
New Sulfanilamide Derivatives Incorporating Heterocyclic Carboxamide Moieties as Carbonic Anhydrase Inhibitors

 , ,
, ,  , , ,
, , ,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
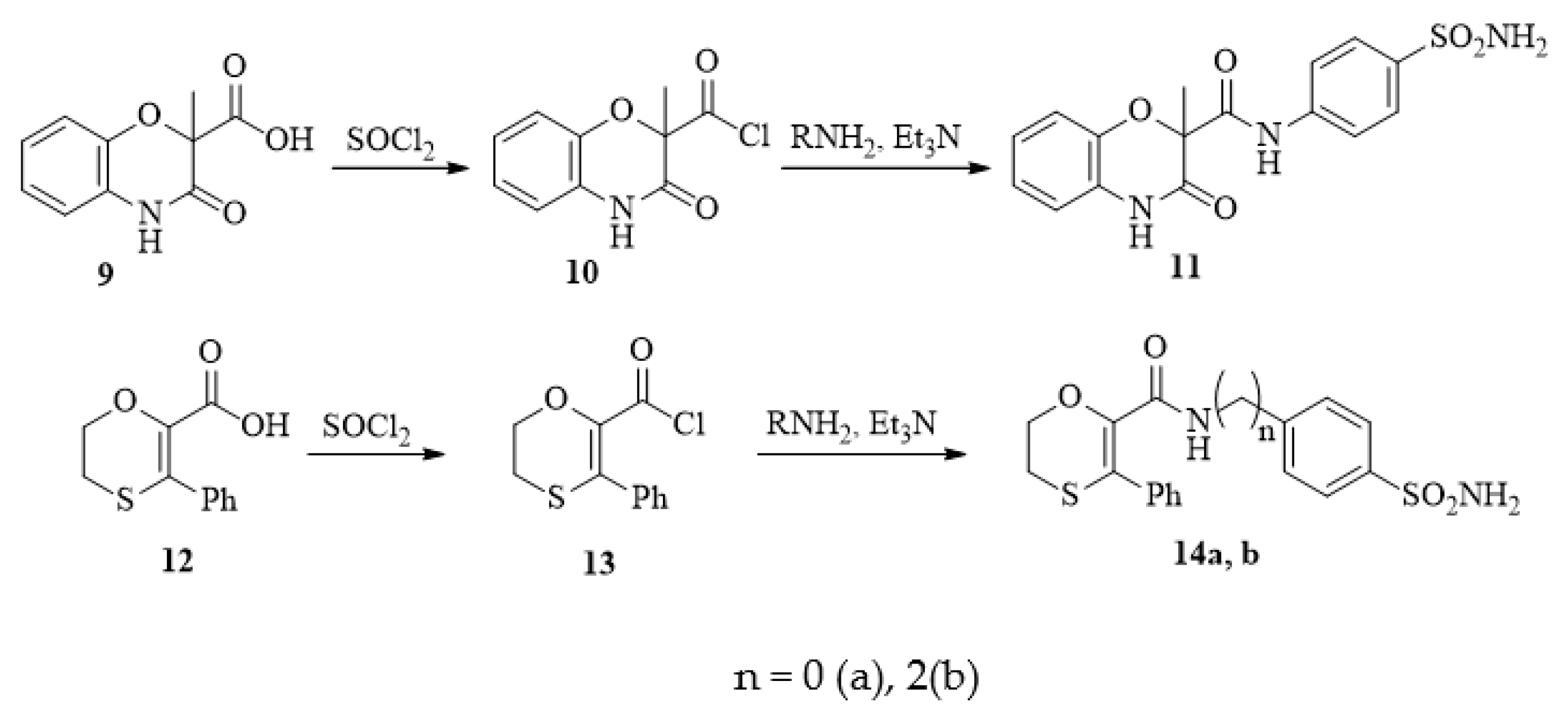
2.1. Design and Synthesis of Compounds
2.2. Carbonic Anhydrase Inhibition
2.3. Molecular Docking Studies
2.4. In Silico Prediction Studies
3. Materials and Methods
3.1. General
3.2. General Procedure for the Synthesis of 4-Alkyl-4H-furo[3,2-b]pyrrole-5-carboxylic Acid 3a,c-f and 4-Alkyl-4H-thieno[3,2-b]pyrrole-5-carboxylic Acid 4a-d, f
3.3. General Procedure for the Synthesis of 4-Alkyl-4H-furo[3,2-b]pyrrole-5-carbonyl Chloride 5a,c-f and 4-Alkyl-4H-thieno[3,2-b]pyrrole-5-carbonyl Chloride 6a-d,f
3.4. General Procedure for the Synthesis of N-[4-(aminosulfonyl)phenyl]-4-alkyl-4H-furo[3,2-b]pyrrole-5-carboxamide 7a,c-f and N-[4-(aminosulfonyl)phenyl]-4-alkyl-4H-thieno[3,2-b]pyrrole-5-carboxamide 8a-d, f
3.5. Synthesis of N-[4-(aminosulfonyl)phenyl]-2-methyl-3-oxo-3,4-dihydro-2H-1,4-benzoxazine-2-carboxamide (11)
3.6. Synthesis of N-[4-(aminosulfonyl)phenyl]-3-phenyl-5,6-dihydro-1,4-oxathiine-2-carboxamide (14a)
3.7. Synthesis of 4-(5-{(Z)-[2-(3-chlorophenyl)-6-oxo[1,3]thiazolo[3,2-b][1,2,4]triazol-5(6H)-ylidene]methyl}-2-furyl)enzenesulfonamide (17a)
3.8. Carbonic Anhydrase Inhibition
3.9. Molecular Modeling Studies
3.10. In-Silico Predictive Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Supuran, C.T. Emerging role of carbonic anhydrase inhibitors. Clin. Sci. 2021, 135, 1233–1249. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Feng, L.; Jeffrey, P.D.; Shi, Y.; Morel, F.M. Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms. Nature 2008, 452, 56–61. [Google Scholar] [CrossRef]
- Capasso, C.; Supuran, C.T. An overview of the alpha-, beta- and gamma-carbonic anhydrases from bacteria: Can bacterial carbonic anhydrases shed new light on evolution of bacteria? J. Enzym. Inhib. Med. Chem. 2015, 30, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Supuran, C.T.; Capasso, C. The eta-class carbonic anhydrases as drug targets for antimalarial agents. Expert. Opin. Ther. Targets 2015, 19, 551–563. [Google Scholar] [CrossRef]
- Del Prete, S.; Vullo, D.; De Luca, V.; Supuran, C.T.; Capasso, C. Biochemical characterization of the δ- carbonic anhydrase from the marine diatom Thalassiosira weissflogii, TweCA. J. Enzym. Inhib. Med. Chem. 2014, 29, 906–911. [Google Scholar] [CrossRef]
