
Synthesis and Human Carbonic Anhydrase I, II, IX, and XII Inhibition Studies of Sulphonamides Incorporating Mono-, Bi- and Tricyclic Imide Moieties

 , and
, and 
Abstract
:1. Introduction

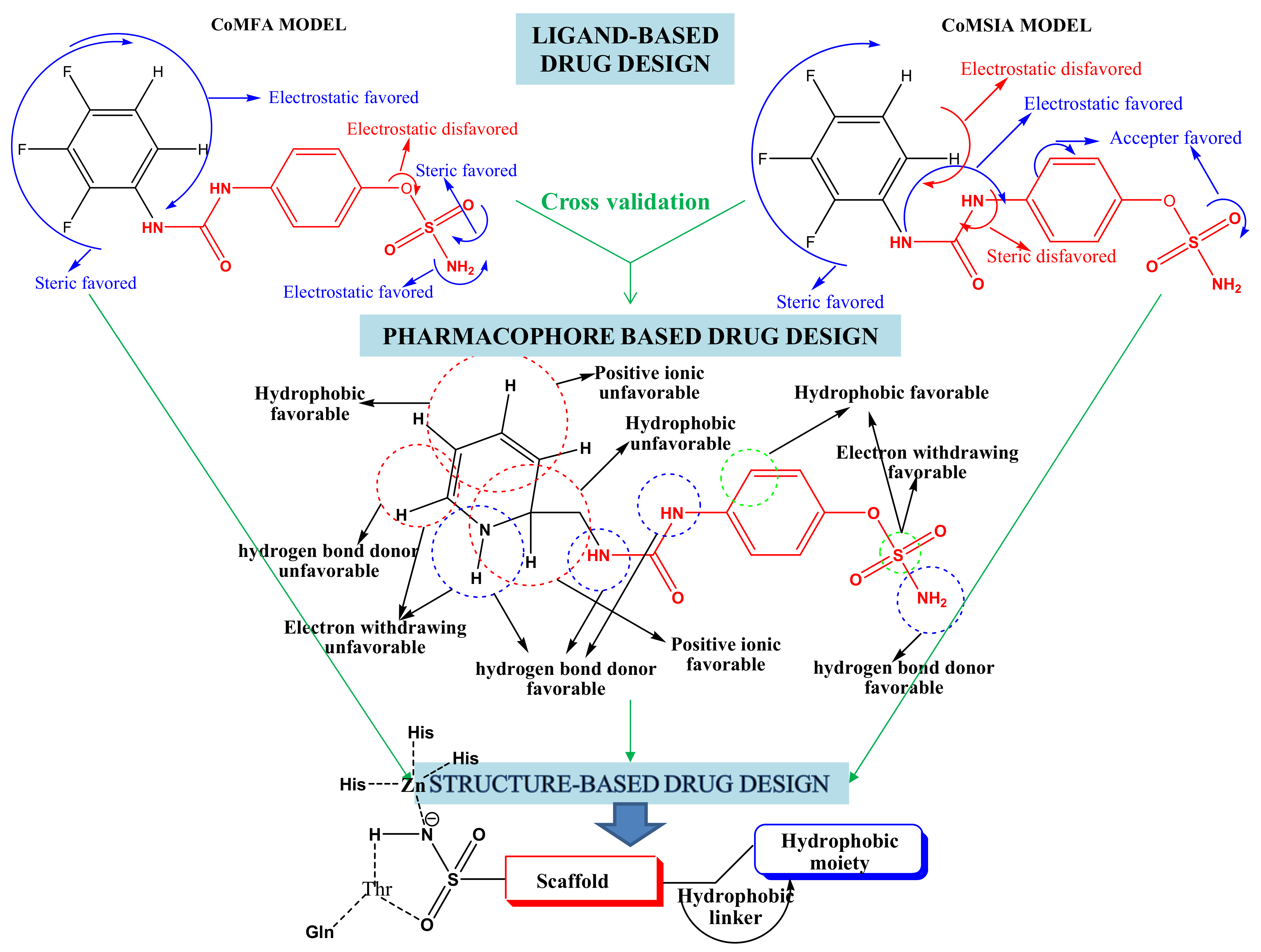
2. Results and Discussion
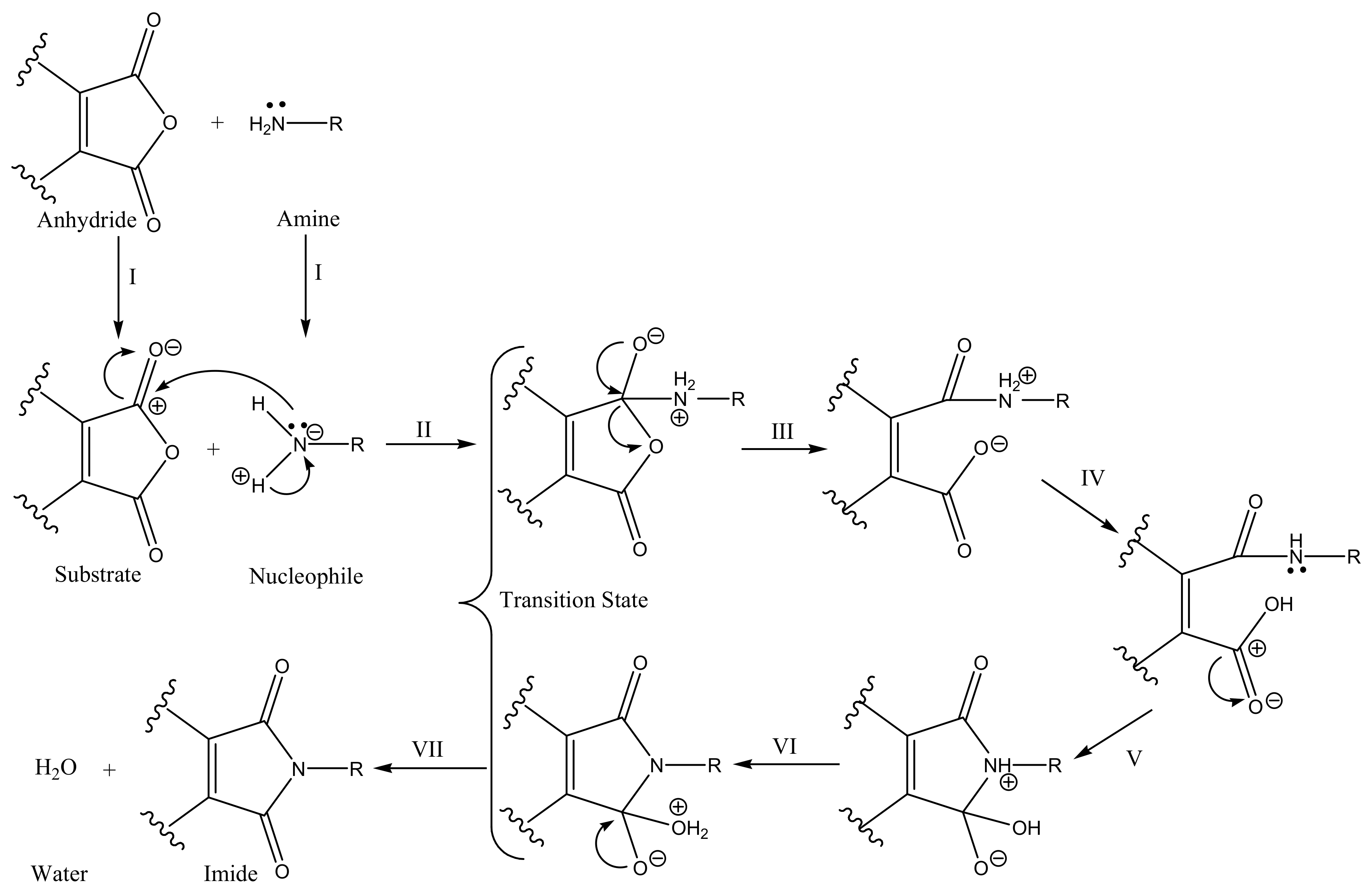
2.1. Chemistry
2.2. Carbonic Anhydrases Inhibition Studies
- i.
- The synthesised sulphonamides inhibited the cytosolic hCA I, with inhibition constants (Ki) ranging from 49 nM to >10,000 nM (Table 2). The 4-(4-sulfo-1,8-napthalic-1,3-dioxopyridine) potassium benzene sulphonamide 1 (Ki of 49 nM) was the most potent inhibitor of the series, whereas the compound 3-chloro-4-(4,5,6,7-tetrachloro-1,3-dioxoisoindolin-2-yl) benzene sulphonamide 12 with an inhibition constant of 159 nM better than AAZ and TPM (Table 2). In comparison to BRZ, CLX, DZR, SLP, and VLX, all the synthesised compounds showed significant inhibition constant (Table 2). The compound 10 is the weakest inhibitor that can be contemplated as a positive feature of hCA I because it is abundantly found in RBCs and is undoubtedly an off-target for other CAIs [2,28]. Increasing the chain length of carbon between the benzene and bulky aromatic group (-CH2CH2- or -CH2-) leads to a decreased activity of compounds 7 and 8 with 427 nM to 332 nM, respectively compared to compound 6. The position of -SO2NH2 plays an important role in increase or decrease of the activities. The decreased activity of compounds 6 > 9 > 10 according to position of -SO2NH2 in the benzene ring are 4th > 3rd > 2nd (Kis 332 nM to >10,000 nM). It is very clear that –SO2NH2 present in the 2nd position comparatively very less active than 3rd than 4th due to steric hindrance. The electronegativity of halogen (F > Cl > Br) to the benzene ring was found to influence the hCA I inhibition activity of compounds 11 (368 nM), 12 (159 nM), and 13 (281 nM).
- ii.
