
Pyrroles as Privileged Scaffolds in the Search for New Potential HIV Inhibitors

Abstract
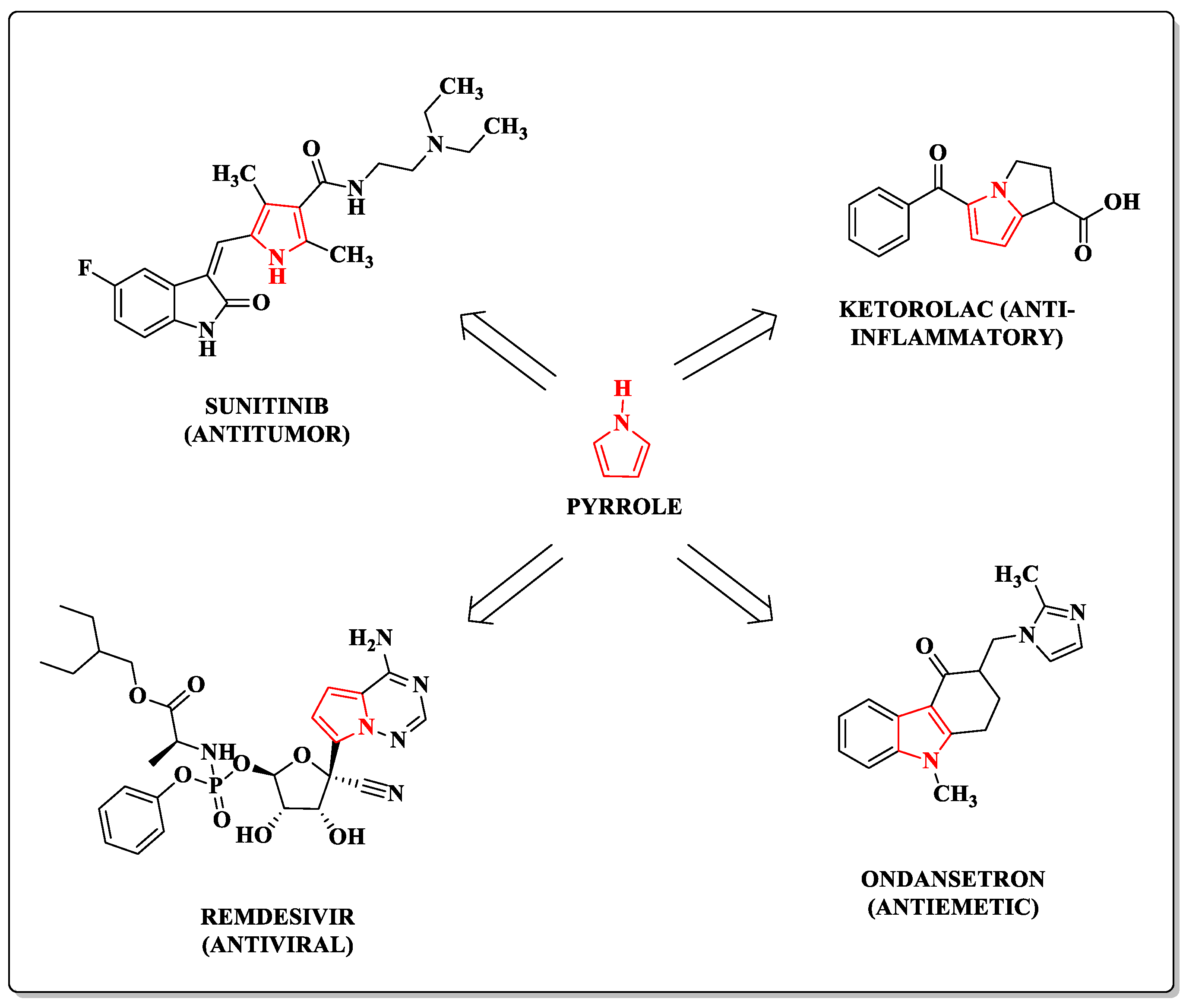
:1. Introduction
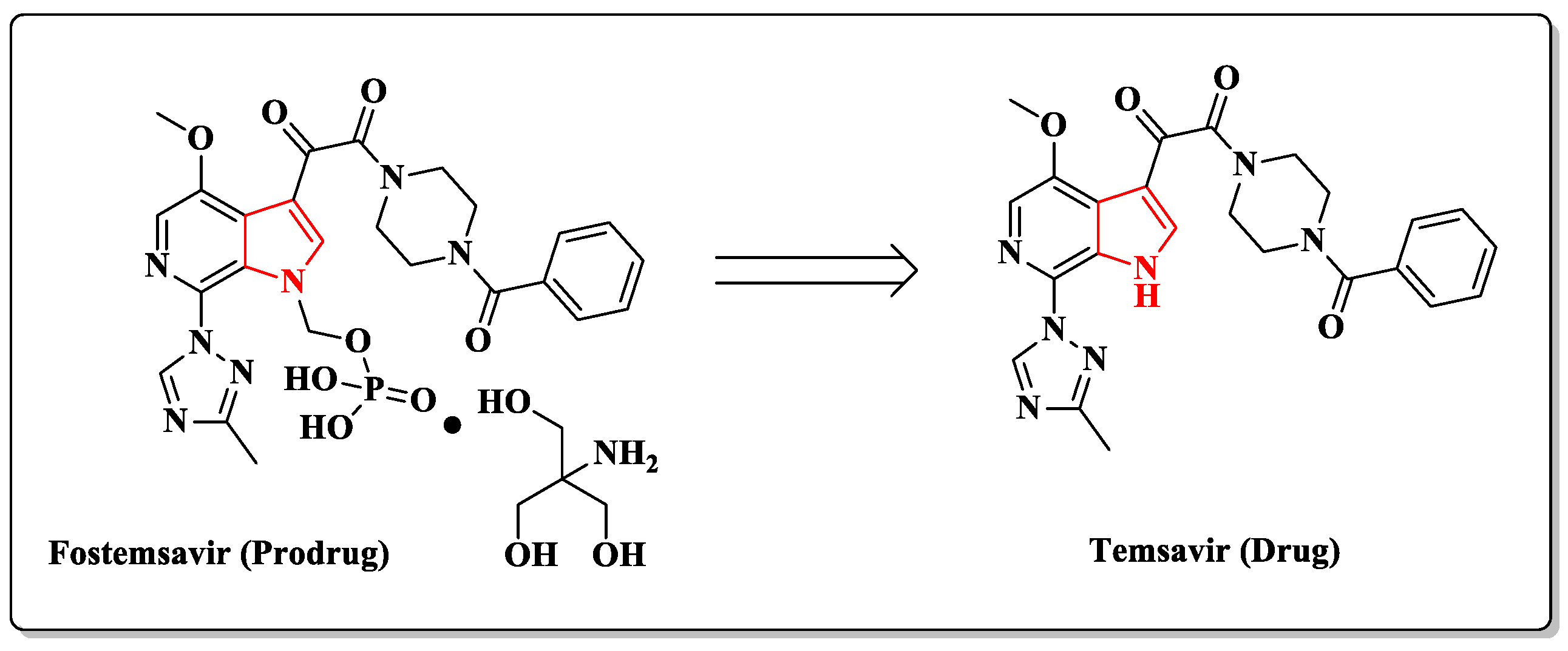
