J.M. Teflon radiolysis as the major source of carrier in fluorine-18. J. Label. Compd. Radiopharm. 2009, 52, 543–548. [Google Scholar] [CrossRef]
- Castner, J.F.; Zdankiewicz, D.D.; Anderson, J.E. Stabilization of Radiopharmaceutical Compositions Using Ascorbic Acid. U.S. Patent. US20130101508A9, 25 April 2013. [Google Scholar]
- Par, I.N.; With, T. Successful Flash Chromatography A White Paper from Biotage Successful Flash Chromatography; Biotage: Uppsala, Sweden, 2018. [Google Scholar]
- Kaufmann, A.; Jegle, U.; Dreyer, A.; Majors, R.E. Using DMSO as an Injection Solvent to Increase Sample Load in Preparative LC Application, Agilent Technologies. 2005. Available online: https://www.agilent.com/cs/library/applications/5989-2485EN.pdf (accessed on 22 June 2021).
- Kim, D.W.; Jeong, H.J.; Lim, S.T.; Sohn, M.H. Recent trends in the nucleophilic [18F]-radiolabeling method with no-carrier-added [18F]fluoride. Nucl. Med. Mol. Imaging 2010, 44, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozmér, Z.; Takács, E.; Wojnárovits, L.; Alapi, T.; Hernádi, K.; Dombi, A. The influence of radical transfer and scavenger materials in various concentrations on the gamma radiolysis of phenol. Radiat. Phys. Chem. 2016, 124, 52–57. [Google Scholar] [CrossRef] [Green Version]
- Wojnarovits, L.; Laverne, J.A. Iodine as a radical scavenger in the radiolysis of cyclopentane. Radiat. Phys. Chem. 1996, 47, 361–363. [Google Scholar] [CrossRef]
- Mock, B.H.; Winkle, W.; Vavrek, M.T. A color spot test for the detection of Kryptofix 2.2.2 in [18F]FDG preparations. Nucl. Med. Biol. 1997, 24, 193–195. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Parameter | Acceptance Criteria | Result | ||
|---|---|---|---|---|
| Specificity | Resolution of >2 between the peaks | Resolution (impurity at the RT of 3.4 and FE-PE2I) = 2.6 | ||
| Linearity | 0.25–1.25 µg/mL (five concentrations in triplicate) | R2 > 0.995 | R2 > 0.998 | |
| Repeatability | 10 repetitions (1 µg/mL standard) | RSD ≤ 5% | RSD = 0.48% | |
| Accuracy | Recovery 90–110% (matrix spiked with FE-PE2I; five different concentrations) | Concentration | Recovery | |
| 0.1 µg/mL 0.2 µg/mL 0.3 µg/mL 0.4 µg/mL 0.5 µg/mL | 97% 96% 106% 103% 101% | |||
| Limit of detection | S/N * ratio ≥ 3 | 0.01 µg/mL | ||
| Limit of quantification | S/N ratio ≥ 10 | 0.05 µg/mL | ||
| Test | Specification | Batch Results | ||
|---|---|---|---|---|
| Batch No. | PE2I-200513-2 | PE2I-200514-2 | PE2I-200520-2 | |
| Radioactivity | 0.2–20 GBq at EOS | 14.1 GBq | 14.8 GBq | 19.8 GBq |
| Radiochemical yield (DC) | Reported value | 13% | 14% | 20% |
| [18F]Fluoride | ≤5% | 0.1% | 0.2% | 0.2% |
| Other 18F impurities | ≤5% | 1.0% | 0.9% | 2.0% |
| Radiochemical purity at EOS | ≥96% | 98.9% | 99.0% | 97.8% |
| Radiochemical purity after 6 h | ≥90% | 94.6% | 94.6% | 92.5% |
| FE-PE2I | ≤1.00 µg/mL | 0.45 µg/mL | 0.44 µg/mL | 0.28 µg/mL |
| Specified impurity (RT of 3.4 min) | ≤1.00 µg/mL | 0.15 µg/mL | 0.20 µg/mL | 0.13 µg/mL |
| Total unspecified impurities | 1.00 µg/mL | n.d. | n.d. | n.d. |
| Total FE-PE2I and impurities | ≤1.00 µg/mL | 0.60 µg/mL | 0.64 µg/mL | 0.41 µg/mL |
| Molar radioactivity | Reported value | 619 GBq/µmol | 664 GBq/µmol | 1393 GBq/µmol |
| Solvent | RCC, 2 min | RCC, 5 min | RCC, 10 min | RCC, 20 min |
|---|---|---|---|---|
| DMSO, 10% tBuOH | 8.5% | 26.8% | 30.4% | 30.8% |
| DMSO, 5% tBuOH | 13.6% | 36.5% | 42.1% | 46.8% |
| DMSO, 2% tBuOH | 37.9% | 75.6% | 77.4% | 83.8% |
| Results of full synthesis | DMSO, 2% tBuOH, 5 min reaction time | DMSO, 2 min reaction time | DMSO, 3.5 min reaction time | DMSO, 5 min reaction time |
| Isolated RCY (45-GBq starting activity) | 32.8% | 32.0% | - | 34.8% |
| Isolated RCY (140-GBq starting activity) | - | 23.0% | 20.8% | 22.9% |
| Sample | K2CO3/K222 | Et4NHCO3 75 mM | Et4NHCO3 50 mM | Et4NHCO3 25 mM | Et4NHCO3 25 mM |
|---|---|---|---|---|---|
| Reaction temperature | 140 °C | 140 °C | 140 °C | 140 °C | 120 °C |
| Reaction solvent | DMSO (0.6 mL) | DMSO (0.6 mL) | DMSO (0.6 mL) | DMSO (0.6 mL) | DMSO (0.6 mL) |