- hCA II was significantly inhibited by many of the new sulphonamides, which exhibited Kis ranging from 2.4 to 4515 nM (Table 2). Most of the compounds showed significant inhibition constants than clinically used standard AAZ and other sulphonamide drugs. Compound 13 significantly inhibits hCA II with a Ki of 2.4 nM than the other sulphonamides of the series. Although the compounds showed excellent to moderate inhibition activity for hCA II. The SAR is straightforward. The increased chain length of carbon between the benzene and bulky aromatic group (-CH2CH2- or -CH2-) results in increased activities of the compounds 1 < 2 < 3 with Kis of 7.1 nM, 5.2 nM, and 2.9 nM, respectively. Due to steric hindrance, the hCA II inhibition activity was lost, i.e., the Ki of ortho, meta, and para position of –SO2NH2 in compound 10 < 9 < 6 was 4515, 27.7, and 7.1 nM, respectively). The electronegativity of halogen (F > Cl > Br) attached to the benzene ring was found to influence the hCA II inhibition of compounds 11 (3.4 nM), 12 (4.9 nM), and 13 (2.4 nM). Similarly, benzthiazole sulphonamide analogues 3 (742 nM) and 4 (44 nM). More electronegativity of halogen lesser is the potency/efficacy. Overall, most of these sulphonamides showed a potent action of inhibition against hCA II. hCA II is the main off-target isoform among several hCAs [28].
- iii.
- hCA IX was moderately to poorly inhibited by the sulphonamide (1–13) with Kis ranging from 9.7 to 7766 nM (Table 2). Compound 6 potentially inhibited hCA IX with a Ki of 9.7 nM than the standard AAZ. Although the sulphonamides are moderate to poorly effective against hCA IX, but the SARs are generated for the future development of more potent hCA IX inhibitors. The increase in the chain length of the carbon between the two bulky groups (-CH2CH2- or -CH2-) results in decreased activities of the compounds, i.e., compound 6 (Ki = 9.7 nM) is more potent than compounds 7 (Ki = 103 nM) and 8 (Ki = 53 nM). The steric hindrance effects cause a significant loss of the hCA IX inhibition properties of sulphonamides, i.e., –SO2NH2 at ortho, meta, and para in compound 9 (Ki ≥ 498 nM) < 10 (Ki = 559 nM) < 6 (Ki = 9.7 nM). The electronegativity of halogen (F > Cl > Br) in the benzene ring was found to be influencing the hCA IX inhibition of compounds 11 (88 nM), 12 (49 nM), and 13 (40 nM). More electronegative halogen atom attached to the benzene ring, reduce the hCA IX inhibition properties.
- iv.
- hCA XII was moderately inhibited by these sulphonamides (Table 2). The inhibition constant showed Kis ranging from 14 to 316 nM. Compound 11 was the most potent hCA XII inhibitor (Ki = 14 nM) of the series. The SARs of these sulphonamides were developed for hCA XII inhibition. The increased carbon chain length (-CH2CH2- or -CH2-) between benzene and anhydride decreased the activities of compounds 6 > 8 > 7 with Kis of 33 nM, 226 nM, and 300 nM, respectively. Steric hindrance responsible for the loss of hCA XII inhibition properties, i.e., –SO2NH2 present in the ortho, para, and meta substituted sulphonamides 9 < 10 < 6 (Kis in the range of 33, 309, and 316 nM), respectively. The electronegativity of halogen (F > Cl > Br) in the benzene ring was found to be influencing the hCA XII inhibition of compounds 11 (14 nM), 12 (22 nM), and 13 (99 nM). More the electronegativity of halogen attached to the benzene ring, increase the hCA XII inhibition properties.
- v.
- The close structural similarities in hCAs were the major challenge in finding novel selective hCA isoenzyme inhibitors. Thus, calculation of the selectivity ratios has been done (Table 2). The selectivity ratios for the tumour-associated isoforms hCA IX and XII over hCA II, ranged from 1 to 8.077 nM and 0.013 to 14.616 nM, respectively for the synthesised sulphonamide reported here (Table 2). Some compounds were observed to have high selectivity ratios for hCA IX and XII over hCA II. Compound 10 was highly selective for hCA IX inhibition over hCA II. Similarly, compounds 3 and 10 were highly selective for hCA XII inhibition over hCA II. However, most of the compounds had a low selectivity ratio, indicating that these are more selective for inhibition of hCA I and II than hCA IX and CA XII.
