
The Psychonauts’ Benzodiazepines; Quantitative Structure-Activity Relationship (QSAR) Analysis and Docking Prediction of Their Biological Activity

 , ,
, ,  ,
,  ,
, 
Abstract
:1. Introduction
2. Results
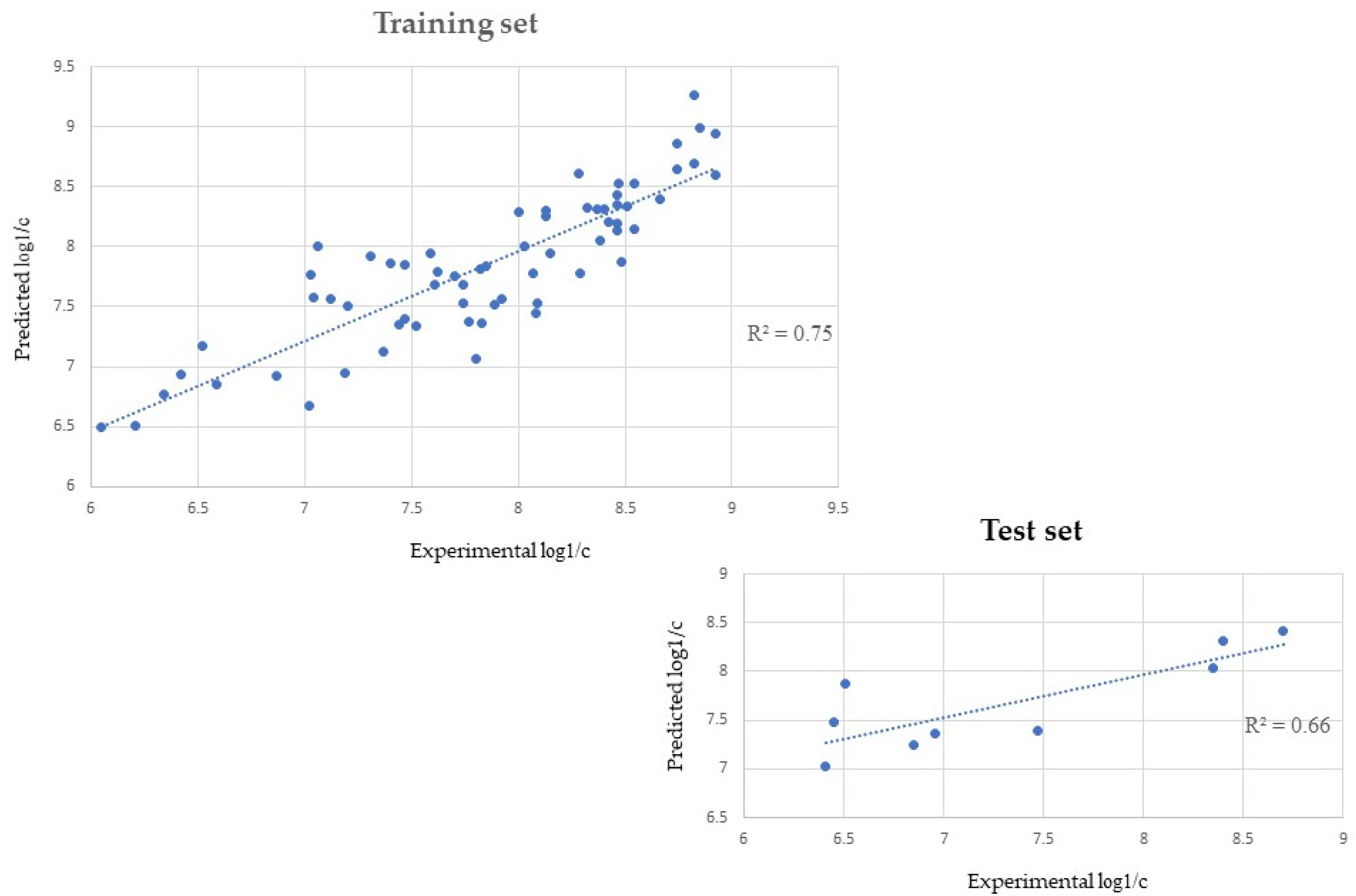
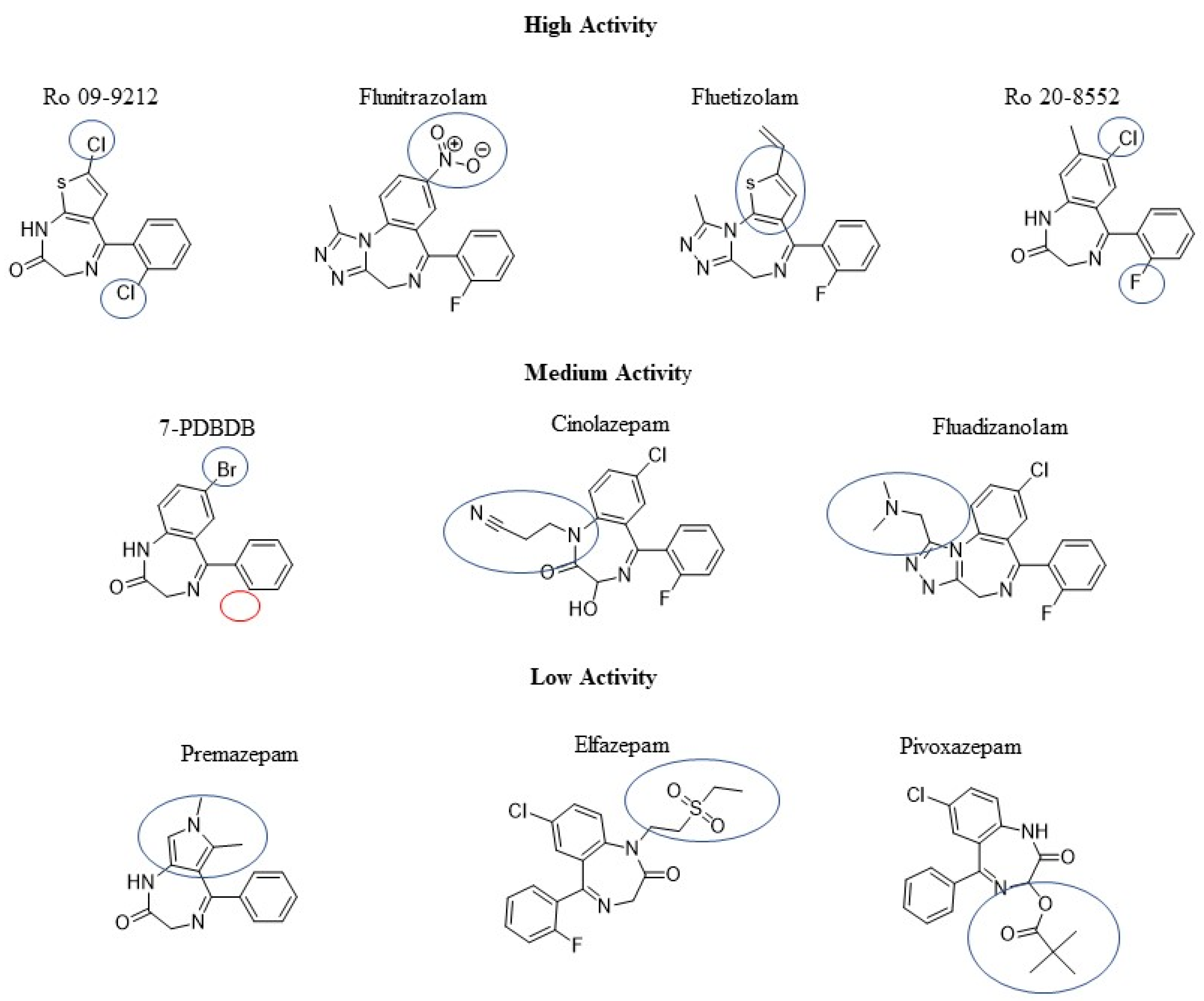
2.1. QSAR
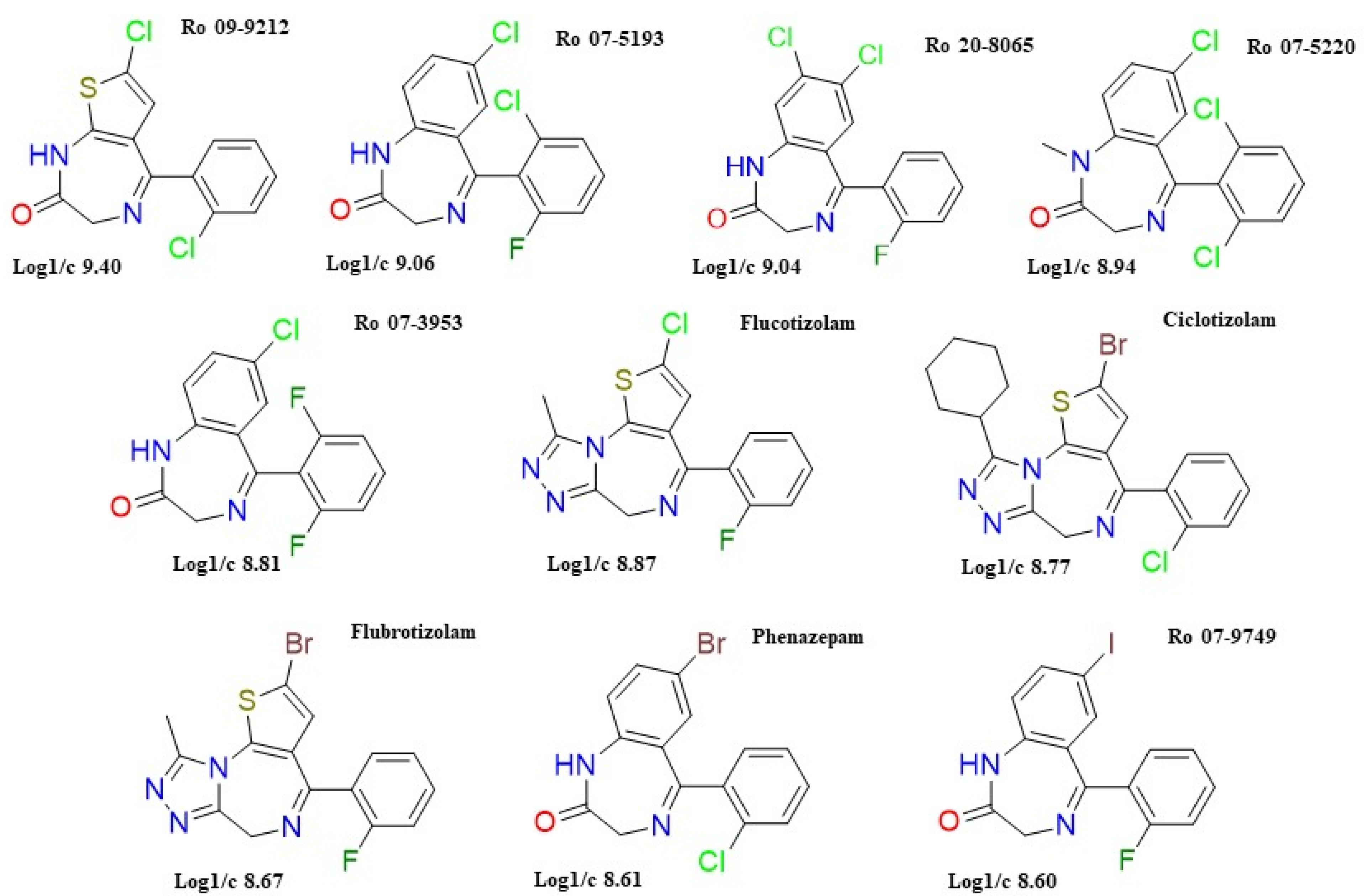
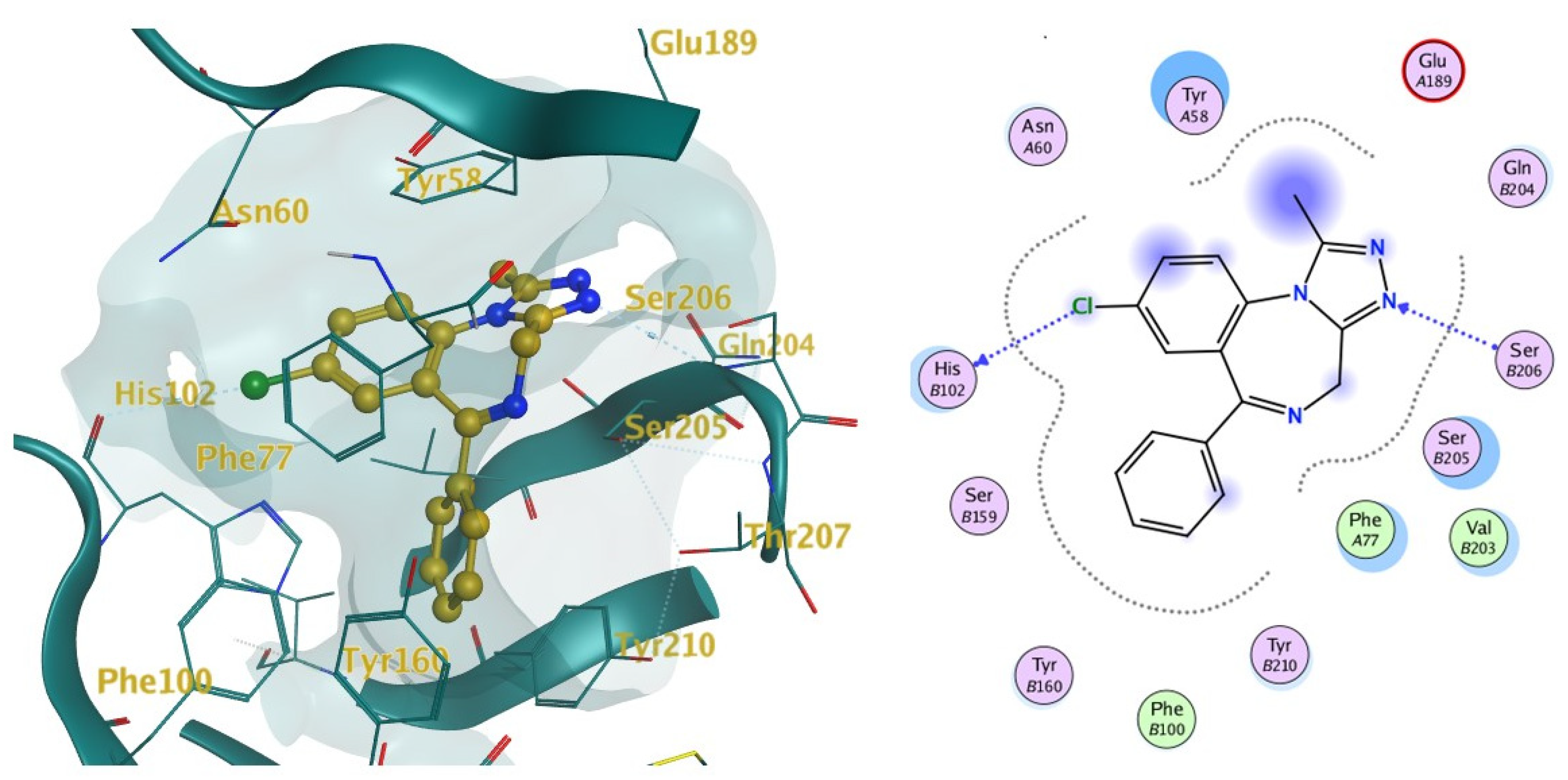
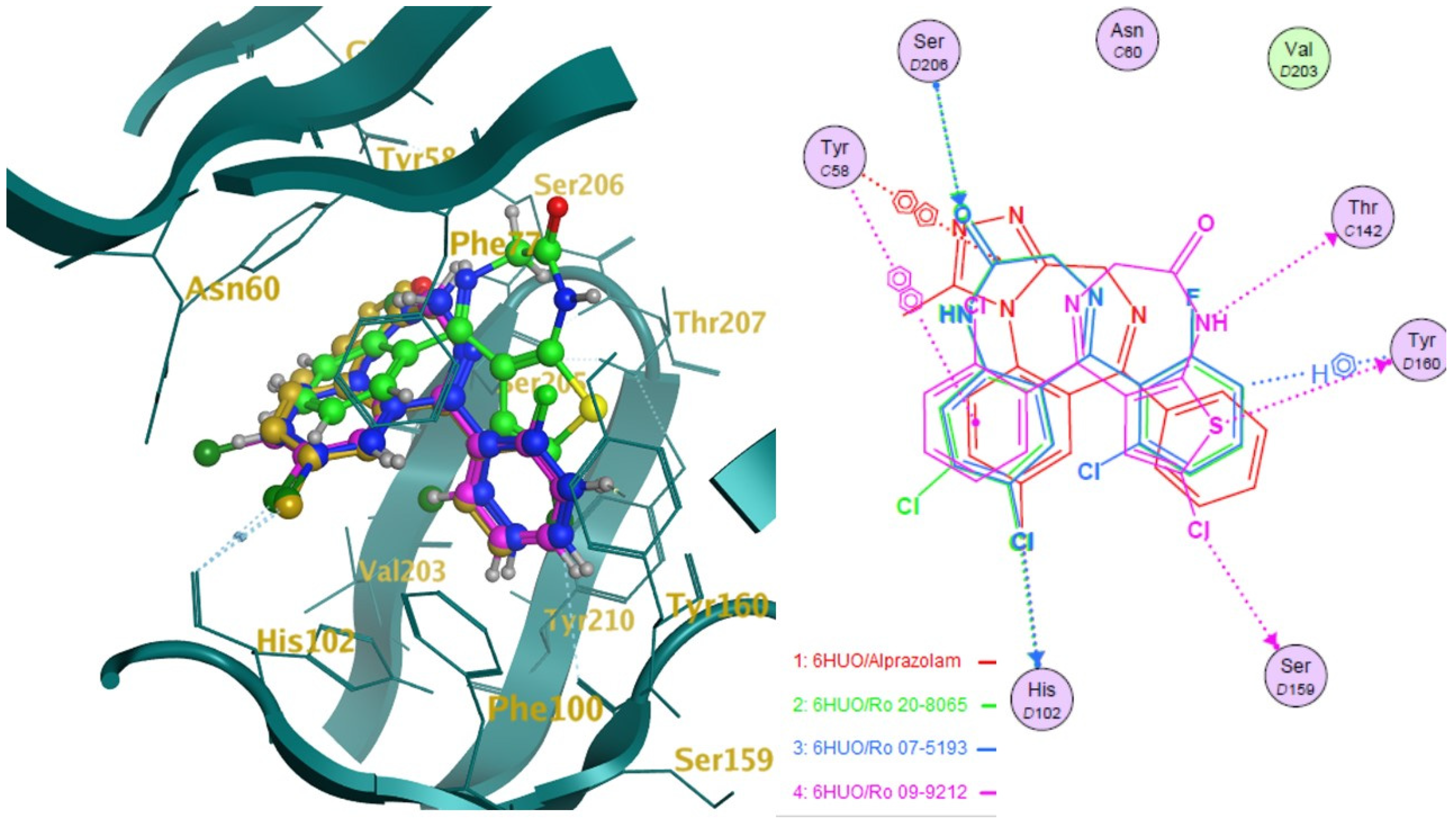
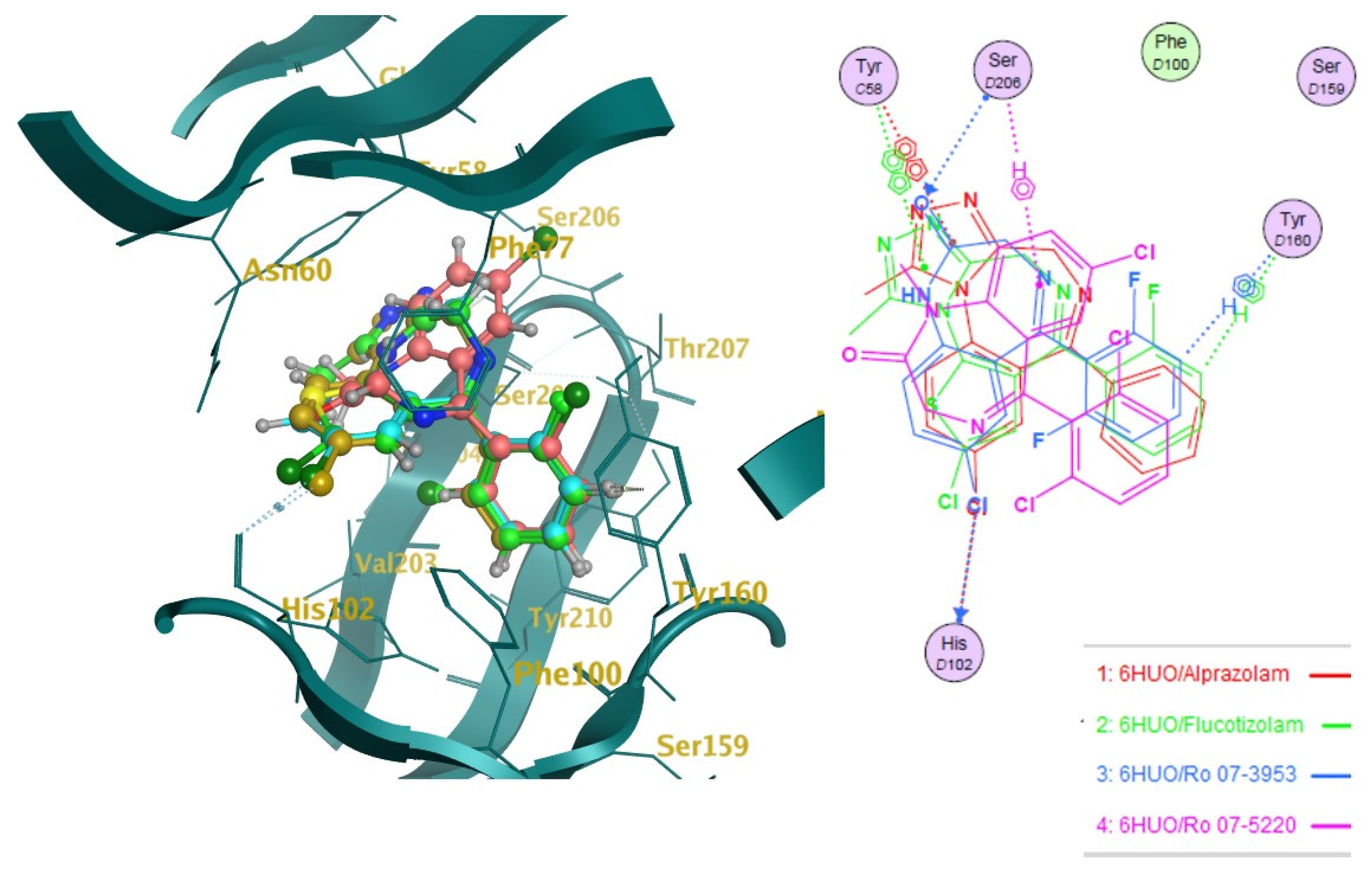
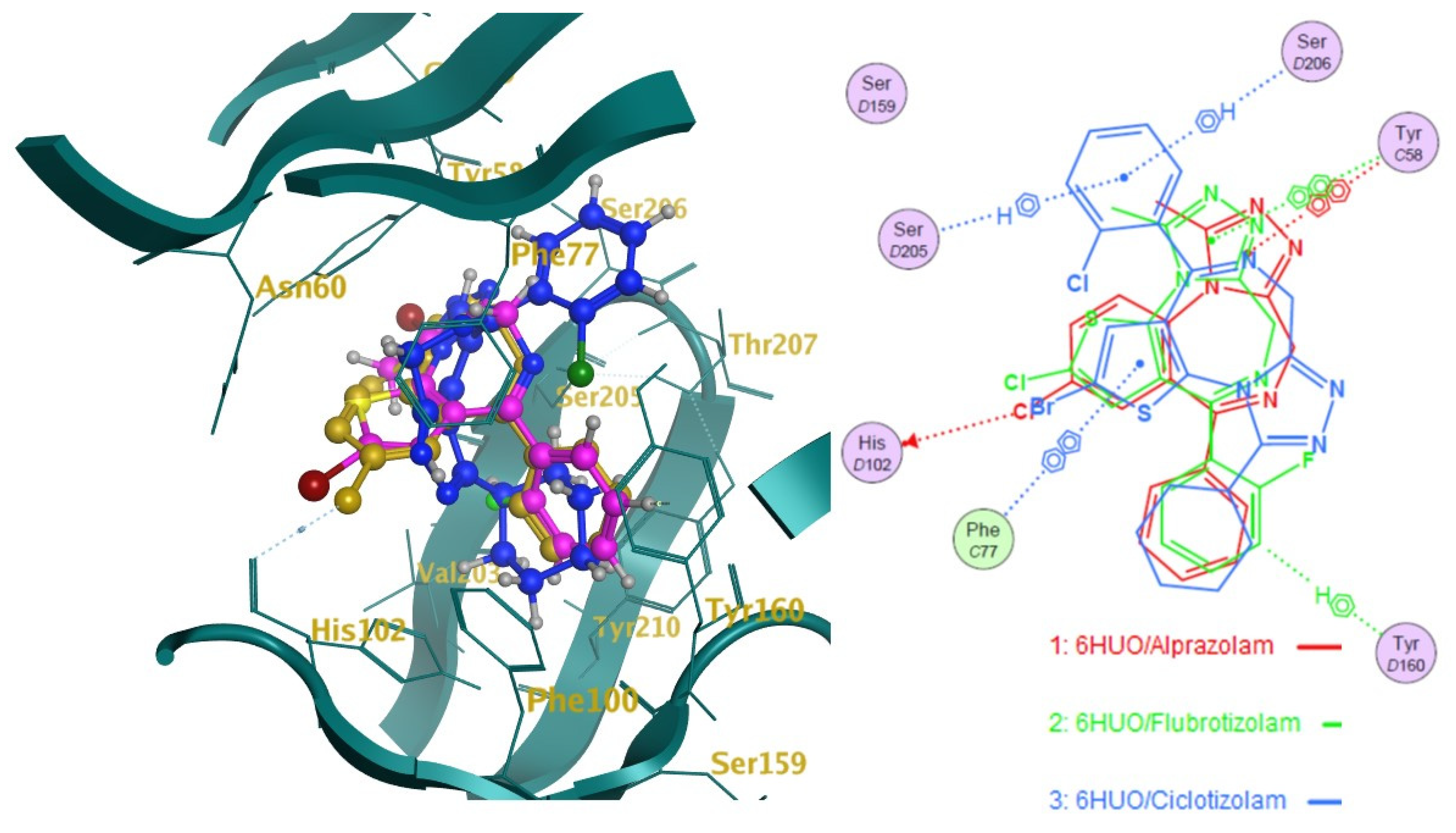
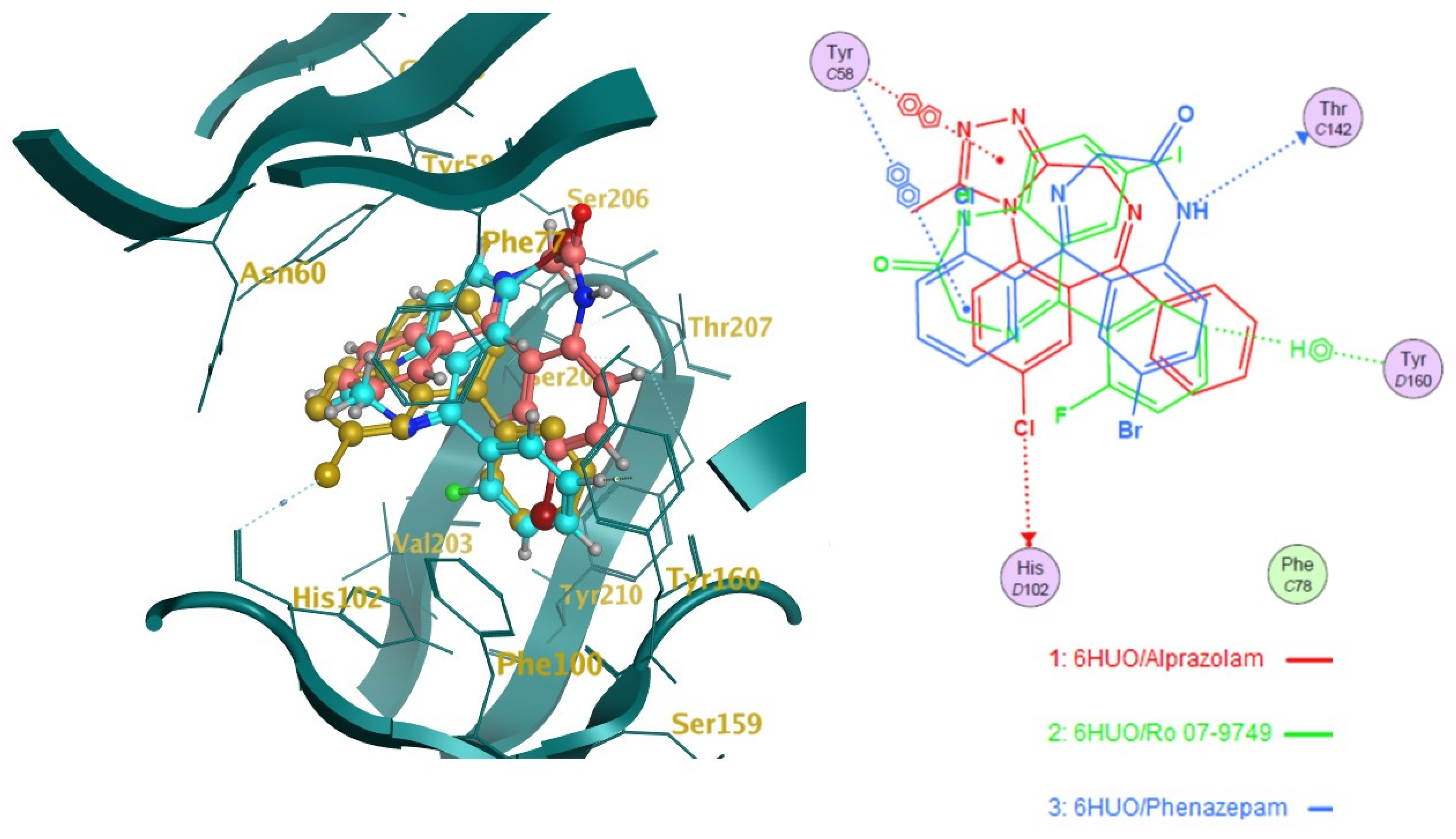
2.2. Docking
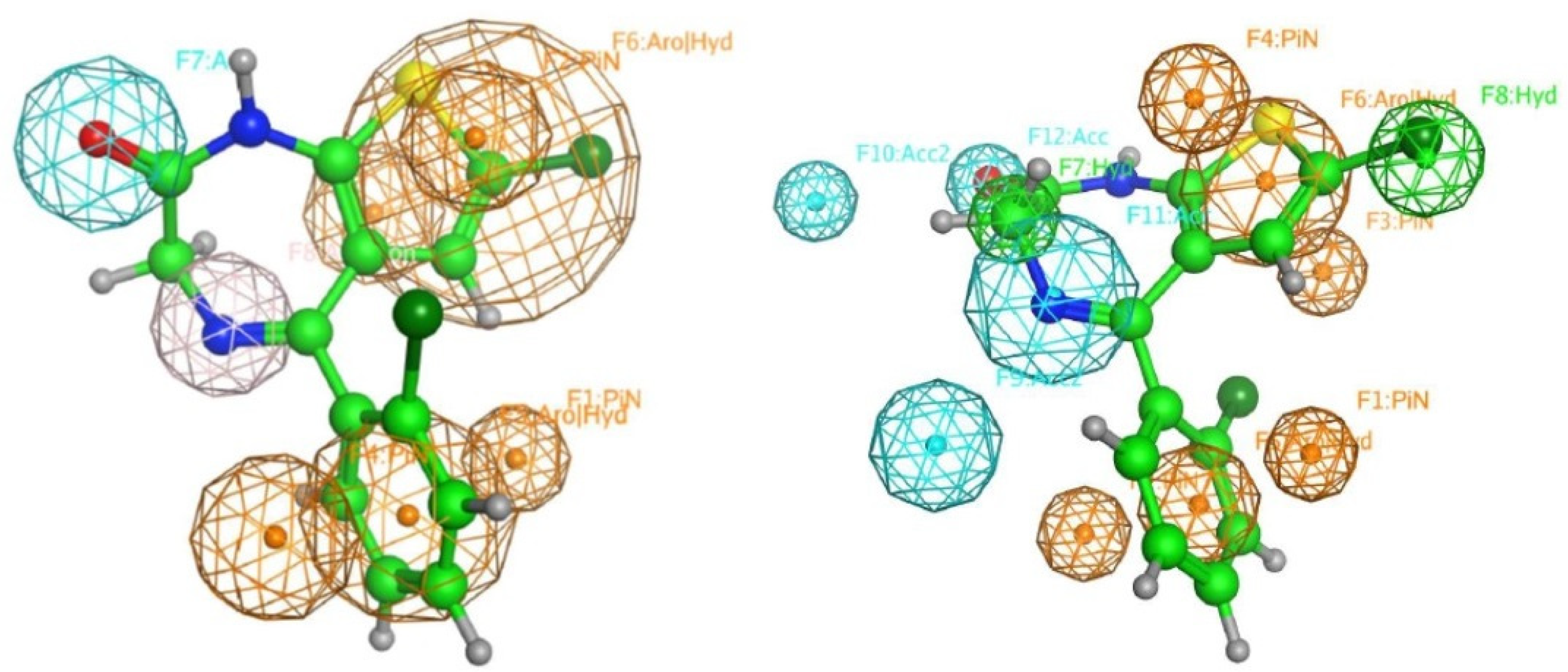
2.3. Pharmacophore
3. Discussion
Limitations
4. Methods
4.1. Identification of Molecules
4.2. Computational Models
4.2.1. QSAR
4.2.2. Docking
4.2.3. Pharmacophore Mapping