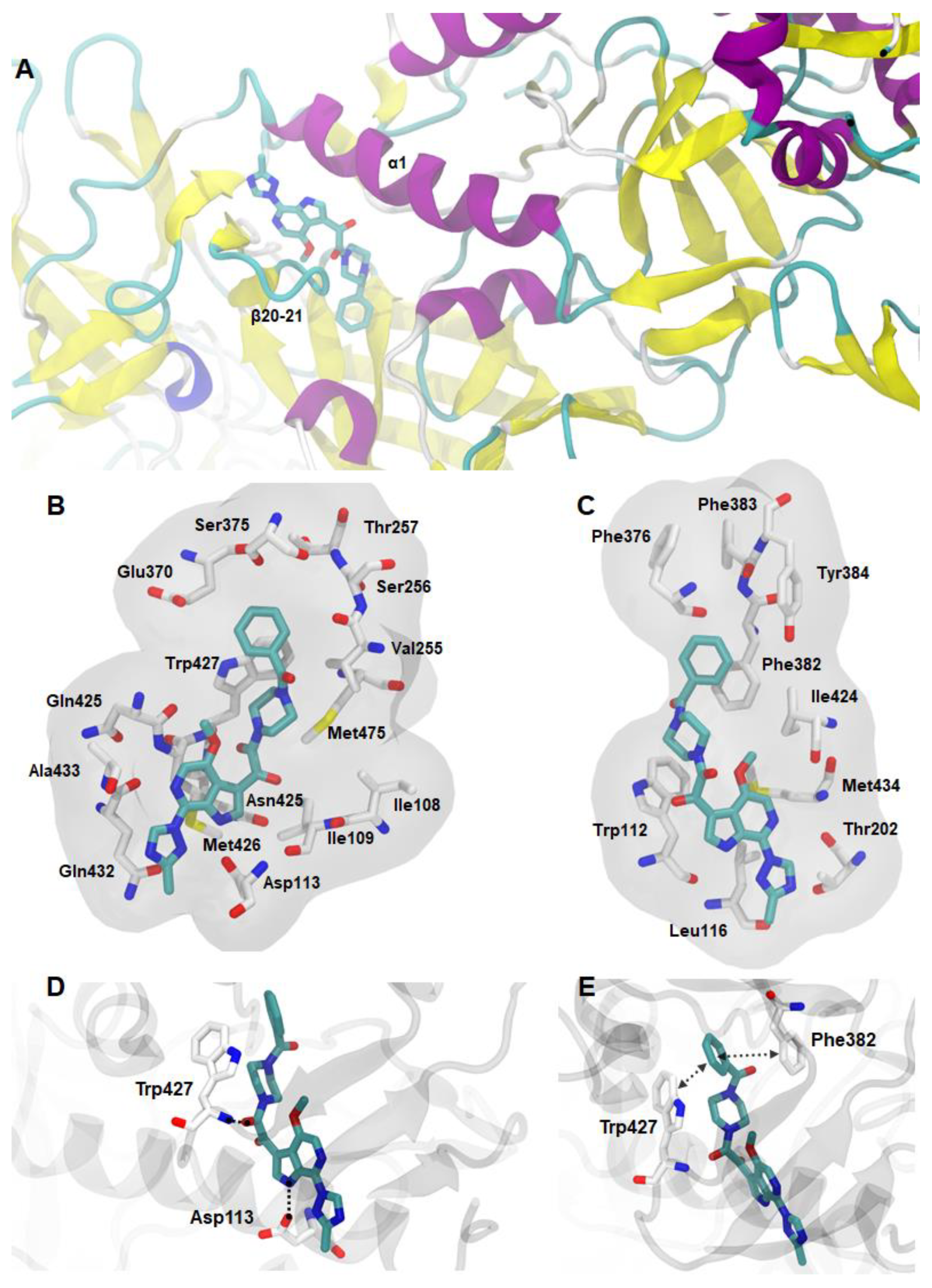
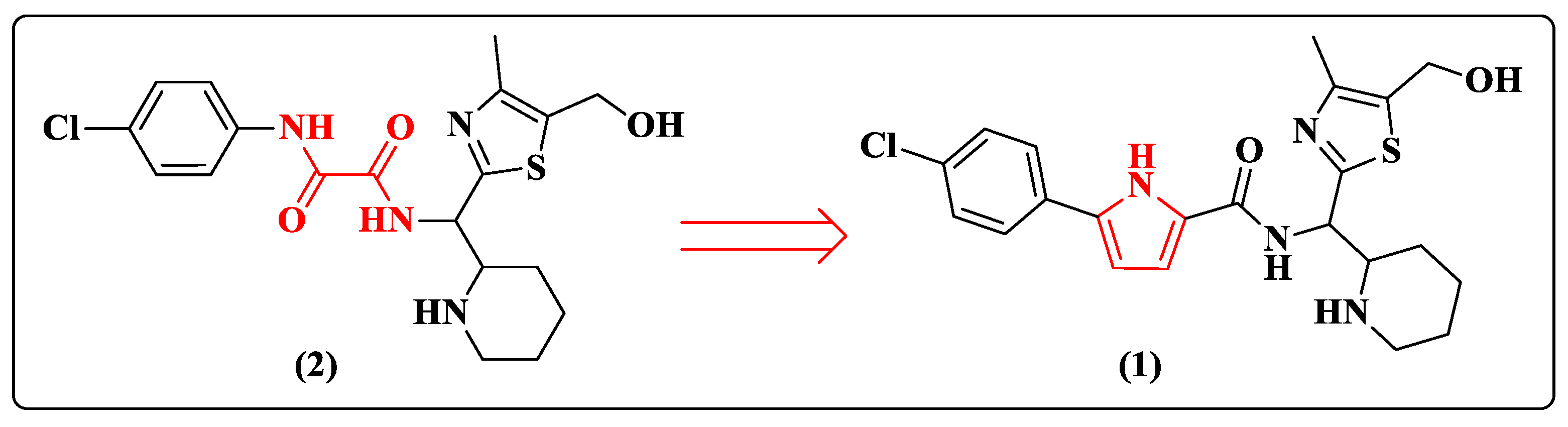
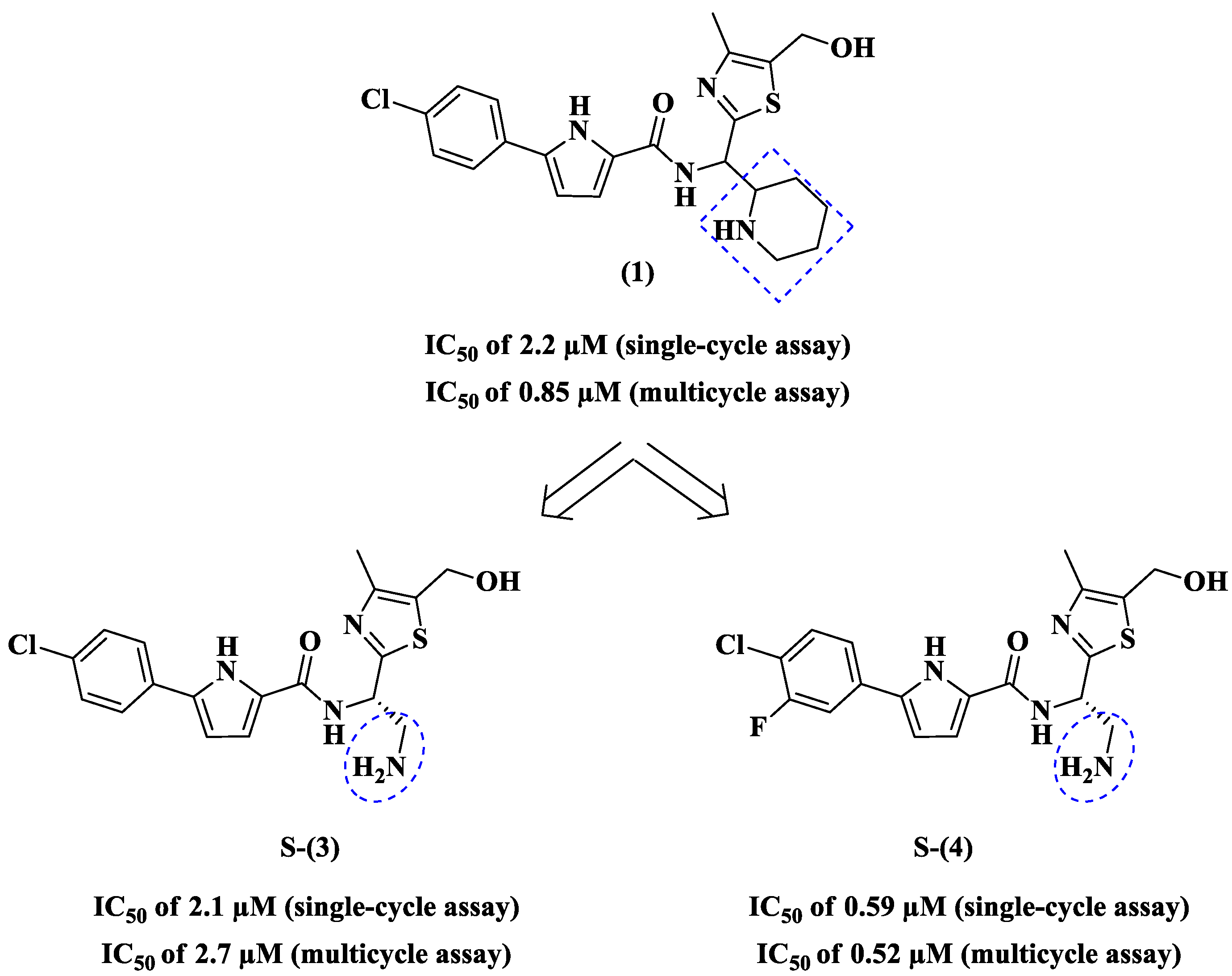
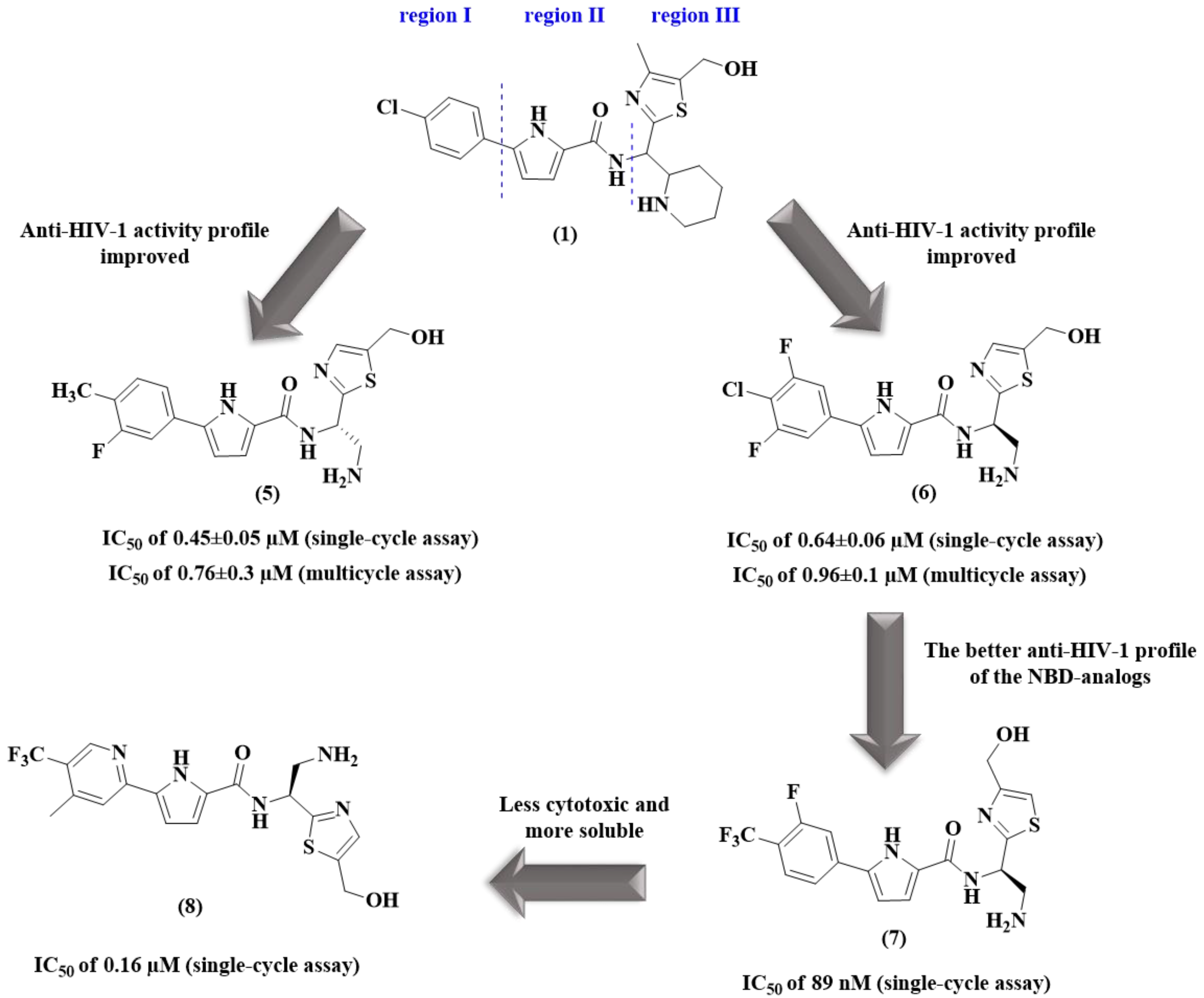
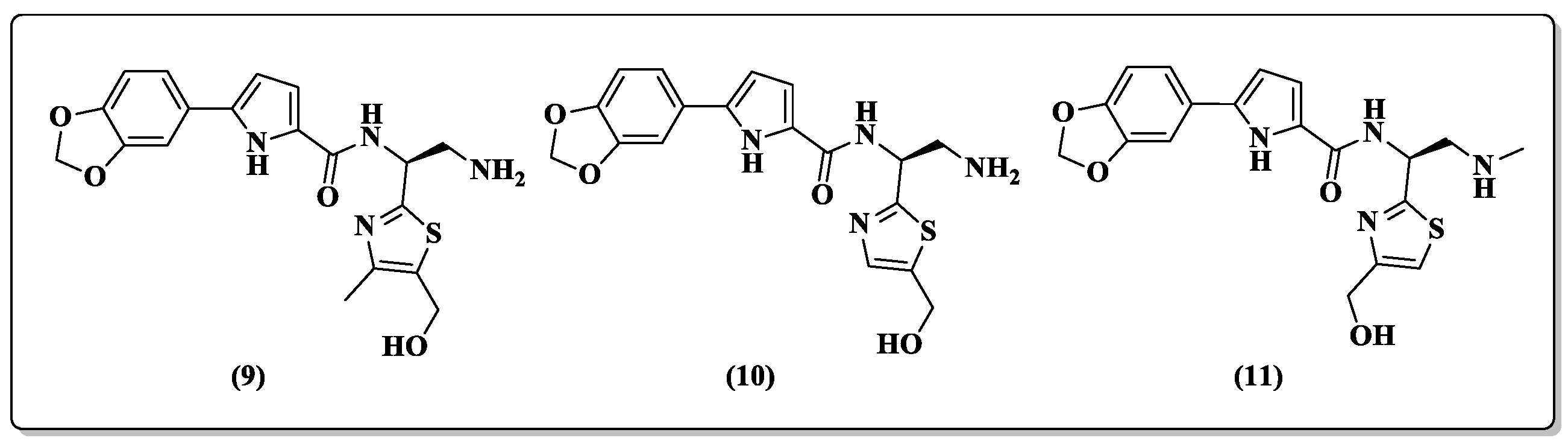