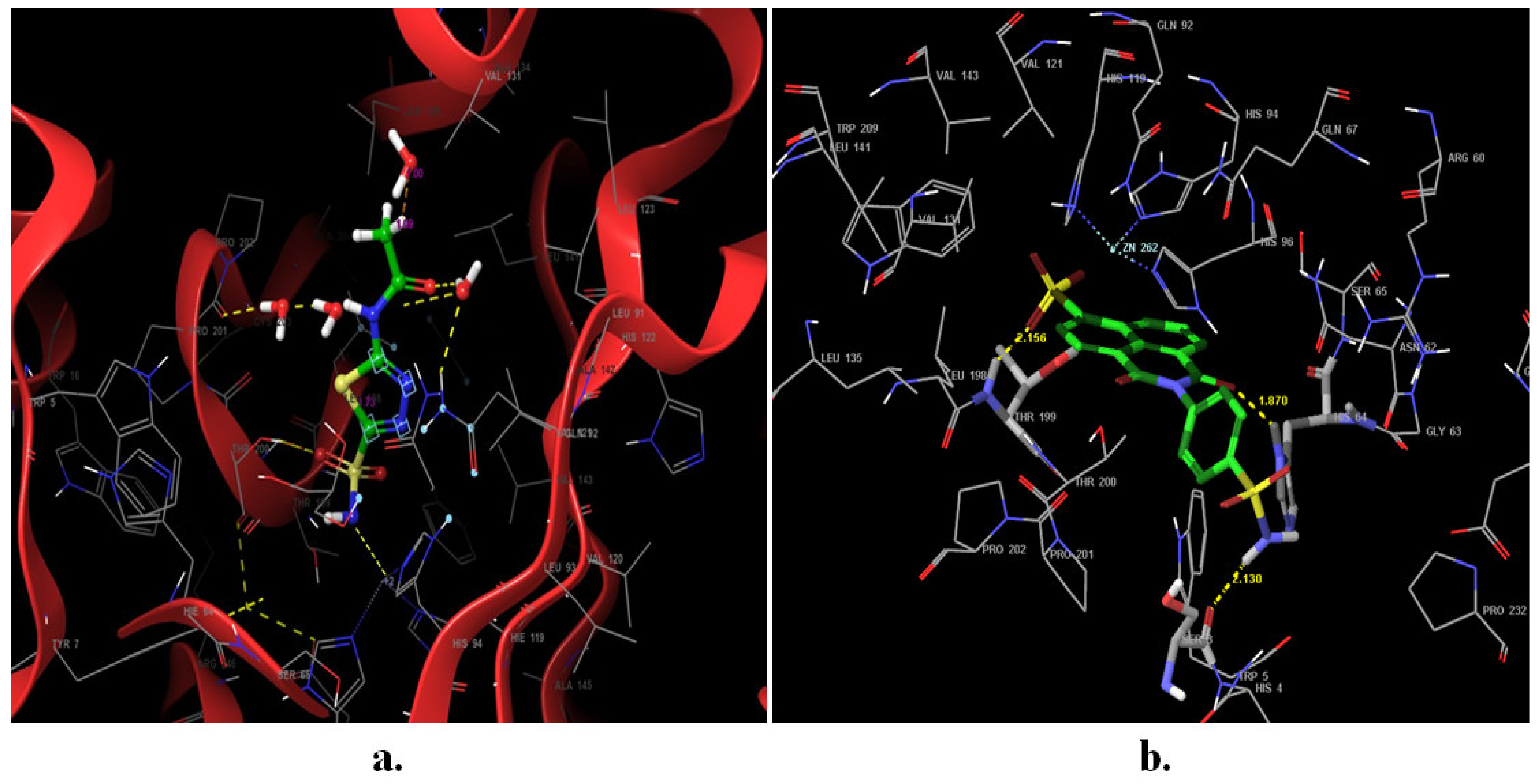
2.3. Docking Studies
3. Materials and Methods
3.1. Reagents and Instruments
3.2. Chemistry
3.2.1. Synthesis of 4-(4-Sulfo-1,8-napthalic-1,3-dioxopyridine) Potassium Benzenesulphonamide (1)
3.2.2. Synthesis of 4-(2-(2,5-Dioxo-2H-pyrrol-1(5H)-yl) Ethyl) Benzenesulphonamide (2)
3.2.3. Synthesis of 2-(4,5,6,7-Tetrachloro-1,3-dioxoisoindolin-2-yl) Benzo[d]Thiazole-5-Sulphonamide (3)
3.2.4. Synthesis of 2-(4,5,6,7-Tetrabromo-1,3-dioxoisoindolin-2-yl) Benzo[d]Thiazole-5-Sulphonamide (4)
3.2.5. Synthesis of 3-Fluoro-4-(5-Nitro-1,3-dioxoisoindolin-2-yl) Benzenesulphonamide (5)
3.2.6. Synthesis of 4-(4,5,6,7-Tetrachloro-1,3-dioxoisoindolin-2-yl) Benzenesulphonamide (6)
3.2.7. Synthesis of 4-((4,5,6,7-Tetrachloro-1,3-dioxoisoindolin-2-yl) Methyl) Benzenesulphonamide (7)
3.2.8. Synthesis of 4-(2-(4,5,6,7-Tetrachloro-1,3-dioxoisoindolin-2-yl) Ethyl) Benzenesulphonamide (8)
3.2.9. Synthesis of 3-(4,5,6,7-Tetrachloro-1,3-dioxoisoindolin-2-yl) Benzenesulphonamide (9)
3.2.10. Synthesis of 2-(4,5,6,7-Tetrachloro-1,3-dioxoisoindolin-2-yl) Benzenesulphonamide (10)
3.2.11. Synthesis of 3-Fluoro-4-(4,5,6,7-Tetrachloro-1,3-dioxoisoindolin-2-yl) Benzenesulphonamide (11)
3.2.12. Synthesis of 3-Chloro-4-(4,5,6,7-Tetrachloro-1,3-Dioxoisoindolin-2-yl) Benzenesulphonamide (12)
3.2.13. Synthesis of 3-Bromo-4-(4,5,6,7-Tetrachloro-1,3-Dioxoisoindolin-2-yl) Benzenesulphonamide (13)
3.3. CA Inhibition Assay
3.4. Molecular Docking Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAZ | Acetazolamide |
| ASN | asparagines |
| BRZ | brinzolamide |
| BZA | benzolamide |
| CA | carbonic anhydrase |
| C:H:N:S:O | carbon:hydrogen:nitrogen:sulphur:oxygen |
| CAI | carbonic anhydrase inhibitor |
| CNS | central nervous system |
| DMSO | dimethyl sulfoxide |
| DZA | Dorzolamide |
| E-I | enzyme-inhibitor |
| EZA | ethoxzolamide |
| FT-IR | Fourier transform infrared |
| GLN | glutamine |
| hCA | human carbonic anhydrase |
| HIS | Histidine |
| HRMS | high-resolution mass spectrometry |
| IND | indisulam |
| mM | milimolar |
| MZA | methazolamide |
| NMR | nuclear magnetic resonance |
| nM | nanomolar |
| PDB | protein data bank |
| RMSD | Root mean square deviation |
| SAR | structure–activity relationship |
| SLP | Sulpiride |
| THR | threonine |
| TRP | tryptophan |
| TPM | topiramate |
| UV | ultraviolet |
| XP | Extra precision |
| Zn | Zinc |
| ZNS | zonisamide |
References
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple binding modes of inhibitors to carbonic anhydrases: How to design specific drugs targeting 15 different isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. How many carbonic anhydrase inhibition mechanisms exist? J. Enzyme Inhib. Med. Chem. 2016, 31, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, A.; Supuran, C.T. Carbonic anhydrase inhibitors as antitumor/anti- metastatic agents: A patent review (2008–2018). Expert Opin. Ther. Pat. 2018, 28, 729–740. [Google Scholar] [CrossRef]
- Supuran, C.T. Emerging role of carbonic anhydrase inhibitors. Clin. Sci. 2021, 135, 1233–1249. [Google Scholar] [CrossRef]
- Winum, J.Y.; Scozzafava, A.; Montero, J.L.; Supuran, C.T. Design of zinc binding functions for carbonic anhydrase inhibitors. Curr. Pharm. Des. 2008, 14, 615–621. [Google Scholar]