- Stefanucci, A.; Angeli, A.; Dimmito, M.P.; Luisi, G.; Del Prete, S.; Capasso, C.; Donald, W.A.; Mollica, A.; Supuran, C.T. Activation of β- and γ-carbonic anhydrases from pathogenic bacteria with tripeptides. J. Enzym. Inhib. Med. Chem. 2018, 33, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Angeli, A.; Del Prete, S.; Alasmary, F.A.S.; Alqahtani, L.S.; AlOthman, Z.; Donald, W.A.; Capasso, C.; Supuran, C.T. The first activation studies of the η-carbonic anhydrase from the malaria parasite Plasmodium falciparum with amines and amino acids. Bioorg. Chem. 2018, 80, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Angeli, A.; Buonanno, M.; Donald, W.A.; Monti, S.M.; Supuran, C.T. The zinc—but not cadmium—containing ζ-carbonic from the diatom Thalassiosira weissflogii is potently activated by amines and amino acids. Bioorg. Chem. 2018, 80, 261–265. [Google Scholar] [CrossRef]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef]
- Supuran, C.T. Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery. Expert Opin. Drug Discov. 2020, 15, 671–686. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrases as drug targets--an overview. Curr. Top. Med. Chem. 2007, 7, 825–833. [Google Scholar] [CrossRef]
- Aggarwal, M.; Boone, C.D.; Kondeti, B.; McKenna, R. Structural annotation of human carbonic anhydrases. J. Enzym. Inhib. Med. Chem. 2013, 28, 267–277. [Google Scholar] [CrossRef]
- Supuran, C.T. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert. Opin. Ther. Pat. 2018, 28, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors. Expert. Opin. Investig. Drugs 2018, 27, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Scozzafava, A.; Supuran, C.T. Glaucoma and the applications of carbonic anhydrase inhibitors. Subcell. Biochem. 2014, 75, 349–359. [Google Scholar]
- Carta, F.; Dumy, P.; Supuran, C.T.; Winum, J.Y. Multivalent Carbonic Anhydrases Inhibitors. Int. J. Mol. Sci. 2019, 20, 5352. [Google Scholar] [CrossRef] [Green Version]
- Bozdag, M.; Altamimi, A.S.A.; Vullo, D.; Supuran, C.T.; Carta, F. State of the Art on Carbonic Anhydrase Modulators for Biomedical Purposes. Curr. Med. Chem. 2019, 26, 2558–2573. [Google Scholar] [CrossRef]
- Peng, W.; Peltier, D.C.; Larsen, M.J.; Kirchhoff, P.D.; Larsen, S.D.; Neubig, R.R.; Miller, D.J. Identification of Thieno[3,2-b]Pyrrole Derivatives as Novel Small Molecule Inhibitors of Neurotropic Alphaviruses. J. Infect. Dis. 2009, 199, 950–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vianello, P.; Sartori, L.; Amigoni, F.; Cappa, A.; Fagá, G.; Fattori, R.; Legnaghi, E.; Ciossani, G.; Mattevi, A.; Meroni, G.; et al. Thieno[3,2-b]pyrrole-5-carboxamides as New Reversible Inhibitors of Histone Lysine Demethylase KDM1A/LSD1. Part 2: Structure-Based Drug Design and Structure-Activity Relationship. J. Med. Chem. 2017, 60, 1693–1715. [Google Scholar] [CrossRef] [PubMed]