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- EMCDDA. Benzodiazepines Drug Profile. Available online: http://www.emcdda.europa.eu/publications/drug-profiles/benzodiazepines_en (accessed on 12 April 2020).
- UNODC. The International Drug Control Conventions; UNODC: New York, NY, USA, 2013. [Google Scholar]
- UNODC. Schedules of the Convention on Psychotropic Substances of 1971, as at 3 November 2020; UNODC: New York, NY, USA, 2020. [Google Scholar]
- EMCDDA. Perspectives on Drugs: The Misuse of Benzodiazepines among High-Risk Opioid Users in Europe; EMCDDA: Lisbon, Portugal, 2018. [Google Scholar]
- EMCDDA. New Psychoactive Substances: Global Markets, Glocal Threats and the COVID-19 Pandemic—An Update from the EU Early Warning System; EMCDDA: Lisbon, Portugal, Lisbon, 2020. [Google Scholar]
- Griffin, C.E.; Kaye, A.M.; Rivera Bueno, F.; Kaye, A.D. Benzodiazepine pharmacology and central nervous system-mediated effects. Ochsner J. 2013, 13, 214–223. [Google Scholar]
- Kelly, M.D.; Smith, A.; Banks, G.; Wingrove, P.; Whiting, P.W.; Atack, J.; Seabrook, G.R.; Maubach, K.A. Role of the histidine residue at position 105 in the human α5 containing GABA A receptor on the affinity and efficacy of benzodiazepine site ligands. Br. J. Pharmacol. 2002, 135, 248–256. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.; Bateson, A.N.; Dunn, S.M.J. Structural Requirements for Ligand Interactions at the Benzodiazepine Recognition Site of the GABAA Receptor. J. Neurochem. 2002, 70, 2188–2194. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.R.; Rudolph, U.; Lüscher, C. Hooked on benzodiazepines: GABAA receptor subtypes and addiction. Trends Neurosci. 