- Winum, J.Y.; Scozzafava, A.; Montero, J.L.; Supuran, C.T. Therapeutic potential of sulfamides as enzyme inhibitors. Med. Res. Rev. 2006, 26, 767–792. [Google Scholar] [CrossRef]
- Supuran, C.T. Diuretics: From classical carbonic anhydrase inhibitors to novel applications of the sulfonamides. Curr. Pharm. Des. 2008, 14, 641–648. [Google Scholar] [CrossRef]
- Fares, M.; Eldehna, W.M.; Bua, S.; Lanzi, C.; Lucarini, L.; Masini, E.; Peat, T.S.; Abdel-Aziz, H.A.; Nocentini, A.; Keller, P.A.; et al. Discovery of potent dual-tailed benzenesulfonamide inhibitors of human carbonic anhydrases implicated in glaucoma and in vivo profiling of their intraocular pressure-lowering action. J. Med. Chem. 2020, 63, 3317–3326. [Google Scholar] [CrossRef]
- Thiry, A.; Dognè, J.M.; Masereel, B.; Supuran, C.T. Carbonic anhydrase inhibitors as anticonvulsant agents. Curr. Top. Med. Chem. 2007, 7, 855–864. [Google Scholar] [CrossRef]
- De Simone, G.; Di Fiore, A.; Supuran, C.T. Are carbonic anhydrase inhibitors suitable for obtaining antiobesity drugs? Curr. Pharm. Des. 2008, 14, 655–660. [Google Scholar]
- Supuran, C.T. Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors. Expert Opin. Investig. Drugs 2018, 27, 963–970. [Google Scholar] [CrossRef]
- Shaldam, M.; Eldehna, W.M.; Nocentini, A.; Elsayed, Z.M.; Ibrahim, T.M.; Salem, R.; El-Domany, R.A.; Capasso, C.; Abdel-Aziz, H.A.; Supuran, C.T. Development of novel benzofuran-based SLC-0111 analogs as selective cancer-associated carbonic anhydrase isoform IX inhibitors. Eur. J. Med. Chem. 2021, 216, 113283. [Google Scholar] [CrossRef]
- Said, M.A.; Eldehna, W.M.; Nocentini, A.; Fahim, S.H.; Bonardi, A.; Elgazar, A.A.; Krystof, V.; Soliman, D.H.; Abdel-Aziz, H.A.; Gratteri, P.; et al. Sulfonamide-based ring-fused analogues for CAN508 as novel carbonic anhydrase inhibitors endowed with antitumor activity: Design, synthesis, and in vitro biological evaluation. Eur. J. Med. Chem. 2020, 189, 112019. [Google Scholar] [CrossRef]
- Buller, F.; Steiner, M.; Frey, K.; Mircsof, D.; Scheuermann, J.; Kalisch, M.; Bühlmann, P.; Supuran, C.T.; Neri, D. Selection of carbonic anhydrase IX inhibitors from one million DNA-encoded compounds. ACS Chem. Biol. 2011, 6, 336. [Google Scholar] [CrossRef] [PubMed]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [PubMed] [Green Version]
- Hackett, P.H.; Roach, R.C. High-altitude illness. N. Eng. J. Med. 2001, 345, 107–114. [Google Scholar] [CrossRef]
- Gao, B.B.; Clermont, A.; Rook, S.; Fonda, S.J.; Srinivasan, V.J.; Wojtkowski, M.; Fujimoto, J.G.; Avery, R.L.; Arrigg, P.G.; Bursell, S.E.; et al. Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrein activation. Nat. Med. 2007, 13, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, J.M.; Ohlemiller, K.K.; Shah, G.N.; Ulmasov, B.; Becker, T.A.; Waheed, A.; Hennig, A.K.; Lukasiewicz, P.D.; Sly, W.S. Carbonic anhydrase XIV deficiency produces a functional defect in the retinal light response. Proc. Natl. Acad. Sci. USA 2007, 104, 8514–8519. [Google Scholar] [CrossRef] [Green Version]
- Alterio, V.; Hilvo, M.; Di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef] [Green Version]