- Ching, K.C.; Tran, T.; Amrun, S.; Kam, Y.; Ng, L.; Chai, C. Structural Optimizations of Thieno[3,2-b]pyrrole Derivatives for the Development of Metabolically Stable Inhibitors of Chikungunya Virus. J. Med. Chem. 2017, 60, 3165–3186. [Google Scholar] [CrossRef]
- Mohamed, M.S.; El-Domany, R.A.; Abd El-Hameed, R.H. Synthesis of certain pyrrole derivatives as antimicrobial agents. Acta Pharm. 2009, 59, 145–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alizadeh, M.; Jalal, M.; Hamed, K.; Saber, A.; Kheirouri, S.; Pourteymour Fard Tabrizi, F.; Kamari, N. Recent Updates on Anti-Inflammatory and Antimicrobial Effects of Furan Natural Derivatives. J. Inflamm. Res. 2020, 13, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Malladi, S.; Nadh, R.V.; Babu, K.S.; Babu, P.S. Synthesis and antibacterial activity studies of 2,4-di substituted furan derivatives. Beni Suef Univ. J. Basic Appl. Sci. 2017, 6, 345–353. [Google Scholar] [CrossRef]
- Fang, B.; Hu, C.; Ding, Y.; Qin, H.; Luo, Y.; Xu, Z.; Meng, J.; Chen, Z. Discovery of 4H-thieno[3,2-b]pyrrole derivatives as potential anticancer agents. J. Heterocyclic Chem. 2021, 58, 1610. [Google Scholar] [CrossRef]
- Archna, P.S.; Chawla, P.A. Thiophene–based derivatives as anticancer agents. An overview on decade’s work. Bioorg. Chem. 2020, 101, 104026. [Google Scholar] [CrossRef]
- Lashin, W.H.; Nassar, I.F.; El Farargy, A.F.; Abdelhamid, A.O. Synthesis of New Furanone Derivatives with Potent Anticancer Activity. Russ. J. Bioorg. Chem. 2020, 46, 1074–1086. [Google Scholar] [CrossRef]
- Kumar, P.R.; Raju, S.; Goud, P.S.; Sailaja, M.; Sarma, M.R.; Reddy, G.O.; Kumar, M.P.; Reddy, V.V.; Suresh, T.; Hegde, P. Synthesis and biological evaluation of thiophene [3,2-b] pyrrole derivatives as potential anti-inflammatory agents. Bioorg. Med. Chem. 2004, 12, 1221–1230. [Google Scholar] [CrossRef]
- Abd El-Hameed, R.H.; Mahgoub, S.; El-Shanbaky, H.M.; Mohamed, M.S.; Ali, S.A. Utility of novel 2-furanones in synthesis of other heterocyclic compounds having anti-inflammatory activity with dual COX2/LOX inhibition. J. Enzym. Inhib. Med. Chem. 2021, 36, 977–986. [Google Scholar] [CrossRef]
- Mohamed, M.S.; Ali, S.A.; Abdelaziz, D.H.; Fathallah, S.S. Synthesis and evaluation of novel pyrroles and pyrrolopyrimidines as anti-hyperglycemic agents. BioMed Res. Int. 2014, 2014, 249780–249793. [Google Scholar] [CrossRef]
