
CPPs to the Test: Effects on Binding, Uptake and Biodistribution of a Tumor Targeting Nanobody

 ,
,  , ,
, , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Production of Nanobody-CPP Conjugates
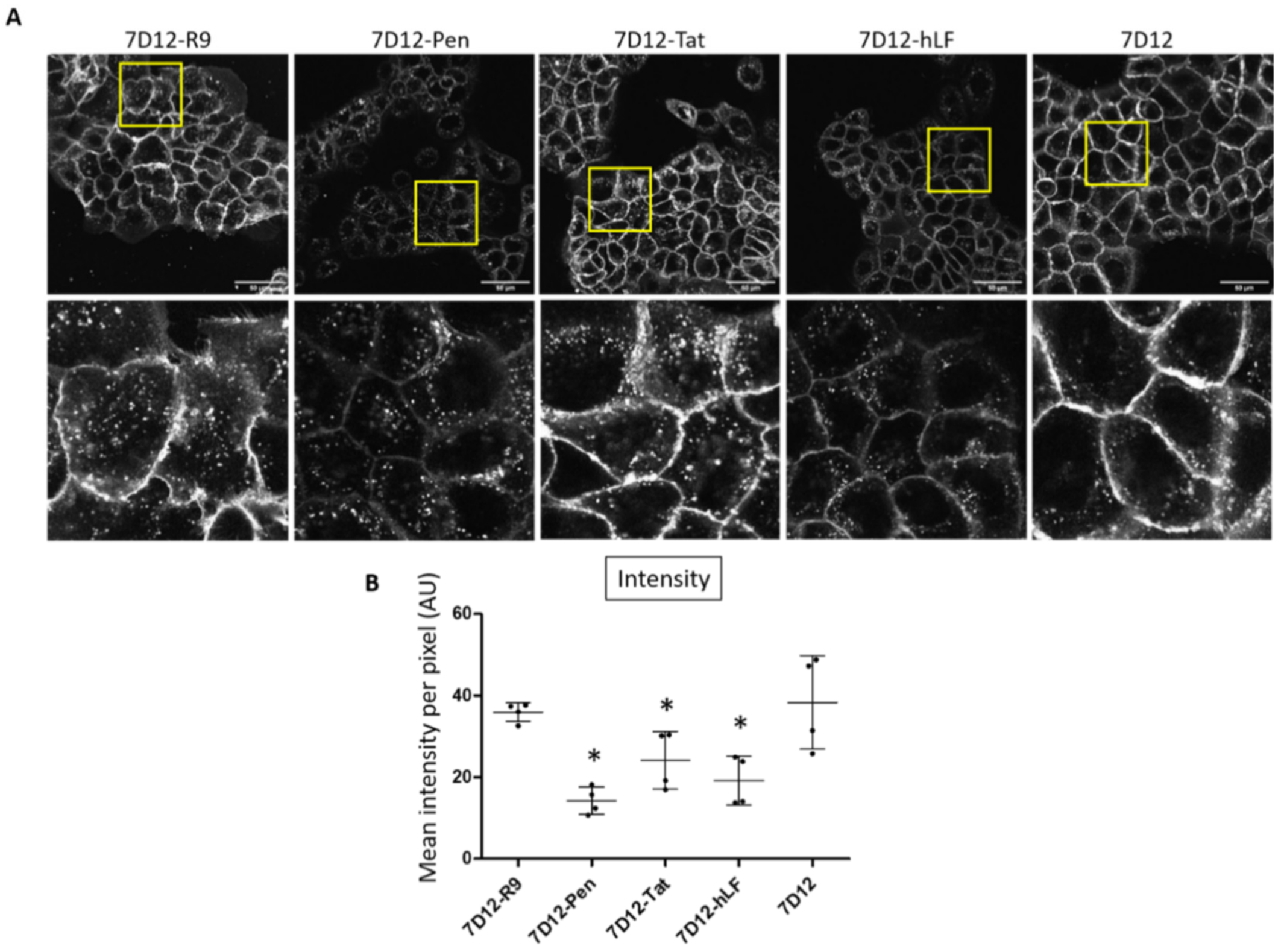
2.2. Differential Uptake of 7D12-CPP Conjugates in EGFR Expressing Cells
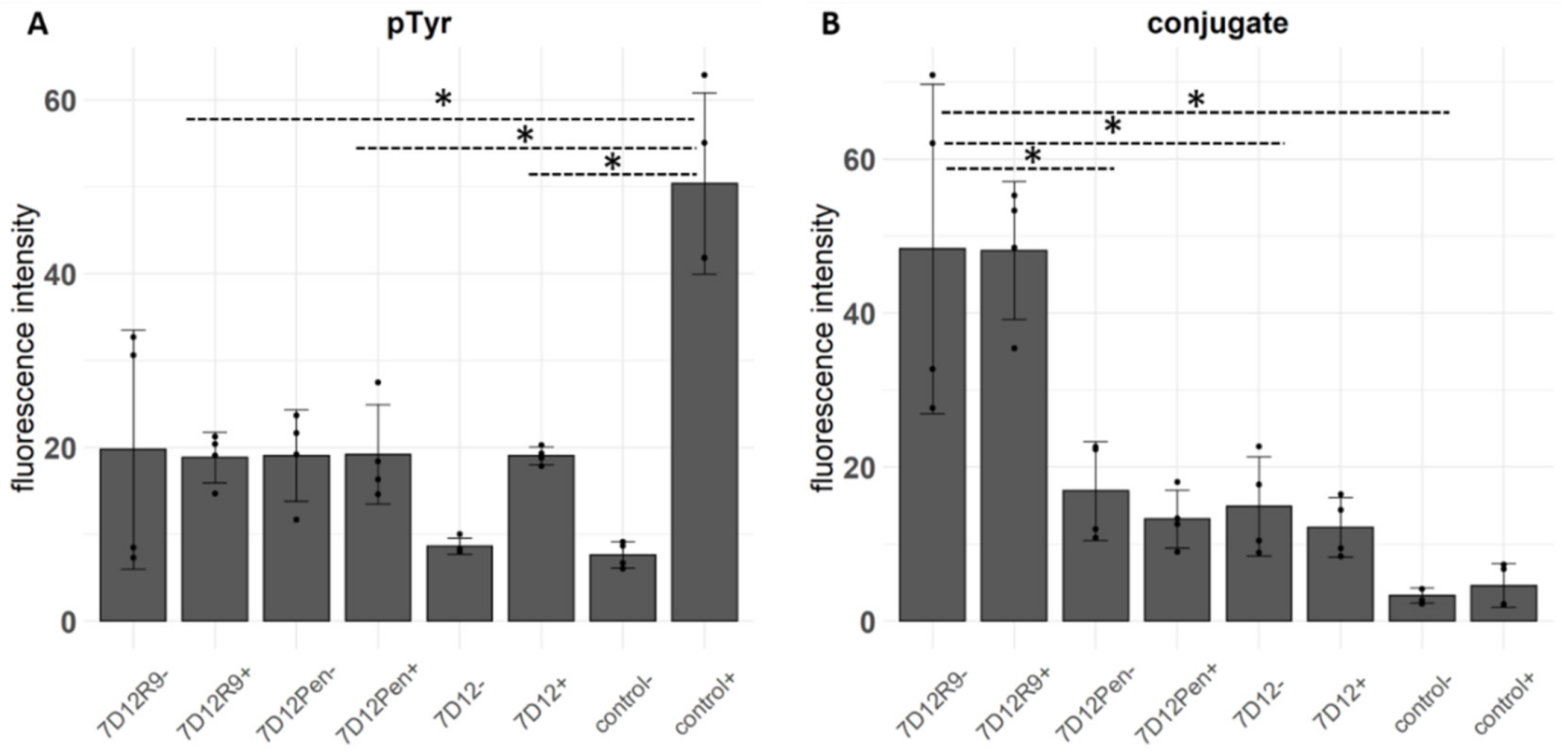
2.3. Competition of EGF Binding by 7D12-R9 and 7D12-Pen
2.4. The Effect of 7D12-R9 and 7D12-Pen on EGFR Activation
2.5. Binding and Uptake of 7D12-DTPA-R9 and 7D12-DTPA-Pen and Their D-Peptide Counterparts at Picomolar Concentrations
2.6. D Tissue Penetration and Retention in Cell Spheroids
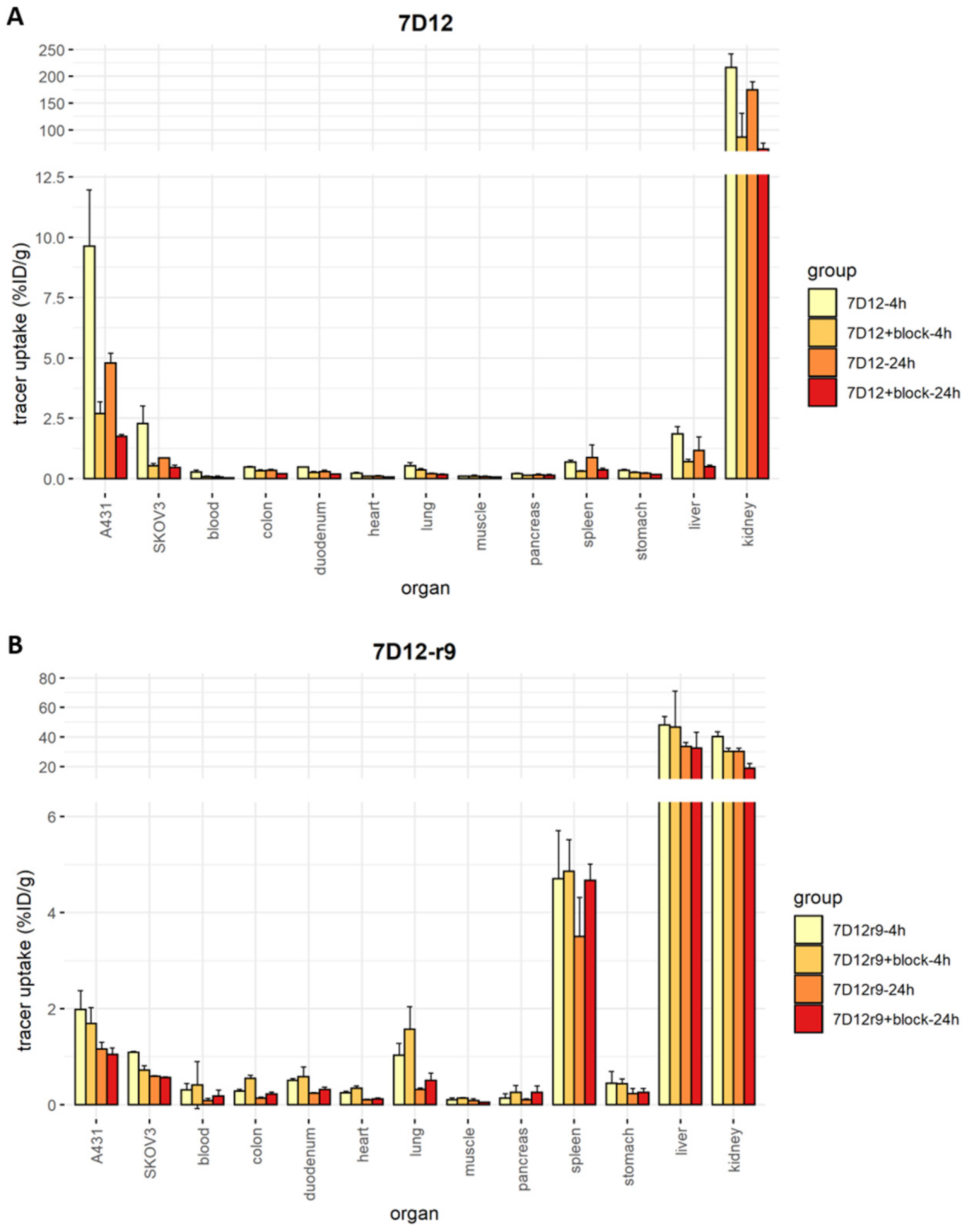
2.7. Biodistribution and SPECT Imaging of the r9 and Pen Conjugates In Vivo
3. Discussion
4. Materials and Methods
4.1. Nanobody Expression and Purification
4.2. WT Sortase A Expression
4.3. Dye and Chelator Coupling to the Nanobody
4.4. Coupling of CPPs to Nanobodies Via Sortase A Transpeptidation
4.5. Mass Spectrometry
4.6. Cell Culture
4.7. Uptake Experiments in Life Cells (Confocal Microscopy)
4.8. Detection of EGF Binding
4.9. pTyr1068 Staining
4.10. Spheroid Culture, Incubation and Optical Clearing
4.11. Radiolabeling of the 7D12 Conjugates and Quality Control
4.12. Animals
4.13. In Vivo Biodistribution and SPECT Imaging
4.14. Data Analysis and Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Desmyter, A.; Transue, T.R.; Ghahroudi, M.A.; Thi, M.H.D.; Poortmans, F.; Hamers, R.; Muyldermans, S.; Wyns, L. Crystal structure of a camel single-domain V(H) antibody fragment in complex with lysozyme. Nat. Struct. Biol. 1996, 3, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Jovčevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Genst, E.; Silence, K.; Decanniere, K.; Conrath, K.; Loris, R.; Kinne, J.; Muyldermans, S.; Wyns, L. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. USA 2006, 103, 4586–4591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández, I.B.; Rompen, R.; Rossin, R.; Xenaki, K.T.; Katrukha, E.A.; Nicolay, K.; Henegouwen, P.v.B.E.; Grüll, H.; Oliveira, S. Imaging of Tumor Spheroids, Dual-Isotope SPECT, and Autoradiographic Analysis to Assess the Tumor Uptake and Distribution of Different