- Sethi, K.K.; Verma, S.M. A systematic quantitative approach to rational drug design and discovery of novel human carbonic anhydrase IX inhibitors. J Enzyme Inhib. Med. Chem. 2014, 29, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Sethi, K.K.; Verma, S.M.; Tanç, M.; Carta, F.; Supuran, C.T. Carbonic anhydrase inhibitors: Synthesis and inhibition of the cytosolic mammalian carbonic anhydrase isoforms I, II and VII with benzene sulfonamides incorporating 4,5,6,7-tetrachlorophthalimide moiety. Bioorg. Med. Chem. 2013, 21, 5168–5174. [Google Scholar] [CrossRef] [PubMed]
- Sethi, K.K.; Vullo, D.; Verma, S.M.; Tanç, M.; Carta, F.; Supuran, C.T. Carbonic anhydrase inhibitors: Synthesis and inhibition of the human carbonic anhydrase isoforms I, II, VII, IX and XII with benzene sulfonamides incorporating 4,5,6,7-tetrabromophthalimide moiety. Bioorg. Med. Chem. 2013, 21, 5973–5982. [Google Scholar] [CrossRef]
- Sethi, K.K.; Verma, S.M.; Tanç, M.; Purper, G.; Calafato, G.; Carta, F.; Supuran, C.T. Carbonic anhydrase inhibitors: Synthesis and inhibition of the human carbonic anhydrase isoforms I, II, IX and XII with benzene sulfonamides incorporating 4- and 3-nitrophthalimide moieties. Bioorg. Med. Chem. 2014, 22, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- Moi, D.; Nocentini, A.; Deplano, A.; Osman, S.M.; AlOthman, Z.A.; Piras, V.; Balboni, G.; Supuran, C.T.; Onnis, V. Appliance of the piperidinyl-hydrazidoureido linker to benzenesulfonamide compounds: Synthesis, in vitro and in silico evaluation of potent carbonic anhydrase II, IX and XII inhibitors. Bioorg. Chem. 2020, 98, 103728. [Google Scholar] [CrossRef]
- Supuran, C.T. Bacterial carbonic anhydrases as drug targets: Toward novel antibiotics? Front. Pharmacol. 2011, 2, 34. [Google Scholar]
- Tarko, L.; Supuran, C.T. QSAR studies of sulfamate and sulfamide inhibitors targeting human carbonic anhydrase isozymes I, II, IX and XII. Bioorg. Med. Chem. 2013, 2, 1404–1409. [Google Scholar] [CrossRef]
- Nocentini, A.; Angeli, A.; Carta, F.; Winum, J.Y.; Zalubovskis, R.; Carradori, S.; Capasso, C.; Donald, W.A.; Supuran, C.T. Reconsidering anion inhibitors in the general context of drug design studies of modulators of activity of the classical enzyme carbonic anhydrase. J. Enzyme Inhib. Med. Chem. 2021, 36, 561–580. [Google Scholar]
- Santos, J.L.; Yamasaki, P.R.; Chin, C.M.; Takashi, C.H.; Pavan, F.R.; Leite, C.Q.F. Synthesis and in vitro anti Mycobacterium tuberculosis activity of a series of phthalimide derivatives. Bioorg. Med. Chem. 2009, 17, 3795–3799. [Google Scholar] [CrossRef]
- Khalifah, R.G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar]
- Torella, D.; Ellison, G.M.; Torella, M.; Vicinanza, C.; Aquila, I.; Iaconetti, C.; Scalise, M.; Marino, F.; Henning, B.J.; Lewis, F.C.; et al. Carbonic Anhydrase Activation Is Associated with Worsened Pathological Remodeling in Human Ischemic Diabetic Cardiomyopathy. J. Am. Heart Assoc. 2014, 3, e000434. [Google Scholar] [CrossRef] [Green Version]
- Leaf, D.E.; Goldfarb, D.S. Mechanisms of action of acetazolamide in the prophylaxis and treatment of acute mountain sickness. J. Appl. Physiol. 2007, 102, 1313–1322. [Google Scholar] [CrossRef]