- Baldwin, J.J.; Ponticello, G.S.; Anderson, P.S.; Christy, M.E.; Murcko, M.A.; Randall, W.C.; Schwam, H.; Sugrue, M.F.; Springer, J.P.; Gautheron, P. Thienothiopyran-2-sulfonamides: Novel topically active carbonic anhydrase inhibitors for the treatment of glaucoma. J. Med. Chem. 1989, 32, 2510–2513. [Google Scholar] [CrossRef] [PubMed]
- Rakesh, K.P.; Wang, S.M.; Leng, J.; Ravindar, L.; Asiri, A.M.; Marwani, H.M.; Qin, H.L. Recent Development of Sulfonyl or Sulfonamide Hybrids as Potential Anticancer Agents: A Key Review. Anticancer Agents Med. Chem. 2018, 18, 488–505. [Google Scholar] [CrossRef] [PubMed]
- Kachaeva, M.V.; Hodyna, D.M.; Semenyuta, I.V.; Pilyo, S.G.; Prokopenko, V.M.; Kovalishyn, V.V.; Metelytsia, L.O.; Brovarets, V.S. Design, synthesis and evaluation of novel sulfonamides as potential anticancer agents. Comput. Biol. Chem. 2018, 74, 294–303. [Google Scholar] [CrossRef]
- Abbas, H.S.; Abd El-Karim, S.S.; Abdelwahed, N.A.M. Synthesis and biological evaluation of sulfonamide derivatives as antimicrobial agents. Acta Pol. Pharm. 2017, 74, 849–860. [Google Scholar]
- Azzam, R.A.; Elsayed, R.E.; Elgemeie, G.H. Design, Synthesis, and Antimicrobial Evaluation of a New Series of N-Sulfonamide 2-Pyridones as Dual Inhibitors of DHPS and DHFR Enzymes. ACS Omega 2020, 5, 10401–10414. [Google Scholar] [CrossRef]
- Hussein, E.M.; Al-Rooqi, M.M.; Abd El-Galil, S.M.; Ahmed, S.A. Design, synthesis, and biological evaluation of novel N4 -substituted sulfonamides: Acetamides derivatives as dihydrofolate reductase (DHFR) inhibitors. BMC Chem. 2019, 13, 91. [Google Scholar] [CrossRef]
- Ingle, R.G.; Marathe, R.P. Sulfonamido Quinoxalines—Search for anti-inflammatory Agents. IJPRAS 2012, 1, 46–51. [Google Scholar]
- Markowicz-Piasecka, M.; Huttunen, K.M.; Broncel, M.; Sikora, I. Sulfenamide and Sulfonamide Derivatives of Metformin—A New Option to Improve Endothelial Function and Plasma Haemostasis. Sci. Rep. 2019, 9, 6573–6592. [Google Scholar] [CrossRef] [Green Version]
- Bhuva, N.H.; Talpara, P.K.; Singala, P.M.; Gothaliya, V.K.; Shah, V.H. Synthesis and biological evaluation of pyrimidinyl sulphonamide derivatives as promising class of antitubercular agents. J. Saudi Chem. Soc. 2017, 21, 517–527. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, A.G.; Lévesque, F.; Seeberger, P.H. Continuous flow thermolysis of azidoacrylates for the synthesis of heterocycles and pharmaceutical intermediates. Chem. Commun. 2011, 47, 2688–2690. [Google Scholar] [CrossRef]
- Yarovenko, V.N.; Semenov, S.L.; Zavarzin, I.V.; Ignatenko, A.V.; Krayushkin, M.M. Regioselective acylation of methyl 2-methyl-4H-thieno[3,2-b]pyrrole-5-carboxylate. Russ. Chem. Bull. 2003, 52, 451–456. [Google Scholar] [CrossRef]