Nanobodies. Mol. Imaging Biol. 2019, 21, 1079–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debie, P.; Lafont, C.; Defrise, M.; Hansen, I.; van Willigen, D.M.; van Leeuwen, F.W.B.; Gijsbers, R.; D’Huyvetter, M.; Devoogdt, N.; Lahoutte, T.; et al. Size and affinity kinetics of nanobodies influence targeting and penetration of solid tumours. J. Control. Release 2020, 317, 34–42. [Google Scholar] [CrossRef]
- Debie, P.; Devoogdt, N.; Hernot, S. Targeted Nanobody-Based Molecular Tracers for Nuclear Imaging and Image-Guided Surgery. Antibodies 2019, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, L.; Gainkam, T.; Huang, L.; Caveliers, V.; Keyaerts, M.; Hernot, S.; Vaneycken, I.; Vanhove, C.; Revets, H.; De Baetselier, P.; et al. Comparison of the Biodistribution and Tumor Targeting of Two 99mTc-Labeled Anti-EGFR Nanobodies in Mice, Using Pinhole SPECT/Micro-CT. J. Nucl. Med. 2008, 49, 788–795. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, S.; Van Dongen, G.A.M.S.; Stigter-Van Walsum, M.; Roovers, R.C.; Stam, J.C.; Mali, W.; Van Diest, P.J.; Henegouwen, P.M.P.v.B.E. Rapid visualization of human tumor xenografts through optical imaging with a near-infrared fluorescent anti-epidermal growth factor receptor nanobody. Mol. Imaging 2012, 11, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, S.I.; Lou, J.; Shaller, C.C.; Tang, Y.; Klein-Szanto, A.J.P.; Weiner, L.M.; Marks, J.D.; Adams, G.P. Influence of affinity and antigen internalization on the uptake and penetration of anti-HER2 Antibodies in Solid Tumors. Cancer Res. 2011, 71, 2250–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Scheltinga, A.G.T.; Lub-de Hooge, M.N.; Abiraj, K.; Schrodër, C.P.; Pot, L.; Bossenmaier, B.; Thomas, M.; Hölzlwimmer, G.; Friess, T.; Kosterink, J.G.; et al. ImmunoPET and biodistribution with human epidermal growth factor receptor 3 targeting antibody89Zr-RG7116. MAbs 2014, 6, 1051–1058. [Google Scholar] [CrossRef] [Green Version]
- Stillebroer, A.B.; Franssen, G.M.; Mulders, P.F.A.; Oyen, W.J.G.; Van Dongen, G.A.M.S.; Laverman, P.; Oosterwijk, E.; Boerman, O.C. ImmunoPET imaging of renal cell carcinoma with 124I- and 89Zr-Labeled Anti-CAIX monoclonal antibody cG250 in mice. Cancer Biother. Radiopharm. 2013, 28, 510–515. [Google Scholar] [CrossRef] [Green Version]