- Ferrari, A.; Tiraferri, I.; Neri, L.; Sternieri, E. Clinical pharmacology of topiramate in migraine prevention. Expert Opin. Drug. Metab. Toxicol. 2011, 7, 1169–1181. [Google Scholar] [CrossRef]
- Grover, S.; Apushkin, M.; Fishman, G. Topical dorzolamide for the treatment of cystoid macular edema in patients with retinitis pigmentosa. Am. J. Ophthalmol. 2006, 141, 850–858. [Google Scholar] [CrossRef]
- March, W.F.; Ochsner, K.I. The long-term safety and efficacy of brinzolamide 1.0% (Azopt) in patients with primary open-angle glaucoma or ocular hypertension. The brinzolamide long-term therapy study group. Am. J. Ophthalmol. 2000, 129, 136–143. [Google Scholar] [CrossRef]
- Canepa, E.; Debure, L.; Vazquez-Torres, R.; Fossati, S. Carbonic anhydrase inhibition ameliorates Aβ-induced neurovascular dysfunction in vivo. Alzheimer Dement. 2020, 16, e044221. [Google Scholar] [CrossRef]
- Deniz, S.; Uysal, T.K.; Capasso, C.; Supuran, C.T.; Guler, O.O. Is carbonic anhydrase inhibition useful as a complementary therapy of Covid-19 infection? J. Enzyme Inhib. Med. Chem. 2021, 36, 1230. [Google Scholar] [CrossRef] [PubMed]
- Sethi, K.K.; Verma, S.; Prasanthi, N.; Annapurna, M.M. Synthesis, neurotoxicity and anticonvulsant study of some benzothiazole analogs. Lett. Drug Des. Discov. 2011, 8, 774–777. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending force field coverage for drug-like small molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Schneider, G.; Fechner, U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug. Discov. 2005, 4, 649–663. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| hCA | Distribution | Localization | Disease Involved | Offtargets | Kcat/Km | Affinity for Sulphonamides |
|---|---|---|---|---|---|---|
| hCA I | erythrocytes, gastrointestinal (GI) tract, eyes | cytosol | retinal/cerebral oedema | All other isoforms | 5.0 × 107 | medium |
| hCA II | eyes, erythrocytes, bone osteoclasts, GI tract, kidneys, testis, lung, brain | cytosol | glaucoma | hCA I | 1.5 × 108 | very high |
| oedema | All other isoforms | |||||
| epilepsy | unknown | |||||
| altitude sickness | unknown | |||||
| cancer | unknown | |||||
| hCA IX | GI mucosa, tumours | transmembrane | cancer | hCA I and II | 5.5 × 107 | high |
| hCA XII | renal, germinal epithelia, intestinal mucosa, eyes, tumours | transmembrane | cancer | hCA I and II All other isoforms except hCA II | 3.5 × 107 | very high |
| glaucoma | ||||||
| retinopathies |
| Comp. Code | Structures/Name | Ki (nM) * | Selectivity Ratio # | ||||
|---|---|---|---|---|---|---|---|
| hCA I | hCA II | hCA IX | hCA XII | A | B | ||
| 1 |  | 49 ± 3.2 | 13 ± 0.9 | 7766 ± 145 | 66 ± 5 | 0.002 | 0.196 |
| 2 |  | 309 ± 23 | 23 ± 1.8 | 7417 ± 348 | 69 ± 4.5 | 0.003 | 0.333 |
| 3 |  | 258 ± 12 | 742 ± 33 | 933 ± 56 | 80 ± 7 | 0.795 | 9.275 |
| 4 |  | 463 ± 29 | 44 ± 1.9 | 4034 ± 237 | 87 ± 6 | 0.011 | 0.506 |
| 5 |  | 400 ± 31 | 4.6 ± 0.3 | 4898 ± 300 | 75 ± 4 | 0.0 | 0.061 |
| 6 |  | 332 ± 17 | 7.1 ± 0.4 | 9.7 ± 0.8 | 33 ± 2 | 0.732 | 0.215 |
| 7 |  | 427 ± 31 | 5.2 ± 0.4 | 103 ± 5 | 300 ± 18 | 0.05 | 0.017 |
| 8 |  | 326 ± 26 | 2.9 ± 0.2 | 53 ± 3 | 226 ± 13 | 0.055 | 0.013 |
| 9 |  | 444 ± 35 | 27.7 ± 1.5 | 498 ± 34 | 316 ± 28 | 0.056 | 0.088 |