- Suhadolc, E.; Urleb, U.; Žbontar, U.; Kikelj, D. A convenient synthesis of 3,4-dihydro-2-methyl-3-oxo-2H-1,4-benzoxazine-2-carboxylic acids and 3,4-dihydro-2-methyl-3-oxo-2H-pyrido[3,2-b]-1, 4-oxazine-2-carboxylic acid. J. Heterocycl. Chem. 1993, 30, 597–602. [Google Scholar] [CrossRef]
- Mamedov, V.A.; Sibgatullina, F.G.; Gubskaya, V.P.; Gainullin, R.M.; Shagidullin, R.R.; Il’yasov, A.V. Synthesis of some new derivatives of dithiene-and oxathienecarboxylic acids. Chem. Heterocycl. Compd. 1994, 30, 1029–1033. [Google Scholar] [CrossRef]
- Murty, M.S.R.; Ram, K.R.; Rao, B.R.; Rao, R.V.; Katiki, M.R.; Rao, J.V.; Pamanji, R.; Velatooru, L.R. Synthesis, characterization, and anticancer studies of S and N alkyl piperazine-substituted positional isomers of 1,2,4-triazole derivatives. Med. Chem. Res. 2014, 23, 1661–1671. [Google Scholar] [CrossRef]
- Kozlov, N.G.; Zhikharko, Y.D.; Lytvyn, R.Z.; Gorak, Y.I.; Skakovskii, E.D.; Baranovskii, A.V.; Basalaeva, L.I.; Obushak, M.D. Synthesis of 12-hetaryl-9,9-dimethyl-7,8,9,10-tetrahydrobenzo[a]acridin-11(12H)-ones. Russ. J. Org. Chem. 2014, 50, 833–839. [Google Scholar] [CrossRef]
- Karthikeyan, M.S.; Holla, B.S. Synthesis, antiinflammatory and antimicrobial activities of some 2,4-dichloro-5-fluorophenyl substituted arylidenetriazolothiazolidinones. Monatsh. Chem. 2008, 139, 691–696. [Google Scholar] [CrossRef]
- Tozkoparan, B.; Kılcıgilb, G.A.; Ertanb, R.; Ertana, M.; Kelicen, P.; Demirdamar, R. Synthesis and evaluation of anti-inflammatory activity of some thiazolo[3,2-b]-1,2,4-triazole-5(6H)-ones and their Michael addition products. Arzneim. Forsch. 1999, 49, 1006–1011. [Google Scholar] [CrossRef]
- Rzhevskii, A.A.; Gerasimova, N.P.; Alov, E.M.; Kozlova, O.S.; Danilova, A.S.; Khapova, S.A.; Suponitskii, K.Y. Condensation of 5-(4-methylphenyl)-2,4-dihydro-3H-1,2,4-triazole-3-thione with haloacetic acids. Russ. Chem. Bull. 2012, 61, 2133–2136. [Google Scholar] [CrossRef]
- Koysal, Y.; Isik, S.; Dogdas, E.; Tozkoparan, B.; Ertan, M. 6-(2-Fluorobenzylidene)-2-[1-(2-fluoro-4-biphenyl)ethyl]thiazolo[3,2-b][1,2,4]triazol-5(6H)-one. Acta Crystallogr. Sect. C 2004, 60, 356–357. [Google Scholar] [CrossRef] [Green Version]
- Ottana‘, R.; Maccari, R.; Barreca, M.L.; Bruno, G.; Rotondo, A.; Rossi, A.; Chiricosta, G.; Di Paola, R.; Sautebin, L.; Cuzzocrea, S.; et al. 5-Arylidene-2-imino-4-thiazolidinones: Design and synthesis of novel anti-inflammatory agents. Bioorg. Med. Chem. 2005, 13, 4243–4252. [Google Scholar] [CrossRef]