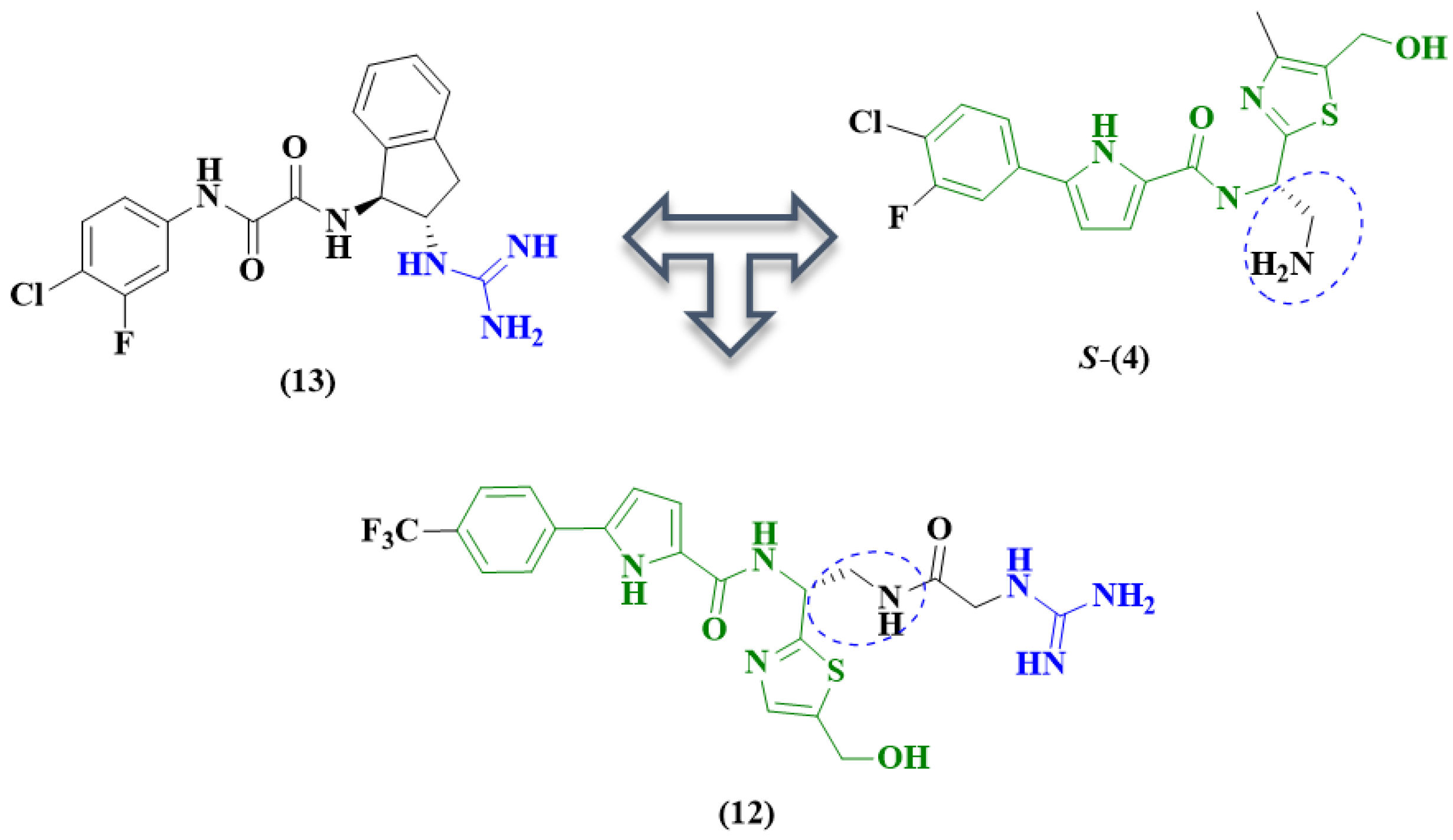
2. Pyrrole-Based Entry Inhibitors of the HIV-1
3. Pyrrole-Based Anti-HIV-1 Inhibitors (Dual Inhibitors, RT Inhibitors and Replication Inhibitors)
4. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 8 March 2021).
- Available online: https://unaids.org.br/estatisticas/#:~:text=38%20milh%C3%B5es%20%5B31%2C6%20milh%C3%B5es,at%C3%A9%20o%20fim%20de%202019 (accessed on 8 March 2021).