2011, 34, 188–197. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, U.; Crestani, F.; Benke, D.; Brünig, I.; Benson, J.A.; Fritschy, J.M.; Martin, J.R.; Bluethmann, H.; Möhler, H. Benzodiazepine actions mediated by specific γ-aminobutyric acid(A) receptor subtypes. Nature 1999, 401, 796–800. [Google Scholar] [CrossRef]
- Tan, K.R.; Brown, M.; Labouébe, G.; Yvon, C.; Creton, C.; Fritschy, J.M.; Rudolph, U.; Lüscher, C. Neural bases for addictive properties of benzodiazepines. Nature 2010, 463, 769–774. [Google Scholar] [CrossRef]
- Manchester, K.R.; Lomas, E.C.; Waters, L.; Dempsey, F.C.; Maskell, P.D. The emergence of new psychoactive substance (NPS) benzodiazepines: A review. Drug Test. Anal. 2018, 10, 37–53. [Google Scholar] [CrossRef] [Green Version]
- LSS/RAB/DPA/UNODC. New Psychoactive Substances: Overview of Trends, Challenges and Legal Approaches; UNODC: Vienna, Austria, 2016. [Google Scholar]
- Drug Enforcement Administration. Benzodiazepines; Drug Enforcement Administration: Springfield, VA, USA, 2019. [Google Scholar]
- Orsolini, L.; Corkery, J.M.; Chiappini, S.; Guirguis, A.; Vento, A.; De Berardis, D.; Papanti, D.; Schifano, F. “New/Designer Benzodiazepines”: An analysis of the literature and psychonauts’ trip reports. Curr. Neuropharmacol. 2020, 18, 809–837. [Google Scholar] [CrossRef]
- EMCDDA. New Benzodiazepines in Europe—A Review; EMCDDA: Lisbon, Portugal, 2021. [Google Scholar]
- EMCDDA. European Drug Report 2021: Trends and Developments; EMCDDA: Lisbon, Portugal, 2021; Available online: www.emcdda.europa.eu (accessed on 1 July 2021).