- Alterio, V.; Hilvo, M.; Di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Fiore, A.; Truppo, E.; Supuran, C.T.; Alterio, V.; Dathan, N.; Bootorabi, F.; Parkkila, S.; Monti, S.M.; De Simone, G. Crystal structure of the C183S/C217S mutant of human CA VII in complex with acetazolamide. Bioorg. Med. Chem. Lett. 2010, 20, 5023–5026. [Google Scholar] [CrossRef]
- Whittington, D.A.; Waheed, A.; Ulmasov, B.; Shah, G.N.; Grubb, J.H.; Sly, W.S.; Christianson, D.W. Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc. Natl. Acad. Sci. USA 2001, 98, 9545–9550. [Google Scholar] [CrossRef] [Green Version]
- Kanamori, K.; Roberts, J.D. Nitrogen-15 nuclear magnetic resonance study of benzenesulfonamide and cyanate binding to carbonic anhydrase. Biochemistry 1983, 22, 2658–2664. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption Using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Wars, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Khalifah, R.G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [CrossRef]
- Bonardi, A.; Falsini, M.; Catarzi, D.; Varano, F.; Di Cesare Mannelli, L.; Tenci, B.; Ghelardini, C.; Angeli, A.; Supuran, C.T.; Colotta, V. Structural investigations on coumarins leading to chromeno[4,3-c]pyrazol-4-ones and pyrano[4,3-c]pyrazol-4-ones: New scaffolds for the design of the tumor-associated carbonic anhydrase isoforms IX and XII. Eur. J. Med. Chem. 2018, 146, 47–59. [Google Scholar] [CrossRef]
- Angeli, A.; Peat, T.S.; Bartolucci, G.; Nocentini, A.; Supuran, C.T.; Carta, F. Intramolecular oxidative deselenization of acylselenoureas: A facile synthesis of benzoxazole amides and carbonic anhydrase inhibitors. Org. Biomol. Chem. 2016, 14, 11353–11356. [Google Scholar] [CrossRef]
- Angeli, A.; Di Cesare Mannelli, L.; Lucarini, E.; Peat, T.S.; Ghelardini, C.; Supuran, C.T. Design, synthesis and X-ray crystallography of selenides bearing benzenesulfonamide moiety with neuropathic pain modulating effects. Eur. J. Med. Chem. 2018, 154, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Angeli, A.; Ferraroni, M.; Supuran, C.T. Famotidine, an antiulcer agent, strongly inhibits helicobacter pylori and human carbonic anhydrases. ACS Med. Chem. Lett. 2018, 9, 1035–1038. [Google Scholar] [CrossRef] [PubMed]
- Angeli, A.; Carta, F.; Bartolucci, G.; Supuran, C.T. Synthesis of novel acyl selenoureido benzensulfonamides as carbonic anhydrase I, II, VII and IX inhibitors. Bioorg. Med. Chem. 2017, 25, 3567–3573. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- A Structural View of Biology. Available online: http://www.rcsb.org/ (accessed on 8 June 2021).