- Roovers, R.C.; Laeremans, T.; Huang, L.; De Taeye, S.; Verkleij, A.J.; Revets, H.; De Haard, H.J.; Henegouwen, P.M.P.v.B.E. Efficient inhibition of EGFR signalling and of tumour growth by antagonistic anti-EGFR Nanobodies. Cancer Immunol. Immunother. 2007, 56, 303–317. [Google Scholar] [CrossRef]
- Roovers, R.C.; Vosjan, M.J.W.D.; Laeremans, T.; El Khoulati, R.; De Bruin, R.C.G.; Ferguson, K.M.; Verkleij, A.J.; Van Dongen, G.A.M.S.; Van Bergen En Henegouwen, P.M.P. A biparatopic anti-EGFR nanobody efficiently inhibits solid tumour growth. Int. J. Cancer 2011, 129, 2013–2024. [Google Scholar] [CrossRef]
- Van Roy, M.; Ververken, C.; Beirnaert, E.; Hoefman, S.; Kolkman, J.; Vierboom, M.; Breedveld, E.; ’t Hart, B.; Poelmans, S.; Bontinck, L.; et al. The preclinical pharmacology of the high affinity anti-IL-6R Nanobody® ALX-0061 supports its clinical development in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 135. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Huang, H.; Sheng, Y.; Shi, H.; Min, Y.; Liu, Y. Transglutaminase mediated PEGylation of nanobodies for targeted nano-drug delivery. J. Mater. Chem. B 2018, 6, 1011–1017. [Google Scholar] [CrossRef]
- Rashidian, M.; LaFleur, M.W.; Verschoor, V.L.; Dongre, A.; Zhang, Y.; Nguyen, T.H.; Kolifrath, S.; Aref, A.R.; Lau, C.J.; Paweletz, C.P.; et al. Immuno-PET identifies the myeloid compartment as a key contributor to the outcome of the antitumor response under PD-1 blockade. Proc. Natl. Acad. Sci. USA 2019, 116, 16971–16980. [Google Scholar] [CrossRef] [Green Version]
- Bruce, V.J.; Lopez-Islas, M.; McNaughton, B.R. Resurfaced cell-penetrating nanobodies: A potentially general scaffold for intracellularly targeted protein discovery. Protein Sci. 2016, 25, 1129–1137. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M.; Kang, Y.-S.; Yang, J.; Buciak, J.L. Enhanced cellular uptake and in vivo biodistribution of a monoclonal antibody following cationization. J. Pharm. Sci. 1995, 84, 943–948. [Google Scholar] [CrossRef]
- Zhang, Z.; Qian, H.; Huang, J.; Sha, H.; Zhang, H.; Yu, L.; Liu, B.; Hua, D.; Qian, X. Anti-EGFR-iRGD recombinant protein modified biomimetic nanoparticles loaded with gambogic acid to enhance targeting and antitumor ability in colorectal cancer treatment. Int. J. Nanomed. 2018, 13, 4961. [Google Scholar] [CrossRef] [Green Version]