- UNODC. Drug Supply World Drug Report 2020; UNODC: Vienna, Austria, 2020. [Google Scholar]
- UNODC. Booklet 2—Global Overview of Drug Demand and Drug Supply; UNODC: Vienna, Austria, 2021. [Google Scholar]
- ACMD. Novel Benzodiazepines A Review of the Evidence of Use and Harms of Novel Benzodiazepines; ACMD: London, UK, 2020. [Google Scholar]
- UNODC. Non-Medical Use of Benzodiazepines: A Growing Threat to Public Health ? UNODC: Vienna, Austria, 2017; Volume 18. [Google Scholar]
- Carpenter, J.E.; Murray, B.P.; Dunkley, C.; Kazzi, Z.N.; Gittinger, M.H. Designer benzodiazepines: A report of exposures recorded in the National Poison Data System, 2014–2017. Clin. Toxicol. 2019, 57, 282–286. [Google Scholar] [CrossRef]
- UNODC. Current NPS Threaths Volume II; UNODC: Vienna, Austria, 2020. [Google Scholar]
- UNODC. Current NPS Threats Volume III; UNODC: Vienna, Austria, 2020. [Google Scholar]
- Greenblatt, H.K.; Greenblatt, D.J. Designer Benzodiazepines: A Review of Published Data and Public Health Significance. Clin. Pharmacol. Drug Dev. 2019, 8, 266–269. [Google Scholar] [CrossRef]
- Brunetti, P.; Giorgetti, R.; Tagliabracci, A.; Huestis, M.A.; Busardò, F.P. Designer Benzodiazepines: A Review of Toxicology and Public Health Risks. Pharmaceuticals 2021, 14, 560. [Google Scholar] [CrossRef]
- World Health Organization. Summary Assessment and Recommendations of the 43rd Summary Assessment and Recommendations of the 43rd Expert Committee on Drug Dependence; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Public Health England. Evidence of Harm from Illicit or Fake Benzodiazepines; Public Health England: London, UK, 2020. [Google Scholar]
- Schifano, F.; Napoletano, F.; Arillotta, D.; Zangani, C.; Gilgar, L.; Guirguis, A.; Corkery, J.M.; Vento, A. The clinical challenges of synthetic cathinones. Br. J. Clin. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Zangani, C.; Schifano, F.; Napoletano, F.; Arillotta, D.; Gilgar, L.; Guirguis, A.; Corkery, J.M.; Gambini, O.; Vento, A. The e-Psychonauts’ ‘Spiced’ World; Assessment of the Synthetic Cannabinoids’ Information Available Online. Curr. Neuropharmacol. 2020, 18. [Google Scholar] [CrossRef]
- Arillotta, D.; Schifano, F.; Napoletano, F.; Zangani, C.; Gilgar, L.; Guirguis, A.; Corkery, J.M.; Aguglia, E.; Vento, A. Novel Opioids: Systematic Web Crawling within the e-Psychonauts’ Scenario. Front. Neurosci. 2020, 14, 149. [Google Scholar] [CrossRef] [Green Version]
- Catalani, V.; Arillotta, D.; Corkery, J.M.; Guirguis, A.; Vento, A.; Schifano, F. Identifying new/emerging psychoactive substances at the time of COVID-19; a web-based approach. Front. Psychiatry 2021, 11, 1638. [Google Scholar] [CrossRef]
- EMCDDA. European Database on New Drugs. Available online: https://ednd2.emcdda.europa.eu (accessed on 4 September 2020).
- UNODC. Early Warning Advisory (EWA) on New Psychoactive Substances (NPS). Available online: https://www.unodc.org/LSS/Home/NPS (accessed on 4 February 2021).
- Corazza, O.; Assi, S.; Simonato, P.; Corkery, J.; Bersani, F.S.; Demetrovics, Z.; Stair, J.; Fergus, S.; Pezzolesi, C.; Pasinetti, M.; et al. Promoting innovation and excellence to face the rapid diffusion of novel psychoactive substances in the EU: The outcomes of the ReDNet project. Hum. Psychopharmacol. 2013, 28, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Schifano, F.; Orsolini, L.; Duccio Papanti, G.; Corkery, J.M. Novel psychoactive substances of interest for psychiatry. World Psychiatry 2015, 14, 15–26. [Google Scholar] [CrossRef] [Green Version]
- El Balkhi, S.; Chaslot, M.; Picard, N.; Dulaurent, S.; Delage, M.; Mathieu, O.; Saint-Marcoux, F. Characterization and identification of eight designer benzodiazepine metabolites by incubation with human liver microsomes and analysis by a triple quadrupole mass spectrometer. Int. J. Legal Med. 2017, 131, 979–988. [Google Scholar] [CrossRef] [PubMed]
- El Balkhi, S.; Monchaud, C.; Herault, F.; Géniaux, H.; Saint-Marcoux, F. Designer benzodiazepines’ pharmacological effects and potencies: How to find the information. J. Psychopharmacol. 2020, 269881119901096. [Google Scholar] [CrossRef]
- Orsolini, L.; Francesconi, G.; Papanti, D.; Giorgetti, A.; Schifano, F. Profiling online recreational/prescription drugs’ customers and overview of drug vending virtual marketplaces. Hum. Psychopharmacol. 2015, 30, 302–318. [Google Scholar] [CrossRef] [Green Version]
- Orsolini, L.; St John-Smith, P.; McQueen, D.; Papanti, D.; Corkery, J.; Schifano, F. Evolutionary Considerations on the Emerging Subculture of the E-psychonauts and the Novel Psychoactive Substances: A Comeback to the Shamanism? Curr. Neuropharmacol. 2017, 15. [Google Scholar] [CrossRef] [Green Version]
- Schifano, F. Coming off prescribed psychotropic medications: Insights from their use as recreational drugs. Psychother. Psychosom. 2020. [Google Scholar] [CrossRef] [PubMed]
- Schifano, F.; Chiappini, S.; Catalani, V.; Napoletano, F.; Arillotta, D.; Zangani, C.; Guirguis, A.; Vento, A.E.; Bonaccorso, S.; Corkery, J.M. Psychobiological, Medical, and Psychiatric Implications of New/Novel Psychoactive Substance (NPS) Use. In Psychobiological Issues in Substance Use and Misuse; Routledge: Abingdon, UK, 2020; pp. 213–233. [Google Scholar]
- De Luca, M.A.; Castelli, M.P.; Loi, B.; Porcu, A.; Martorelli, M.; Miliano, C.; Kellett, K.; Davidson, C.; Stair, J.L.; Schifano, F.; et al. Native CB1 receptor affinity, intrinsic activity and accumbens shell dopamine stimulant properties of third generation SPICE/K2 cannabinoids: BB-22, 5F-PB-22, 5F-AKB-48 and STS-135. Neuropharmacology 2016, 105, 630–638. [Google Scholar] [CrossRef] [Green Version]
- Valerio, L.G.; Choudhuri, S. Chemoinformatics and chemical genomics: Potential utility of in silico methods. J. Appl. Toxicol. 2012, 32, 880–889. [Google Scholar] [CrossRef]
- Artemenko, A.G.; Kuz’Min, V.E.; Muratov, E.N.; Polishchuk, P.G.; Borisyuk, I.Y.; Golovenko, N.Y. Influence of the structure of substituted benzodiazepines on their pharmacokinetic properties. Pharm. Chem. J. 2009, 43, 454–462. [Google Scholar] [CrossRef]