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KI (nM) * | ||||
|---|---|---|---|---|
| Cmp | hCA I | hCA II | hCA IX | hCA XII |
| 7a | 717.9 | 8.4 | 103.2 | 6.9 |
| 7c | 3563 | 37.5 | 275.6 | 4.6 |
| 7e | 858.3 | 23.8 | 239.5 | 19.6 |
| 7f | 878.1 | 34.3 | 187.2 | 37.2 |
| 8a | 664.6 | 8.8 | 66.6 | 30.7 |
| 8b | 269.7 | 6.8 | 250.8 | 5.1 |
| 8c | 399.5 | 8.9 | 140.5 | 7.8 |
| 8d | 656.3 | 8.7 | 171.3 | 8.2 |
| 8f | 699.2 | 68.1 | 81.4 | 8.9 |
| 11 | 70.6 | 6.5 | 259.8 | 5.3 |
| 14a | 56.5 | 6.7 | 280.5 | 6.1 |
| 14b | 71.2 | 7.4 | 155.2 | 6.6 |
| 17a | 2925 | 44.7 | 320.0 | 7.6 |
| 17b | 918.5 | 59.4 | 252.9 | 40.2 |
| AAZ | 250 | 12.1 | 25.7 | 5.7 |
| No | hCA Isoform | Estimated Free Binding Energy (Kcal/mol) | Chelating the Zn (II) Ion | Residues Involved in H-Bond Interactions | Residues Involved in Hydrophobic Interactions |
|---|---|---|---|---|---|
| 7c | hCA I | −5.47 | No | - | Ala121, Ala135 |
| hCA II | −7.11 | Yes | Thr199 | Ile91 | |
| hCA IX | −6.80 | Yes | Thr199 | Val121, Thr200 | |
| hCA XII | −11.32 | Yes | His67, Gln92, Thr200 | Leu198 | |
| 8a | hCA I | −6.88 | Yes | Thr199 | Ala135, Leu198 |
| hCA II | −8.95 | Yes | His94, Thr199 | Ile91, Phe131, Thr200 | |
| hCA IX | −7.91 | Yes | Thr199 | Val121, Thr200 | |
| hCA XII | −6.42 | Yes | - | Val121, Leu198 | |
| 8b | hCA I | −8.17 | Yes | Gln92 | Ala121, Leu198 |
| hCA II | −9.24 | Yes | His94, Thr199 | Val121, Phe131, Leu198 | |
| hCA IX | −6.54 | Yes | Thr199 | Val121, Leu198 | |
| hCA XII | −9.80 | Yes | Gln92, His94, Thr200 | Val121, Ala131, Leu198 | |
| 11 | hCA I | −9.91 | Yes | Gln92, Thr199 | Leu131, Ala132, Thr202 |
| hCA II | −9.24 | Yes | His94, Thr200 | Val121, Phe131, Leu198 | |
| hCA IX | −6.20 | Yes | - | Val121, Val131, Leu198 | |
| hCA XII | −9.78 | Yes | His94, Thr200 | Val121, Val143, Ala131, Leu198 | |
| 14a | hCA I | −11.02 | Yes | His67, Thr199 | Ala121, Ala132, Leu198 |
| hCA II | −9.15 | Yes | Asn67, Thr199 | Ile191, Val121, Phe131, Leu198, Thr200 | |
| hCA IX | −6.51 | Yes | Thr199 | Val121, Thr200 | |
| hCA XII | −9.42 | Yes | Thr200 | Val121, Leu198 | |
| AAZ | hCA I | −8.28 | Yes | Gln92 | Leu198, Thr199, His200, Pro201, Trp209 |
| hCA II | −8.87 | Yes | Thr199, Thr200 | Val121, Phe131, Leu198, Trp209 | |
| hCA IX | −9.02 | Yes | Thr199, Thr200 | Val121, Val143, Val131, Leu198, Trp209 | |
| hCA XII | −9.14 | Yes | Thr199, Thr200 | Val121, Val143, Leu198, Trp209 |
| No | MW | Number of HBA a | Number of HBD b | Log Po/w (iLOGP) c | Log S d | TPSA e | BBB permeant f | Lipinski, Ghose, Veber, Egan, and Muegge Violations | Bioavailability Score | Drug-Likeness Model Score |