- Jin, P.; Sha, R.; Zhang, Y.; Liu, L.; Bian, Y.; Qian, J.; Qian, J.; Lin, J.; Ishimwe, N.; Hu, Y.; et al. Blood Circulation-Prolonging Peptides for Engineered Nanoparticles Identified via Phage Display. Nano Lett. 2019, 19, 1467–1478. [Google Scholar] [CrossRef]
- Ji, F.; Sha, H.; Meng, F.; Zhu, A.; Ding, N.; Zhang, H.; Xu, H.; Qian, H.; Yu, L.; Liu, Q.; et al. Tumor-penetrating recombinant protein anti-EGFR-iRGD enhance the extravasation and tumor penetration of lymphocytes in gastric cancer. J. Immunother. Cancer 2017, 40, 1583–1591. [Google Scholar] [CrossRef]
- Lingasamy, P.; Laarmann, A.-H.; Teesalu, T. Tumor Penetrating Peptide-Functionalized Tenascin-C Antibody for Glioblastoma Targeting. Curr. Cancer Drug Targets 2020, 21, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.B.; Braun, G.B.; Ruoslahti, E. Neuropilin-1 and heparan sulfate proteoglycans cooperate in cellular uptake of nanoparticles functionalized by cationic cell-penetrating peptides. Sci. Adv. 2015, 1, e1500821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [CrossRef]
- Vivès, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [Green Version]
- Derossi, D. Antennapedia homeodomain third helix as a peptide and oligonucleotide vector. Restor. Neurol. Neurosci. 1995, 8, 17–18. [Google Scholar] [CrossRef]
- Gurney, L.R.I.; Taggart, J.J.; Tong, W.-C.; Jones, A.T.; Robson, S.C.; Taggart, M.J. Inhibition of Inflammatory Changes in Human Myometrial Cells by Cell Penetrating Peptide and Small Molecule Inhibitors of NFκB. Front. Immunol 2018, 9, 2966. [Google Scholar] [CrossRef] [Green Version]
- Sauter, M.; Strieker, M.; Kleist, C.; Wischnjow, A.; Daniel, V.; Altmann, A.; Haberkorn, U.; Mier, W. Improving antibody-based therapies by chemical engineering of antibodies with multimeric cell-penetrating peptides for elevated intracellular delivery. J. Control. Release 2020, 322, 200–208. [Google Scholar] [CrossRef]
- Knight, J.C.; Topping, C.; Mosley, M.; Kersemans, V.; Falzone, N.; Fernández-Varea, J.M.; Cornelissen, B. PET imaging of DNA damage using 89Zr-labelled anti-γH2AX-TAT immunoconjugates. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1707–1717. [Google Scholar] [CrossRef]