- Waters, L.; Manchester, K.R.; Maskell, P.D.; Haegeman, C.; Haider, S. The use of a quantitative structure-activity relationship (QSAR) model to predict GABA-A receptor binding of newly emerging benzodiazepines. Sci. Justice 2018, 58, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Durdagi, S.; Kapou, A.; Kourouli, T.; Andreou, T.; Nikas, S.P.; Nahmias, V.R.; Papahatjis, D.P.; Papadopoulos, M.G.; Mavromoustakos, T. The application of 3D-QSAR studies for novel cannabinoid ligands substituted at the C1′ position of the alkyl side chain on the structural requirements for binding to cannabinoid receptors CB1 and CB2. J. Med. Chem. 2007, 50, 2875–2885. [Google Scholar] [CrossRef]
- Durdagi, S.; Papadopoulos, M.G.; Papahatjis, D.P.; Mavromoustakos, T. Combined 3D QSAR and molecular docking studies to reveal novel cannabinoid ligands with optimum binding activity. Bioorganic Med. Chem. Lett. 2007, 17, 6754–6763. [Google Scholar] [CrossRef]
- Floresta, G.; Apirakkan, O.; Rescifina, A.; Abbate, V. Discovery of high-affinity cannabinoid receptors ligands through a 3D-QSAR ushered by scaffold-hopping analysis. Molecules 2018, 23, 2183. [Google Scholar] [CrossRef] [Green Version]
- Floresta, G.; Rescifina, A.; Abbate, V. Structure-based approach for the prediction of mu-opioid binding affnity of unclassified designer fentanyl-like molecules. Int. J. Mol. Sci. 2019, 20, 2311. [Google Scholar] [CrossRef] [Green Version]
- Jia, X.; Ciallella, H.L.; Russo, D.P.; Zhao, L.; James, M.H.; Zhu, H. Construction of a Virtual Opioid Bioprofile: A Data-Driven QSAR Modeling Study to Identify New Analgesic Opioids. ACS Sustain. Chem. Eng. 2021, 9, 3909–3919. [Google Scholar] [CrossRef]
- Zhang, Z.; An, L.; Hu, W.; Xiang, Y. 3D-QSAR study of hallucinogenic phenylalkylamines by using CoMFA approach. J. Comput. Aided. Mol. Des. 2007, 21, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Alexandru, M.; Kovar, K.-A.; Vedani, A. Quasi-atomistic Receptor Surrogates for the 5-HT2A Receptor: A 3D-QSAR Study on Hallucinogenic Substances. Mol. Inform. 1999, 18, 548–560. [Google Scholar] [CrossRef]
- Wu, N.; Feng, Z.; He, X.; Kwon, W.; Wang, J.; Xie, X.Q. Insight of Captagon Abuse by Chemogenomics Knowledgebase-guided Systems Pharmacology Target Mapping Analyses. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Guariento, S.; Tonelli, M.; Espinoza, S.; Gerasimov, A.S.; Gainetdinov, R.R.; Cichero, E. Rational design, chemical synthesis and biological evaluation of novel biguanides exploring species-specificity responsiveness of TAAR1 agonists. Eur. J. Med. Chem. 2018, 146, 171–184. [Google Scholar] [CrossRef]
- Schifano, F.; Chiappini, S.; Miuli, A.; Corkery, J.M.; Scherbaum, N.; Napoletano, F.; Arillotta, D.; Zangani, C.; Catalani, V.; Vento, A.; et al. New psychoactive substances (NPS) and serotonin syndrome onset: A systematic review. Exp. Neurol. 2021, 339, 113638. [Google Scholar] [CrossRef]
- Hadjipavlou-Litina, D.; Hansch, C. Quantitative Structure‒Activity Relationships of the Benzodiazepines. A Review and Reevaluation. Chem. Rev. 1994, 94, 1483–1505. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. Mol. Divers. 2000, 5, 231–243. [Google Scholar] [CrossRef]
- Roy, K.; Das, R.N.; Ambure, P.; Aher, R.B. Be aware of error measures. Further studies on validation of predictive QSAR models. Chemom. Intell. Lab. Syst. 2016, 152, 18–33. [Google Scholar] [CrossRef]
- Masiulis, S.; Desai, R.; Uchański, T.; Serna Martin, I.; Laverty, D.; Karia, D.; Malinauskas, T.; Zivanov, J.; Pardon, E.; Kotecha, A.; et al. GABAA receptor signalling mechanisms revealed by structural pharmacology. Nature 2019, 565, 454–459. [Google Scholar] [CrossRef]
- RCSB PDB 6HUO: CryoEM Structure of Human Full-Length Heteromeric alpha1beta3gamma2L GABA(A)R in Complex with Alprazolam (Xanax), GABA and Megabody Mb38. Available online: https://www.rcsb.org/structure/6HUO (accessed on 8 March 2021).
- RCSB PDB 6HUP: CryoEM Structure of Human Full-Length alpha1beta3gamma2L GABA(A)R in Complex with Diazepam (Valium), GABA and megabody Mb38. Available online: https://www.rcsb.org/structure/6HUP (accessed on 8 March 2021).
- Chemical Computing Group ULC. Molecular Operating Enviroment (MOE), 2019.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2021. [Google Scholar]
- Consonni, V.; Ballabio, D.; Todeschini, R. Evaluation of model predictive ability by external validation techniques. J. Chemom. 2010, 24, 194–201. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Alexander, D.L.J.; Tropsha, A.; Winkler, D.A. Beware of R2: Simple, Unambiguous Assessment of the Prediction Accuracy of QSAR and QSPR Models. J. Chem. Inf. Model. 2015, 55, 1316–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worachartcheewan, A.; Toropova, A.P.; Toropov, A.A.; Siriwong, S.; Prapojanasomboon, J.; Prachayasittikul, V.; Nantasenamat, C. Quantitative Structure-activity Relationship Study of Betulinic Acid Derivatives Against HIV using SMILES-based Descriptors. Curr. Comput. Aided. Drug Des. 2018, 14, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.P.; Hansch, C. An approach toward the problem of outliers in QSAR. Bioorganic Med. Chem. 2005, 13, 4597–4621. [Google Scholar] [CrossRef]
- Furusjö, E.; Svenson, A.; Rahmberg, M.; Andersson, M. The importance of outlier detection and training set selection for reliable environmental QSAR predictions. Chemosphere 2006, 63, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Gunja, N. The Clinical and Forensic Toxicology of Z-drugs. J. Med. Toxicol. 2013, 9, 155–162. [Google Scholar] [CrossRef] [Green Version]
- Thakur, M.; Thakur, A.; Sudele, P. Comparative QSAR and QPAR study of benzodiazepines. Indian J. Chem. 2004, 43, 976–982. [Google Scholar]
- Maddalena, D.J.; Johnston, G.A.R. Prediction of Receptor Properties and Binding Affinity of Ligands to Benzodiazepine/Gm& Receptors Using Artificial Neural Networks. J. Med. Chem 1995, 38, 715–724. [Google Scholar]
- Wildman, S.A.; Crippen, G.M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Hall, L.H.; Kier, L.B. The Molecular Connectivity Chi Indexes and Kappa Shape Indexes in Structure-Property Modeling; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 1991; pp. 367–422. [Google Scholar]
- Wang, Y.; Liu, H.; Fan, Y.; Chen, X.; Yang, Y.; Zhu, L.; Zhao, J.; Chen, Y.; Zhang, Y. In Silico Prediction of Human Intravenous Pharmacokinetic Parameters with Improved Accuracy. J. Chem. Inf. Model. 2019, 59, 3968–3980. [Google Scholar] [CrossRef]
- UNODC. EWA March 2020- Recently Scheduled Benzodiazepines Flualprazolam and Etizolam Associated with Multiple Post-mortem and DUID Cases in UNODC EWA. Available online: https://www.unodc.org/LSS/Announcement/Details/ad0c279b-b4d4-49f3-b638-cd87755d2d42 (accessed on 10 April 2020).