- Becerra, J.C.; Bildstein, L.S.; Gach, J.S. Recent Insights into the HIV/AIDS Pandemic. Microb. Cell 2016, 3, 450–474. [Google Scholar] [CrossRef] [PubMed]
- Moranguinho, I.; Valente, S.T. Block-and-Lock: New Horizons for a Cure for HIV-1. Viruses 2020, 12, 1443. [Google Scholar] [CrossRef]
- Rossi, E.; Meuser, M.; Cunanan, C.; Cocklin, S. Structure, Function, and Interactions of the HIV-1 Capsid Protein. Life 2021, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Himmel, D.M.; Arnold, E. Non-Nucleoside Reverse Transcriptase Inhibitors Join Forces with Integrase Inhibitors to Combat HIV. Pharmacy 2020, 13, 122. [Google Scholar] [CrossRef]
- Larijani, M.S.; Ramezani, A.; Sadat, S.M. Updated Studies on the Development of HIV Therapeutic Vaccine. Curr. HIV Res. 2019, 17, 75–84. [Google Scholar] [CrossRef]
- Patel, R.V.; Park, S.W. Pyrroloaryls and pyrroloheteroaryls: Inhibitors of the HIV fusion/attachment, reverse transcriptase and integrase. Bioorg. Med. Chem. 2015, 23, 5247–5263. [Google Scholar] [CrossRef]
- Costi, R.; Métifiot, M.; Esposito, F.; Crucitti, G.C.; Pescatori, L.; Messore, A.; Scipione, L.; Tortorella, S.; Zinzula, L.; Novellino, E.; et al. 6-(1-Benzyl-1H-pyrrol-2-yl)-2,4-dioxo-5-hexenoic Acids as Dual Inhibitors of Recombinant HIV-1 Integrase and Ribonuclease H, Synthesized by a Parallel Synthesis Approach. J. Med. Chem. 2013, 56, 8588–8598. [Google Scholar] [CrossRef] [PubMed]
- Petri, G.L.; Spanò, V.; Spatola, R.; Holl, R.; Raimondi, M.V.; Barraja, P.; Montalbano, A. Bioactive pyrrole-based compounds with target selectivity. Eur. J. Med. Chem. 2020, 208, 112783. [Google Scholar] [CrossRef]
- Walsh, C.T.; Garneau-Tsodikova, S.; Howard-Jones, A.R. Biological formation of pyrroles: Nature’s logic and enzymatic machinery. Nat. Prod. Rep. 2006, 23, 517–531. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Rees, C.W.; Scriven, E.F.V. Comprehensive Heterocyclic Chemistry II: A Review of the Literature 1982-1995, 2nd ed.; Pergamon: São Paulo, Brazil, 1996. [Google Scholar]
- Sruthi, P.R.; Sankar, P.U.; Saranya, T.V.; Anas, S. Facile Synthesis of Dihydroquinolines via Palladium Catalyzed Sequential Amination and Cyclisation of Morita-Baylis-Hillman Alcohols. ChemistrySelect 2020, 5, 13598–13602. [Google Scholar] [CrossRef]
- Iqbal, S.; Rasheed, H.; Awan, R.J.; Awan, R.J.; Mukhtar, A.; Moloney, M.G. Recent Advances in the Synthesis of Pyrroles. Curr. Org. Chem. 2020, 24, 1196–1229. [Google Scholar] [CrossRef]
- Bhardwaj, V.; Gumber, D.; Abbot, V.; Dhiman, S.; Sharma, P. Pyrrole: A resourceful small molecule in key medicinal hetero-aromatics. RSC Adv. 2015, 5, 15233–15266. [Google Scholar] [CrossRef]
- Gholap, S.S. Pyrrole: An emerging scaffold for construction of valuable therapeutic agents. Eur. J. Med. Chem. 2016, 110, 13–31. [Google Scholar] [CrossRef]
- Kaur, R.; Rani, V.; Abbot, V.; Kapoor, Y.; Konar, D.; Kumar, K. Recent synthetic and medicinal perspectives of pyrroles: An overview. J. Pharm. Chem. Chem. Sci. 2017, 1, 17–32. [Google Scholar]
- Ahmad, S.; Alam, O.; Naim, M.J.; Shaquiquzzaman, M.; Alam, M.M.; Iqbal, M. Pyrrole: An insight into recent pharmacological advances with structure activity relationship. Eur. J. Med. Chem. 2018, 157, 527–561. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-M.; Alharbi, N.S.; Sun, B.; Shantharam, C.; Rakesh, K.; Qin, H.-L. Synthetic routes and structure-activity relationships (SAR) of anti-HIV agents: A key review. Eur. J. Med. Chem. 2019, 181, 111566. [Google Scholar] [CrossRef]
- Motati, D.R.; Uredi, D.; Watkins, E.B. The Discovery and Development of Oxalamide and Pyrrole Small Molecule Inhibitors of gp120 and HIV Entry—A Review. Curr. Top. Med. Chem. 2019, 19, 1650–1675. [Google Scholar] [CrossRef]
- Sing, T.; Chou, K.-C.; Sirois, S. HIV-1 gp120 V3 Loop for Structure-Based Drug Design. Curr. Protein Pept. Sci. 2005, 6, 413–422. [Google Scholar] [CrossRef]
- Tintori, C.; Selvaraj, M.; Badia, R.; Clotet, B.; Esté, J.A.; Botta, M. Computational Studies Identifying Entry Inhibitor Scaffolds Targeting the Phe 43 Cavity of HIV-1 gp120. ChemMedChem 2013, 8, 475–483. [Google Scholar] [CrossRef]
- Esté, J.A.; Telenti, A. HIV entry inhibitors. Lancet 2007, 370, 81–88. [Google Scholar] [CrossRef]
- Gulick, R.M. Investigational Antiretroviral Drugs: What is Coming Down the Pipeline. Top. Antivir. Med. 2018, 25, 127–132. [Google Scholar] [PubMed]
- Zhang, Y.; Chapman, J.H.; Ulcay, A.; Sutton, R.E. Neutralization Synergy between HIV-1 Attachment Inhibitor Fostemsavir and Anti-CD4 Binding Site Broadly Neutralizing Antibodies against HIV. J. Virol. 2019, 93, e01446-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-hiv-treatment-patients-limited-treatment-options (accessed on 10 March 2021).
- Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/rukobia (accessed on 10 March 2021).