| 10 |  | >10,000 | 4515 ± 325 | 559 ± 26 | 309 ± 24 | 8077 | 14.616 |
| 11 |  | 368 ± 15 | 3.4 ± 0.2 | 88 ± 7 | 14 ± 1 | 0.039 | 0.243 |
| 12 |  | 159 ± 12 | 4.9 ± 0.3 | 49 ± 0.4 | 22 ± 1.7 | 0.1 | 0.223 |
| 13 |  | 281±15 | 2.4±0.1 | 40±1.5 | 99±3.5 | 0.06 | 0.024 |
| AAZ |  | 250 ± 14 | 12 ± 0.8 | 25 ± 1.1 | 5.7 ± 0.2 | 0.5 | 2.10 |
| BRZ |  | 45,000 ± 3000 | 3 ± 0.1 | 37 ± 1.4 | 3 ± 0.2 | 0.1 | 1 |
| DZA |  | 50,000 ± 4500 | 9 ± 0.4 | 52 ± 1.1 | 3.5 ± 0.2 | 0.2 | 2.57 |
| EZA |  | 25 ± 1.8 | 8 ± 0.4 | 34 ± 1.9 | 22 ± 0.9 | 0.2 | 0.36 |
| IND |  | 31 ± 1.6 | 15 ± 0.7 | 24 ± 1.4 | 3.4 ± 0.2 | 0.6 | 4.41 |
| MZA |  | 50 ± 3 | 14 ± 0.9 | 27 ± 1.5 | 3.4 ± 0.1 | 0.5 | 4.11 |
| SLP |  | 1200 ± 69 | 40 ± 2.1 | 46 ± 3.2 | 3.9 ± 0.2 | 0.9 | 10.25 |
| TPM |  | 250 ± 16 | 10 ± 0.6 | 58 ± 3.5 | 3.8 ± 0.2 | 0.2 | 2.63 |
| ZNS |  | 56 ± 1.5 | 35 ± 1.2 | 5.1 ± 0.3 | 11,000 ± 680 | 6.9 | 0.003 |
| Comp. Code | Docking Score (Glide XP) | Comp. Code | Docking Score (Glide XP) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1AZM | 1ZFQ | 3IAI | 1JD0 | 1AZM | 1ZFQ | 3IAI | 1JD0 | ||
| 1 | −8.124 | −9.218 | −7.749 | −4.209 | 12 | −5.886 | −4.679 | −5.134 | −3.626 |
| 2 | −5.822 | −5.079 | −5.264 | −4.948 | 13 | −6.072 | −4.654 | −4.595 | −3.647 |
| 3 | −4.331 | −4.148 | −3.922 | −4.013 | AAZ | −4.8 | −3.8 | −4.4 | −4.1 |
| 4 | −4.156 | −3.94 | −4.067 | −4.257 | BRZ | −5.5 | −5.3 | −4.9 | −5.1 |
| 5 | −5.760 | −4.542 | −5.293 | −4.159 | DZA | −5.0 | −4.5 | −4.8 | −4.4 |
| 6 | −5.827 | −4.597 | −4.771 | −4.533 | EZA | −5.7 | −4.1 | −5.1 | −4.2 |
| 7 | −5.314 | −4.599 | −5.118 | −3.241 | IND | −6.5 | −5.0 | −6.8 | −5.2 |
| 8 | −6.118 | −4.661 | −4.900 | −4.870 | MZA | −4.9 | −3.8 | −4.9 | −3.5 |
| 9 | −5.922 | −5.149 | −4.951 | −4.467 | SLP | −6.5 | −6.0 | −5.7 | −4.9 |
| 10 | −4.138 | −3.853 | −4.842 | −1.699 | TPM | −6.1 | −4.7 | −5.0 | −4.4 |
| 11 | −5.999 | −4.713 | −5.392 | −3.343 | ZNS | −5.7 | −3.7 | −4.7 | −4.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sethi, K.K.; Mishra, K.A.; Verma, S.M.; Vullo, D.; Carta, F.; Supuran, C.T. Synthesis and Human Carbonic Anhydrase I, II, IX, and XII Inhibition Studies of Sulphonamides Incorporating Mono-, Bi- and Tricyclic Imide Moieties. Pharmaceuticals 2021, 14, 693. https://doi.org/10.3390/ph14070693
Sethi KK, Mishra KA, Verma SM, Vullo D, Carta F, Supuran CT. Synthesis and Human Carbonic Anhydrase I, II, IX, and XII Inhibition Studies of Sulphonamides Incorporating Mono-, Bi- and Tricyclic Imide Moieties. Pharmaceuticals. 2021; 14(7):693. https://doi.org/10.3390/ph14070693
Chicago/Turabian StyleSethi, Kalyan K., KM Abha Mishra, Saurabh M. Verma, Daniela Vullo, Fabrizio Carta, and Claudiu T. Supuran. 2021. "Synthesis and Human Carbonic Anhydrase I, II, IX, and XII Inhibition Studies of Sulphonamides Incorporating Mono-, Bi- and Tricyclic Imide Moieties" Pharmaceuticals 14, no. 7: 693. https://doi.org/10.3390/ph14070693
APA StyleSethi, K. K., Mishra, K. A., Verma, S. M., Vullo, D., Carta, F., & Supuran, C. T. (2021). Synthesis and Human Carbonic Anhydrase I, II, IX, and XII Inhibition Studies of Sulphonamides Incorporating Mono-, Bi- and Tricyclic Imide Moieties. Pharmaceuticals, 14(7), 693. https://doi.org/10.3390/ph14070693