| Anion exchange cartridge | QMA | PS-30 | PS-30 | PS-30 | PS-30 |
| Preconditioning anion | CO32− | HCO3− | HCO3− | HCO3− | HCO3− |
| Eluting anion | K2CO3/K222 * | Et4NHCO3 | Et4NHCO3 | Et4NHCO3 | Et4NHCO3 |
| Eluting anion concentration | 1.1 mg (7.8 µmol) | 14.3 mg (75 µmol) | 9.6 mg (50 µmol) | 4.8 mg (25 µmol) | 4.8 mg (25 µmol) |
| Eluting anion solvent | H2O/MeOH (180 µL/820 µL) | H2O/MeOH (180 µL/820 µL) | H2O/MeOH (180 µL/820 µL) | H2O/MeOH (180 µL/820 µL) | H2O/MeOH (180 µL/820 µL) |
| Sample | Bu4NH2PO4 | Bu4NH2PO4 | Bu4NH2PO4 |
|---|---|---|---|
| Reaction temperature | 120 °C | 100 °C | 120 °C |
| Reaction solvent | DMSO (1.0 mL) | DMSO (1.0 mL) | MeCN (1.0 mL) |
| Anion exchange cartridge | QMA | QMA | QMA |
| Preconditioning anion | CO32− | CO32− | CO32− |
| Eluting anion | Bu4NH2PO4 | Bu4NH2PO4 | Bu4NH2PO4 |
| Eluting anion concentration | 6.8 mg (20.0 µmol) | 6.8 mg (20.0 µmol) | 6.8 mg (20.0 µmol) |
| Eluting anion solvent | H2O/MeCN (500 µL/500µL) | H2O/MeCN (500 µL/500 µL) | H2O/MeCN (500 µL/500 µL) |
| Test | Specifications | Analytical Methods |
|---|---|---|
| Identification of [18F]FE-PE2I | Radioactive half-life: 105–115 min Gamma spectrum shows only the 511 and 1022 keV peaks. The labeled product corresponds in RT to an authentic reference standard of FE-PE2I | T1/2 measurement using a dose calibrator Gamma spectrum using a NaI well counter Product ID using HPLC |
| Radioactivity | 0.2–20 GBq at EOS | Dose calibrator |
| Volume | 32 ± 1 mL | Visual check |
| Appearance | Clear and colorless solution free from visible particulates or cloudiness | Visual inspection |
| pH | 4.0–5.0 | pH-meter |
| Residual Kryptofix | < 0.075 mg/mL | Color spot test |
| Free [18F]fluoride | ≤ 5% | HPLC with a radiodetector |
| Other 18F-labeled impurities | ≤ 5% | HPLC with a radiodetector |
| Radiochemical purity (at EOS) | ≥ 96% | HPLC with a radiodetector |
| Radiochemical purity after 6 h (end of the shelf life) | ≥ 90% | HPLC with a radiodetector |
| FE-PE2I content | ≤ 1.00 µg/mL | HPLC (UV, 220 nm) |
| Specified impurity (HPLC, RT of 3.4 min) | ≤ 1.00 µg/mL | HPLC (UV, 220 nm) |
| Total unspecified organic impurities | ≤ 1.00 µg/mL | HPLC (UV, 220 nm) |
| Total FE-PE2I and organic impurities | ≤ 1.00 µg/mL(maximum injected dose of 10 µg) | HPLC (UV, 220 nm) |
| Radionuclidic purity | ≥ 99.9% (< 0.1% radionuclidic impurities) | Gamma spectrum using an HPGe detector after the complete decay of fluorine-18 (minimum 48 h after EOS) |
| Ethanol | 3–7% (w/v) | Gas chromatography |
| Acetonitrile | ≤ 273 ppm | Gas chromatography |
| Methanol | ≤ 2000 ppm | Gas chromatography |
| DMSO | ≤ 3333 ppm | Gas chromatography |
| Microbiology | Passes the test for sterility (Ph. Eur.) | Test for sterility (Ph. Eur.), filtration method |
| Bacterial endotoxins | ≤ 0.5 EU/mL | Quantitative LAL analysis (Ph. Eur.) |
| Microbiology | Bioburden: < 10 CFU/100 mL | Membrane filtration and media growth |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bratteby, K.; Denholt, C.L.; Lehel, S.; Petersen, I.N.; Madsen, J.; Erlandsson, M.; Ohlsson, T.; Herth, M.M.; Gillings, N. Fully Automated GMP-Compliant Synthesis of [18F]FE-PE2I. Pharmaceuticals 2021, 14, 601. https://doi.org/10.3390/ph14070601
Bratteby K, Denholt CL, Lehel S, Petersen IN, Madsen J, Erlandsson M, Ohlsson T, Herth MM, Gillings N. Fully Automated GMP-Compliant Synthesis of [18F]FE-PE2I. Pharmaceuticals. 2021; 14(7):601. https://doi.org/10.3390/ph14070601
Chicago/Turabian StyleBratteby, Klas, Charlotte Lund Denholt, Szabolcs Lehel, Ida Nymann Petersen, Jacob Madsen, Maria Erlandsson, Tomas Ohlsson, Matthias Manfred Herth, and Nic Gillings. 2021. "Fully Automated GMP-Compliant Synthesis of [18F]FE-PE2I" Pharmaceuticals 14, no. 7: 601. https://doi.org/10.3390/ph14070601
APA StyleBratteby, K., Denholt, C. L., Lehel, S., Petersen, I. N., Madsen, J., Erlandsson, M., Ohlsson, T., Herth, M. M., & Gillings, N. (2021). Fully Automated GMP-Compliant Synthesis of [18F]FE-PE2I. Pharmaceuticals, 14(7), 601. https://doi.org/10.3390/ph14070601