- Markham, A. Fostemsavir: First Approval. Drugs 2020, 80, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Ho, H.-T.; Dicker, I.; Fan, L.; Zhou, N.; Friborg, J.; Wang, T.; McAuliffe, B.V.; Wang, H.-G.H.; Rose, R.E.; et al. Biochemical and Genetic Characterizations of a Novel Human Immunodeficiency Virus Type 1 Inhibitor That Blocks gp120-CD4 Interactions. J. Virol. 2003, 77, 10528–10536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pancera, M.; Lai, Y.-T.; Bylund, T.; Druz, A.; Narpala, S.; O’Dell, S.; Schön, A.; Bailer, R.T.; Chuang, G.-Y.; Geng, H.; et al. Crystal structures of trimeric HIV envelope with entry inhibitors BMS-378806 and BMS-626529. Nat. Chem. Biol. 2017, 13, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Langley, D.R.; Kimura, S.R.; Sivaprakasam, P.; Zhou, N.; Dicker, I.; McAuliffe, B.; Wang, T.; Kadow, J.F.; Meanwell, N.; Krystal, M. Homology models of the HIV-1 attachment inhibitor BMS-626529 bound to gp120 suggest a unique mechanism of action. Proteins Struct. Funct. Bioinform. 2015, 83, 331–350. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, N.; Sun, Y.; Ray, N.; Lataillade, M.; Hanna, G.; Krystal, M. Activity of the HIV-1 Attachment Inhibitor BMS-626529, the Active Component of the Prodrug BMS-663068, against CD4-Independent Viruses and HIV-1 Envelopes Resistant to other Entry Inhibitors. Antimicrob. Agents Chemother. 2013, 57, 4172–4180. [Google Scholar] [CrossRef] [Green Version]
- Nowicka-Sans, B.; Gong, Y.-F.; McAuliffe, B.; Dicker, I.; Ho, H.-T.; Zhou, N.; Eggers, B.; Lin, P.-F.; Ray, N.; Wind-Rotolo, M.; et al. In Vitro Antiviral Characteristics of HIV-1 Attachment Inhibitor BMS-626529, the Active Component of the Prodrug BMS-663068. Antimicrob. Agents Chemother. 2012, 56, 3498–3507. [Google Scholar] [CrossRef] [Green Version]
- Ray, N.; Hwang, C.; Healy, M.; Whitcomb, J.; Lataillade, M.; Wind-Rotolo, M.; Krystal, M.; Hanna, G. Prediction of Virological Response and Assessment of Resistance Emergence to the HIV-1 Attachment Inhibitor BMS-626529 During 8-Day Monotherapy with Its Prodrug BMS-663068. JAIDS J. Acquir. Immune Defic. Syndr. 2013, 64, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Zhou, N.; Nowicka-Sans, B.; McAuliffe, B.; Ray, N.; Eggers, B.; Fang, H.; Fan, L.; Healy, M.; Langley, D.R.; Hwang, C.; et al. Genotypic correlates of susceptibility to HIV-1 attachment inhibitor BMS-626529, the active agent of the prodrug BMS-663068. J. Antimicrob. Chemother. 2013, 69, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Seval, N.; Frank, C.; Kozal, M. Fostemsavir for the treatment of HIV. Expert Rev. Anti-Infect. Ther. 2021, 19, 961–966. [Google Scholar] [CrossRef]
- Curreli, F.; Kwon, Y.D.; Zhang, H.; Scacalossi, D.; Belov, D.S.; Tikhonov, A.A.; Andreev, I.; Altieri, A.; Kurkin, A.V.; Kwong, P.D.; et al. Structure-Based Design of a Small Molecule CD4-Antagonist with Broad Spectrum Anti-HIV-1 Activity. J. Med. Chem. 2015, 58, 6909–6927. [Google Scholar] [CrossRef] [Green Version]
- Curreli, F.; Belov, D.S.; Ramesh, R.R.; Patel, N.; Altieri, A.; Kurkin, A.V.; Debnath, A.K. Design, synthesis and evaluation of small molecule CD4-mimics as entry inhibitors possessing broad spectrum anti-HIV-1 activity. Bioorg. Med. Chem. 2016, 24, 5988–6003. [Google Scholar] [CrossRef] [Green Version]
- Curreli, F.; Kwon, Y.D.; Belov, D.S.; Ramesh, R.R.; Kurkin, A.V.; Altieri, A.; Kwong, P.D.; Debnath, A.K. Synthesis, Antiviral Potency, in Vitro ADMET, and X-ray Structure of Potent CD4 Mimics as Entry Inhibitors that Target the Phe43 Cavity of HIV-1 gp120. J. Med. Chem. 2017, 60, 3124–3153. [Google Scholar] [CrossRef] [PubMed]
- Curreli, F.; Belov, D.S.; Kwon, Y.D.; Ramesh, R.; Furimsky, A.M.; O’Loughlin, K.; Byrge, P.C.; Iyer, L.V.; Mirsalis, J.C.; Kurkin, A.; et al. Structure-based lead optimization to improve antiviral potency and ADMET properties of phenyl-1H-pyrrole-carboxamide entry inhibitors targeted to HIV-1 gp120. Eur. J. Med. Chem. 2018, 154, 367–391. [Google Scholar] [CrossRef]
- Curreli, F.; Ahmed, S.; Victor, S.M.B.; Iusupov, I.R.; Belov, D.S.; Markov, P.O.; Kurkin, A.V.; Altieri, A.; Debnath, A.K. Preclinical Optimization of gp120 Entry Antagonists as anti-HIV-1 Agents with Improved Cytotoxicity and ADME Properties through Rational Design, Synthesis, and Antiviral Evaluation. J. Med. Chem. 2020, 63, 1724–1749. [Google Scholar] [CrossRef]
- Hamada, Y. Role of Pyridines in Medicinal Chemistry and Design of BACE1 Inhibitors Possessing a Pyridine Scaffold. Pyridine 2018, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Yin, Z.; Zhang, Z.; Bender, J.A.; Yang, Z.; Johnson, G.; Zadjura, L.M.; D’Arienzo, C.J.; Parker, D.D.; Gesenberg, C.; et al. Inhibitors of Human Immunodeficiency Virus Type 1 (HIV-1) Attachment. 5. An Evolution from Indole to Azaindoles Leading to the Discovery of 1-(4-Benzoylpiperazin-1-yl)-2-(4,7-dimethoxy-1H-pyrrolo[2,3-c]pyridin-3-yl)ethane-1,2-dione (BMS-488043), a Drug Candidate that Demonstrates Antiviral Activity in HIV-1-Infected Subjects. J. Med. Chem. 2009, 52, 7778–7787. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.D.; LaLonde, J.M.; Yang, Y.; Elban, M.A.; Sugawara, A.; Courter, J.R.; Jones, D.M.; Smith, A.B.; Debnath, A.K.; Kwong, P.D. Crystal Structures of HIV-1 gp120 Envelope Glycoprotein in Complex with NBD Analogues That Target the CD4-Binding Site. PLoS ONE 2014, 9, e85940. [Google Scholar] [CrossRef]