- Van Oppen, L.M.P.E.; Pille, J.; Stuut, C.; van Stevendaal, M.; van der Vorm, L.; Smeitink, J.A.M.; Koopman, W.J.H.; Willenms, W.J.H.; van Hest, J.C.M.; Brock, R. Octa-arginine boosts the penetration of elastin-like polypeptide nanoparticles in 3D cancer models. Eur. J. Pharm. Biopharm. 2019, 13, 175–184. [Google Scholar] [CrossRef]
- Gessner, I.; Neundorf, I. Nanoparticles modified with cell-penetrating peptides: Conjugation mechanisms, physicochemical properties, and application in cancer diagnosis and therapy. Int. J. Mol. Sci. 2020, 21, 2536. [Google Scholar] [CrossRef] [Green Version]
- Duchardt, F.; Ruttekolk, I.R.; Verdurmen, W.P.R.; Lortat-Jacob, H.; Bürck, J.; Hufnagel, H.; Fischer, R.; van den Heuvel, M.; Löwik, D.W.P.; Vuister, G.W.; et al. A cell-penetrating peptide derived from human lactoferrin with conformation-dependent uptake efficiency. J. Biol. Chem. 2009, 284, 36099–36108. [Google Scholar] [CrossRef] [Green Version]
- van Lith, S.A.M.; van den Brand, D.; Wallbrecher, R.; van Duijnhoven, S.M.J.; Brock, R.; Leenders, W.P.J. A Conjugate of an Anti-Epidermal Growth Factor Receptor (EGFR) VHH and a Cell-Penetrating Peptide Drives Receptor Internalization and Blocks EGFR Activation. ChemBioChem 2017, 18, 2390–2394. [Google Scholar] [CrossRef]
- van den Brand, D.; Veelken, C.; Massuger, L.; Brock, R. Penetration in 3D tumor spheroids and explants: Adding a further dimension to the structure-activity relationship of cell-penetrating peptides. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1342–1349. [Google Scholar] [CrossRef]
- Collado Camps, E.; Brock, R. An opportunistic route to success: Towards a change of paradigm to fully exploit the potential of cell-penetrating peptides. Bioorganic Med. Chem. 2018, 26, 2780–2787. [Google Scholar] [CrossRef]
- Sarko, D.; Beijer, B.; Boy, R.G.; Nothelfer, E.M.; Leotta, K.; Eisenhut, M.; Altmann, A.; Haberkorn, U.; Mier, W. The pharmacokinetics of cell-penetrating peptides. Mol. Pharm. 2010, 7, 2224–2231. [Google Scholar] [CrossRef]
- Duchardt, F.; Fotin-Mleczek, M.; Schwarz, H.; Fischer, R.; Brock, R. A Comprehensive Model for the Cellular Uptake of Cationic Cell-penetrating Peptides. Traffic 2007, 8, 848–866. [Google Scholar] [CrossRef]