- Moosmann, B.; Auwärter, V. Designer benzodiazepines: Another class of new psychoactive substances. In Handbook of Experimental Pharmacology; Springer: New York, NY, USA, 2018; Volume 252, pp. 383–410. [Google Scholar]
- WHO. Phenazepam Pre-Review Report Agenda Item 5.8 Expert Committee on Drug Dependence Thirty-seventh Meeting; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- UNODC. WHO: World Health Organization Recommends 8 NPS for Scheduling. Available online: https://www.unodc.org/LSS/Announcement/Details/0d68dc5f-a17e-4edc-83f0-6705aca1e5b3 (accessed on 24 February 2021).
- Chen, Y.C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Sigel, E.; Ernst, M. The Benzodiazepine Binding Sites of GABAA Receptors. Trends Pharmacol. Sci. 2018, 39, 659–671. [Google Scholar] [CrossRef]
- Sigel, E.; Luscher, B.P. A Closer Look at the High Affinity Benzodiazepine Binding Site on GABAA Receptors. Curr. Top. Med. Chem. 2012, 11, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Golbraikh, A.; Muratov, E.; Fourches, D.; Tropsha, A. Data set modelability by QSAR. J. Chem. Inf. Model. 2014, 54, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but verify: On the importance of chemical structure curation in cheminformatics and QSAR modeling research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef]
- Orsolini, L.; Papanti, D.; Corkery, J.; Schifano, F. An insight into the deep web; why it matters for addiction psychiatry? Hum. Psychopharmacol. 2017, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schifano, F.; Napoletano, F.; Chiappini, S.; Guirguis, A.; Corkery, J.M.; Bonaccorso, S.; Ricciardi, A.; Scherbaum, N.; Vento, A. New/emerging psychoactive substances and associated psychopathological consequences. Psychol. Med. 2019, 51, 30–42. [Google Scholar] [CrossRef] [Green Version]
- Heller, S.; McNaught, A.; Stein, S.; Tchekhovskoi, D.; Pletnev, I. InChI - The worldwide chemical structure identifier standard. J. Cheminform. 2013, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Mcnaught, A. The IUPAC International Chemical Identifier: InChl-A New Standard for Molecular Informatics. Chem. Int. 2006. [Google Scholar] [CrossRef]
- Gad, S.C. QSAR. In Encyclopedia of Toxicology: Third Edition; Wexler, P., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 1–9. ISBN 9780123864543. [Google Scholar]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. cheminformatics 2015, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [Green Version]
- Beebe, R.K.; Pell, J.R.; Seasholts, M.B. Chemometrics: A Practical Guide; Wiley: Hoboken, NJ, USA, 1998; ISBN 978-0-471-12451-1. [Google Scholar]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- RCSB PDB: Homepage. Available online: https://www.rcsb.org/ (accessed on 4 February 2021).
- EMDataResource EMDR: EMD-0283. Available online: https://www.emdataresource.org/EMD-0283 (accessed on 21 May 2021).
- EMDataResource EMDR: EMD-0282. Available online: https://www.emdataresource.org/EMD-0282 (accessed on 21 May 2021).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Description | RI |
|---|---|---|
| h_log_pbo | Sum of log (1 + pi bond order) for all bonds | 0.65 |
| KierFlex | Kier molecular flexibility index: (KierA1) (KierA2)/n | 0.68 |
| Q_VSA_HYD | Total hydrophobic van der Waals surface area | 1.00 |
| SlogP_VSA7 | approximate accessible van der Waals surface area contribution to logP(o/w) | 0.25 |
| vsa_pol | Approximation to the sum of VDW surface areas (Å2) of polar atoms | 0.29 |
| KierFlex | h_log_pbo | Q_VSA_HYD | vsa_pol | SlogP_VSA7 | |
|---|---|---|---|---|---|
| KierFlex | 1.00 | ||||
| h_log_pbo | 0.13 | 1.00 | |||
| Q_VSA_HYD | 0.65 | 0.30 | 1.00 | ||
| vsa_pol | 0.07 | 0.11 | −0.12 | 1.00 | |
| SlogP_VSA7 | −0.25 | 0.53 | −0.09 | 0.07 | 1.00 |
| Mol. | Pred. log1/c | S 6HUP (kcal/mol) | S 6HUO (Kcal/mol) |
|---|---|---|---|
| Ro 09-9212 | 9.40 | –6.7 | –6.4 |
| Ro 07-5193 | 9.06 | –6.7 | –6.7 |
| Ro 20-8065 | 9.04 | –6.8 | –6.6 |
| Ro 07-5220 | 8.95 | –6.7 | –6.6 |
| Ro 07-3953 | 8.81 | –6.4 | –6.5 |
| Flucotizolam | 8.77 | –6.6 | –6.8 |
| Ciclotizolam | 8.77 | –7.7 | –7.4 |
| Desmethylflunitrazepam (Ro 05-4435) | 8.70 | –7.0 | –6.6 |
| Flubrotizolam | 8.67 | –6.5 | –6.5 |
| Phenazepam | 8.61 | –6.7 | –6.6 |
| Alprazolam | 7.70 | –7.0 | –7.0 |
| Diazepam | 7.50 | –6.6 | –6.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catalani, V.; Botha, M.; Corkery, J.M.; Guirguis, A.; Vento, A.; Scherbaum, N.; Schifano, F. The Psychonauts’ Benzodiazepines; Quantitative Structure-Activity Relationship (QSAR) Analysis and Docking Prediction of Their Biological Activity. Pharmaceuticals 2021, 14, 720. https://doi.org/10.3390/ph14080720
Catalani V, Botha M, Corkery JM, Guirguis A, Vento A, Scherbaum N, Schifano F. The Psychonauts’ Benzodiazepines; Quantitative Structure-Activity Relationship (QSAR) Analysis and Docking Prediction of Their Biological Activity. Pharmaceuticals. 2021; 14(8):720. https://doi.org/10.3390/ph14080720
Chicago/Turabian StyleCatalani, Valeria, Michelle Botha, John Martin Corkery, Amira Guirguis, Alessandro Vento, Norbert Scherbaum, and Fabrizio Schifano. 2021. "The Psychonauts’ Benzodiazepines; Quantitative Structure-Activity Relationship (QSAR) Analysis and Docking Prediction of Their Biological Activity" Pharmaceuticals 14, no. 8: 720. https://doi.org/10.3390/ph14080720
APA StyleCatalani, V., Botha, M., Corkery, J. M., Guirguis, A., Vento, A., Scherbaum, N., & Schifano, F. (2021). The Psychonauts’ Benzodiazepines; Quantitative Structure-Activity Relationship (QSAR) Analysis and Docking Prediction of Their Biological Activity. Pharmaceuticals, 14(8), 720. https://doi.org/10.3390/ph14080720