- LaLonde, J.M.; Kwon, Y.D.; Jones, D.M.; Sun, A.W.; Courter, J.R.; Soeta, T.; Kobayashi, T.; Princiotto, A.M.; Wu, X.; Schön, A.; et al. Structure-Based Design, Synthesis, and Characterization of Dual Hotspot Small-Molecule HIV-1 Entry Inhibitors. J. Med. Chem. 2012, 55, 4382–4396. [Google Scholar] [CrossRef] [Green Version]
- Hartz, R.A.; Ahuja, V.T.; Zhuo, X.; Mattson, R.J.; Denhart, D.J.; Deskus, J.A.; Vrudhula, V.M.; Pan, S.; Ditta, J.L.; Shu, Y.-Z.; et al. A Strategy to Minimize Reactive Metabolite Formation: Discovery of (S)-4-(1-Cyclopropyl-2-methoxyethyl)-6-[6-(difluoromethoxy)-2,5-dimethylpyridin-3-ylamino]-5-oxo-4,5-dihydropyrazine-2-carbonitrile as a Potent, Orally Bioavailable Corticotropin-Releasing Factor-1 Receptor Antagonist. J. Med. Chem. 2009, 52, 7653–7668. [Google Scholar] [CrossRef]
- Curreli, F.; Belov, D.S.; Ahmed, S.; Ramesh, R.R.; Kurkin, A.V.; Altieri, A.; Debnath, A.K. Synthesis, Antiviral Activity, and Structure–Activity Relationship of 1,3-Benzodioxolyl Pyrrole-Based Entry Inhibitors Targeting the Phe43 Cavity in HIV-1 gp120. ChemMedChem 2018, 13, 2332–2348. [Google Scholar] [CrossRef] [PubMed]
- Belov, D.S.; Curreli, F.; Kurkin, A.V.; Altieri, A.; Debnath, A.K. Guanidine-Containing Phenyl-Pyrrole Compounds as Probes for Generating HIV Entry Inhibitors Targeted to gp120. ChemistrySelect 2018, 3, 6450–6453. [Google Scholar] [CrossRef]
- Ohashi, N.; Harada, S.; Mizuguchi, T.; Irahara, Y.; Yamada, Y.; Kotani, M.; Nomura, W.; Matsushita, S.; Yoshimura, K.; Tamamura, H. Small-Molecule CD4 Mimics Containing Mono-cyclohexyl Moieties as HIV Entry Inhibitors. ChemMedChem 2016, 11, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Liu, J.; Bess, J., Jr.; Chertova, E.; Lifson, J.D.; Grisé, H.; Ofek, G.A.; Taylor, K.A.; Roux, K.H. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 2006, 441, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Yu, F.; Cai, L.; Debnath, A.K.; Jiang, S. Development of Small-molecule HIV Entry Inhibitors Specifically Targeting gp120 or gp41. Curr. Top. Med. Chem. 2015, 16, 1074–1090. [Google Scholar] [CrossRef] [Green Version]
- Yi, H.A.; Fochtman, B.C.; Rizzo, R.C.; Jacobs, A. Inhibition of HIV Entry by Targeting the Envelope Transmembrane Subunit gp41. Curr. HIV Res. 2016, 14, 283–294. [Google Scholar] [CrossRef]
- Qiu, J.; Liang, T.; Wu, J.; Yu, F.; He, X.; Tian, Y.; Xie, L.; Jiang, S.; Liu, S.; Li, L. N-Substituted Pyrrole Derivative 12m Inhibits HIV-1 Entry by Targeting Gp41 of HIV-1 Envelope Glycoprotein. Front. Pharmacol. 2019, 10, 859. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Lu, H.; Liu, S.; Zhao, Q.; He, Y.; Debnath, A.K. N-Substituted Pyrrole Derivatives as Novel Human Immunodeficiency Virus Type 1 Entry Inhibitors That Interfere with the gp41 Six-Helix Bundle Formation and Block Virus Fusion. Antimicrob. Agents Chemother. 2004, 48, 4349–4359. [Google Scholar] [CrossRef] [Green Version]
- Prati, F.; Uliassi, E.; Bolognesi, M.L. Two diseases, one approach: Multitarget drug discovery in Alzheimer’s and neglected tropical diseases. MedChemComm 2014, 5, 853–861. [Google Scholar] [CrossRef]
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef] [Green Version]
- Crucitti, G.C.; Métifiot, M.; Pescatori, L.; Messore, A.; Madia, V.N.; Pupo, G.; Saccoliti, F.; Scipione, L.; Tortorella, S.; Esposito, F.; et al. Structure—Activity Relationship of Pyrrolyl Diketo Acid Derivatives as Dual Inhibitors of HIV-1 Integrase and Reverse Transcriptase Ribonuclease H Domain. J. Med. Chem. 2015, 58, 1915–1928. [Google Scholar] [CrossRef]
- Messore, A.; Corona, A.; Madia, V.N.; Saccoliti, F.; Tudino, V.; De Leo, A.; Scipione, L.; De Vita, D.; Amendola, G.; Di Maro, S.; et al. Pyrrolyl Pyrazoles as Non-Diketo Acid Inhibitors of the HIV-1 Ribonuclease H Function of Reverse Transcriptase. ACS Med. Chem. Lett. 2020, 11, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zheng, G.; Fu, T.; Li, X.; Tu, G.; Li, Y.H.; Yao, X.; Xue, W.; Zhu, F. Prediction of the binding mode and resistance profile for a dual-target pyrrolyl diketo acid scaffold against HIV-1 integrase and reverse-transcriptase-associated ribonuclease H. Phys. Chem. Chem. Phys. 2018, 20, 23873–23884. [Google Scholar] [CrossRef]
- Poongavanam, V.; Corona, A.; Steinmann, C.; Scipione, L.; Grandi, N.; Pandolfi, F.; Di Santo, R.; Costi, R.; Esposito, F.; Tramontano, E.; et al. Structure-guided approach identifies a novel class of HIV-1 ribonuclease H inhibitors: Binding mode insights through magnesium complexation and site-directed mutagenesis studies. MedChemComm 2018, 9, 562–575. [Google Scholar] [CrossRef] [Green Version]
- Corona, A.; Di Leva, F.S.; Rigogliuso, G.; Pescatori, L.; Madia, V.N.; Subra, F.; Delelis, O.; Esposito, F.; Cadeddu, M.; Costi, R.; et al. New insights into the interaction between pyrrolyl diketoacids and HIV-1 integrase active site and comparison with RNase H. Antivir. Res. 2016, 134, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Krieger, I.V.; Freundlich, J.S.; Gawandi, V.B.; Roberts, J.P.; Gawandi, V.B.; Sun, Q.; Owen, J.L.; Fraile, M.T.; Huss, S.I.; Lavandera, J.-L.; et al. Structure-Guided Discovery of Phenyl-diketo Acids as Potent Inhibitors of M. tuberculosis Malate Synthase. Chem. Biol. 2012, 19, 1556–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menéndez-Arias, L.; Sebastián-Martín, A.; Alvarez, M. Viral reverse transcriptases. Virus Res. 2017, 234, 153–176. [Google Scholar] [CrossRef]