- Schmitz, K.R.; Bagchi, A.; Roovers, R.C.; Henegouwen, P.M.P.v.B.E.; Ferguson, K.M. Structural evaluation of EGFR inhibition mechanisms for nanobodies/VHH domains. Structure 2013, 21, 1214–1224. [Google Scholar] [CrossRef] [Green Version]
- DeWitt, A.; Iida, T.; Lam, H.-Y.; Hill, V.; Wiley, H.S.; Lauffenburger, D.A. Affinity Regulates Spatial Range of EGF Receptor Autocrine Ligand Binding. Dev. Biol. 2002, 250, 305–316. [Google Scholar] [CrossRef]
- Verdurmen, W.P.R.; Bovee-Geurts, P.H.; Wadhwani, P.; Ulrich, A.S.; Hällbrink, M.; Van Kuppevelt, T.H.; Brock, R. Preferential uptake of L-versus D-amino acid cell-penetrating peptides in a cell type-dependent manner. Chem. Biol. 2011, 18, 1000–1010. [Google Scholar] [CrossRef] [Green Version]
- Saga, T.; Neumann, R.D.; Heya, T.; Sato, J.; Kinuya, S.; Le, N.; Paik, C.H.; Weinstein, J.N. Targeting cancer micrometastases with monoclonal antibodies: A binding- site barrier. Proc. Natl. Acad. Sci. USA 1995, 92, 8999–9003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dom, G.; Shaw-Jackson, C.; Matis, C.; Bouffioux, O.; Picard, J.J.; Prochiantz, A.; Mingeot-Leclerq, M.P.; Brasseur, R.; Rezsohazy, R. Cellular uptake of Antennapedia Penetratin peptides is a two-step process in which phase transfer precedes a tryptophan-dependent translocation. Nucleic Acids Res. 2003, 31, 556–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsulays, B.B.; Anwer, M.K.; Soliman, G.A.; Alshehri, S.M.; Khafagy, E.S. Impact of penetratin stereochemistry on the oral bioavailability of insulin-loaded solid lipid nanoparticles. Int. J. Nanomed. 2019, 14, 9127–9138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofström, C.; Altai, M.; Honarvar, H.; Strand, J.; Malmberg, J.; Hosseinimehr, S.J.; Orlova, A.; Gräslund, T.; Tolmachev, V. HAHAHA, HEHEHE, HIHIHI, or HKHKHK: Influence of position and composition of histidine containing tags on biodistribution of [99mTc(CO)3]+-labeled affibody molecules. J. Med. Chem. 2013, 14, 9127. [Google Scholar] [CrossRef] [Green Version]