|---|---|---|---|---|---|---|---|---|---|---|
| 7a | 319.3 | 5 | 2 | 1.30 | Moderately soluble | 115.71 | No | 0 | 0.55 | 0.57 |
| 7c | 395.4 | 6 | 2 | 1.79 | Poorly soluble | 115.71 | No | 0 | 0.55 | 0.44 |
| 7e | 429.9 | 5 | 2 | 2.05 | Poorly soluble | 115.71 | No | 0 | 0.55 | 0.36 |
| 7f | 429.9 | 5 | 2 | 2.26 | Poorly soluble | 115.71 | No | 0 | 0.55 | 0.97 |
| 8a | 335.4 | 4 | 2 | 1.72 | Moderately soluble | 130.81 | No | 0 | 0.55 | 0.59 |
| 8b | 349.4 | 4 | 2 | 2.02 | Moderately soluble | 130.81 | No | 0 | 0.55 | 0.80 |
| 8c | 411.5 | 4 | 2 | 2.17 | Poorly soluble | 130.81 | No | 0 | 0.55 | 0.47 |
| 8d | 429,5 | 5 | 2 | 1.84 | Poorly soluble | 130.81 | No | 0 | 0.55 | 0.21 |
| 8f | 445.9 | 4 | 2 | 2.49 | Poorly soluble | 130.81 | No | 0 | 0.55 | 1.00 |
| 11 | 484.9 | 7 | 1 | 2.90 | Poorly soluble | 157.18 | No | 0 | 0.55 | -0.58 |
| 14a | 510.6 | 9 | 1 | 3.10 | Poorly soluble | 175.64 | No | 1 (MW > 500) | 0.55 | -0.53 |
| 14b | 36.,4 | 6 | 3 | 1.19 | Moderately soluble | 135.97 | No | 0 | 0.55 | 0.14 |
| 17a | 376.5 | 5 | 2 | 1.74 | Moderately soluble | 132.17 | No | 0 | 0.55 | 0.45 |
| 17b | 404.5 | 5 | 2 | 2.50 | Poorly soluble | 132.17 | No | 0 | 0.55 | 0.37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angeli, A.; Kartsev, V.; Petrou, A.; Pinteala, M.; Vydzhak, R.M.; Panchishin, S.Y.; Brovarets, V.; De Luca, V.; Capasso, C.; Geronikaki, A.; et al. New Sulfanilamide Derivatives Incorporating Heterocyclic Carboxamide Moieties as Carbonic Anhydrase Inhibitors. Pharmaceuticals 2021, 14, 828. https://doi.org/10.3390/ph14080828
Angeli A, Kartsev V, Petrou A, Pinteala M, Vydzhak RM, Panchishin SY, Brovarets V, De Luca V, Capasso C, Geronikaki A, et al. New Sulfanilamide Derivatives Incorporating Heterocyclic Carboxamide Moieties as Carbonic Anhydrase Inhibitors. Pharmaceuticals. 2021; 14(8):828. https://doi.org/10.3390/ph14080828
Chicago/Turabian StyleAngeli, Andrea, Victor Kartsev, Anthi Petrou, Mariana Pinteala, Roman M. Vydzhak, Svitlana Y. Panchishin, Volodymyr Brovarets, Viviana De Luca, Clemente Capasso, Athina Geronikaki, and et al. 2021. "New Sulfanilamide Derivatives Incorporating Heterocyclic Carboxamide Moieties as Carbonic Anhydrase Inhibitors" Pharmaceuticals 14, no. 8: 828. https://doi.org/10.3390/ph14080828
APA StyleAngeli, A., Kartsev, V., Petrou, A., Pinteala, M., Vydzhak, R. M., Panchishin, S. Y., Brovarets, V., De Luca, V., Capasso, C., Geronikaki, A., & Supuran, C. T. (2021). New Sulfanilamide Derivatives Incorporating Heterocyclic Carboxamide Moieties as Carbonic Anhydrase Inhibitors. Pharmaceuticals, 14(8), 828. https://doi.org/10.3390/ph14080828