- Gainkam, L.O.T.; Caveliers, V.; Devoogdt, N.; Vanhove, C.; Xavier, C.; Boerman, O.; Muyldermans, S.; Bossuyt, A.; Lahoutte, T. Localization, mechanism and reduction of renal retention of technetium-99m labeled epidermal growth factor receptor-specific nanobody in mice. Contrast Media Mol. Imaging 2011, 6, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Lecocq, Q.; Zeven, K.; De Vlaeminck, Y.; Martens, S.; Massa, S.; Goyvaerts, C.; Raes, G.; Keyaerts, M.; Breckpot, K.; Devoogdt, N. Noninvasive imaging of the immune checkpoint LAG-3 using nanobodies, from development to pre-clinical use. Biomolecules 2019, 9, 548. [Google Scholar] [CrossRef] [Green Version]
- Behr, T.M.; Goldenberg, D.M.; Becker, W. Reducing the renal uptake of radiolabeled antibody fragments and peptides for diagnosis and therapy: Present status, future prospects and limitations. Eur. J. Nucl. Med. 1998, 25, 201–212. [Google Scholar] [CrossRef]
- Herce, H.D.; Deng, W.; Helma, J.; Leonhardt, H.; Cardoso, M.C. Visualization and targeted disruption of protein interactions in living cells. Nat. Commun. 2013, 4, 2660. [Google Scholar] [CrossRef] [Green Version]
- Tabtimmai, L.; Suphakun, P.; Srisook, P.; Kiriwan, D.; Phanthong, S.; Kiatwuthinon, P.; Chaicumpa, W.; Choowongkomon, K. Cell-penetrable nanobodies (transbodies) that inhibit the tyrosine kinase activity of EGFR leading to the impediment of human lung adenocarcinoma cell motility and survival. J. Cell. Biochem. 2019, 120, 18077–18087. [Google Scholar] [CrossRef]
- Jain, M.; Chauhan, S.C.; Singh, A.P.; Venkatraman, G.; Colcher, D.; Batra, S.K. Penetratin improves tumor retention of single-chain antibodies: A novel step toward optimization of radioimmunotherapy of solid tumors. Cancer Res. 2005, 65, 7840–7846. [Google Scholar] [CrossRef] [Green Version]
- Ramaker, K.; Henkel, M.; Krause, T.; Röckendorf, N.; Frey, A. Cell penetrating peptides: A comparative transport analysis for 474 sequence motifs. Drug Deliv. 2018, 25, 928–937. [Google Scholar] [CrossRef]
- Adachi, Y.; Sakamoto, K.; Umemoto, T.; Fukuda, Y.; Tani, A.; Asami, T. Investigation on cellular uptake and pharmacodynamics of DOCK2-inhibitory peptides conjugated with cell-penetrating peptides. Bioorganic Med. Chem. 2017, 25, 2148–2155. [Google Scholar] [CrossRef]
- Mueller, J.; Kretzschmar, I.; Volkmer, R.; Boisguerin, P. Comparison of cellular uptake using 22 CPPs in 4 different cell lines. Bioconjug. Chem. 2008, 19, 2363–2374. [Google Scholar] [CrossRef]
- Zhang, W.; Song, J.; Zhang, B.; Liu, L.; Wang, K.; Wang, R. Design of acid-activated cell penetrating peptide for delivery of active molecules into cancer cells. Bioconjug. Chem. 2011, 22, 1410–1415. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, L.; Chang, L.; Liu, H.; Song, J.; Liu, Y.; Bao, H.; Liu, B.; Wang, R.; Ni, J. Design of a new pH-activatable cell-penetrating peptide for drug delivery into tumor cells. Chem. Biol. Drug Des. 2019, 94, 1884–1893. [Google Scholar] [CrossRef]
- Aguilera, T.A.; Olson, E.S.; Timmers, M.M.; Jiang, T.; Tsien, R.Y. Systemic in vivo distribution of activatable cell penetrating peptides is superior to that of cell penetrating peptides. Integr. Biol. 2009, 1, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Ke, M.T.; Fujimoto, S.; Imai, T. SeeDB: A simple and morphology-preserving optical clearing agent for neuronal circuit reconstruction. Nat. Neurosci. 2013, 16, 1154–1161. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Collado Camps, E.; van Lith, S.A.M.; Frielink, C.; Lankhof, J.; Dijkgraaf, I.; Gotthardt, M.; Brock, R. CPPs to the Test: Effects on Binding, Uptake and Biodistribution of a Tumor Targeting Nanobody. Pharmaceuticals 2021, 14, 602. https://doi.org/10.3390/ph14070602
Collado Camps E, van Lith SAM, Frielink C, Lankhof J, Dijkgraaf I, Gotthardt M, Brock R. CPPs to the Test: Effects on Binding, Uptake and Biodistribution of a Tumor Targeting Nanobody. Pharmaceuticals. 2021; 14(7):602. https://doi.org/10.3390/ph14070602
Chicago/Turabian StyleCollado Camps, Estel, Sanne A. M. van Lith, Cathelijne Frielink, Jordi Lankhof, Ingrid Dijkgraaf, Martin Gotthardt, and Roland Brock. 2021. "CPPs to the Test: Effects on Binding, Uptake and Biodistribution of a Tumor Targeting Nanobody" Pharmaceuticals 14, no. 7: 602. https://doi.org/10.3390/ph14070602
APA StyleCollado Camps, E., van Lith, S. A. M., Frielink, C., Lankhof, J., Dijkgraaf, I., Gotthardt, M., & Brock, R. (2021). CPPs to the Test: Effects on Binding, Uptake and Biodistribution of a Tumor Targeting Nanobody. Pharmaceuticals, 14(7), 602. https://doi.org/10.3390/ph14070602