- Corona, A.; Di Leva, F.S.; Thierry, S.; Pescatori, L.; Crucitti, G.C.; Subra, F.; Delelis, O.; Esposito, F.; Rigogliuso, G.; Costi, R.; et al. Identification of Highly Conserved Residues Involved in Inhibition of HIV-1 RNase H Function by Diketo Acid Derivatives. Antimicrob. Agents Chemother. 2014, 58, 6101–6110. [Google Scholar] [CrossRef] [Green Version]
- Tramontano, E.; Esposito, F.; Badas, R.; Di Santo, R.; Costi, R.; La Colla, P. 6-[1-(4-Fluorophenyl)methyl-1H-pyrrol-2-yl)]-2,4-dioxo-5-hexenoic acid ethyl ester a novel diketo acid derivative which selectively inhibits the HIV-1 viral replication in cell culture and the ribonuclease H activity in vitro. Antivir. Res. 2005, 65, 117–124. [Google Scholar] [CrossRef]
- Massari, S.; Corona, A.; Distinto, S.; Desantis, J.; Caredda, A.; Sabatini, S.; Manfroni, G.; Felicetti, T.; Cecchetti, V.; Pannecouque, C.; et al. From cycloheptathiophene-3-carboxamide to oxazinone-based derivatives as allosteric HIV-1 ribonuclease H inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 55–74. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Sarafianos, S.G.; Wang, Z. Cutting into the Substrate Dominance: Pharmacophore and Structure-Based Approaches toward Inhibiting Human Immunodeficiency Virus Reverse Transcriptase-Associated Ribonuclease H. Acc. Chem. Res. 2020, 53, 218–230. [Google Scholar] [CrossRef]
- Beilhartz, G.L.; Ngure, M.; Johns, B.A.; DeAnda, F.; Gerondelis, P.; Götte, M. Inhibition of the Ribonuclease H Activity of HIV-1 Reverse Transcriptase by GSK5750 Correlates with Slow Enzyme-Inhibitor Dissociation. J. Biol. Chem. 2014, 289, 16270–16277. [Google Scholar] [CrossRef] [Green Version]
- Himmel, D.M.; Maegley, K.A.; Pauly, T.A.; Bauman, J.D.; Das, K.; Dharia, C.; Clark, A.D.; Ryan, K.; Hickey, M.J.; Love, R.A.; et al. Structure of HIV-1 Reverse Transcriptase with the Inhibitor β-Thujaplicinol Bound at the RNase H Active Site. Structure 2009, 17, 1625–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billamboz, M.; Bailly, F.; Lion, C.; Touati, N.; Vezin, H.; Calmels, C.; Andréola, M.-L.; Christ, F.; Debyser, Z.; Cotelle, P. Magnesium Chelating 2-Hydroxyisoquinoline-1,3(2H,4H)-diones, as Inhibitors of HIV-1 Integrase and/or the HIV-1 Reverse Transcriptase Ribonuclease H Domain: Discovery of a Novel Selective Inhibitor of the Ribonuclease H Function. J. Med. Chem. 2011, 54, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- Himmel, D.M.; Myshakina, N.S.; Ilina, T.; Van Ry, A.; Ho, W.C.; Parniak, M.A.; Arnold, E. Structure of a Dihydroxycoumarin Active-Site Inhibitor in Complex with the RNase H Domain of HIV-1 Reverse Transcriptase and Structure—Activity Analysis of Inhibitor Analogs. J. Mol. Biol. 2014, 426, 2617–2631. [Google Scholar] [CrossRef] [Green Version]
- Velthuisen, E.J.; Johns, B.A.; Gerondelis, P.; Chen, Y.; Li, M.; Mou, K.; Zhang, W.; Seal, J.W.; Hightower, K.E.; Miranda, S.R.; et al. Pyridopyrimidinone inhibitors of HIV-1 RNase H. Eur. J. Med. Chem. 2014, 83, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.; Onnis, V.; Deplano, A.; Bianco, G.; Demurtas, M.; Distinto, S.; Cheng, Y.-C.; Alcaro, S.; Esposito, F.; Tramontano, E. Design, synthesis and antiviral evaluation of novel heteroarylcarbothioamide derivatives as dual inhibitors of HIV-1 reverse transcriptase-associated RNase H and RDDP functions. Pathog. Dis. 2017, 75, ftx078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corona, A.; Schneider, A.; Schweimer, K.; Rösch, P.; Wöhrl, B.M.; Tramontano, E. Inhibition of Foamy Virus Reverse Transcriptase by Human Immunodeficiency Virus Type 1 RNase H Inhibitors. Antimicrob. Agents Chemother. 2014, 58, 4086–4093. [Google Scholar] [CrossRef] [Green Version]
- Meleddu, R.; Cannas, V.; Distinto, S.; Sarais, G.; DEL Vecchio, C.; Esposito, F.; Bianco, G.; Corona, A.; Cottiglia, F.; Alcaro, S.; et al. Design, Synthesis, and Biological Evaluation of 1,3-Diarylpropenones as Dual Inhibitors of HIV-1 Reverse Transcriptase. ChemMedChem 2014, 9, 1869–1879. [Google Scholar] [CrossRef]
- Shanty, A.; Raghu, K.; Mohanan, P. Synthesis, characterization: Spectral and theoretical, molecular docking and in vitro studies of copper complexes with HIV RT enzyme. J. Mol. Struct. 2019, 1197, 154–163. [Google Scholar] [CrossRef]
- Rogolino, D.; Carcelli, M.; Sechi, M.; Neamati, N. Viral enzymes containing magnesium: Metal binding as a successful strategy in drug design. Coord. Chem. Rev. 2012, 256, 3063–3086. [Google Scholar] [CrossRef]
- Barry, N.P.E.; Sadler, P.J. Exploration of the medical periodic table: Towards new targets. Chem. Commun. 2013, 49, 5106–5131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Song, W.; Clercq, E.; Zhan, P.; Liu, X. Recent Progress in the Research of Small Molecule HIV-1 RNase H Inhibitors. Curr. Med. Chem. 2014, 21, 1956–1967. [Google Scholar] [CrossRef]
- Stanton, R.A.; Lu, X.; Detorio, M.; Montero, C.; Hammond, E.T.; Ehteshami, M.; Domaoal, R.A.; Nettles, J.; Feraud, M.; Schinazi, R.F. Discovery, characterization, and lead optimization of 7-azaindole non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 4101–4105. [Google Scholar] [CrossRef] [PubMed]
- Schinazi, R.F.; Sommadossi, J.P.; Saalmann, V.; Cannon, D.L.; Xie, M.Y.; Hart, G.C.; Smith, G.A.; Hahn, E.F. Activities of 3′-azido-3′-deoxythymidine nucleotide dimers in primary lymphocytes infected with human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 1990, 34, 1061–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, H.-A.; Nam, H.-J.; Kim, U.-I.; Kim, K.; Kim, B.J. Synthesis and Antiviral Activity of Some 1H-Pyrrolo[3,2-b]pyridin-6-yl)acetic Acid Derivatives. Asian J. Chem. 2017, 29, 1199–1205. [Google Scholar] [CrossRef]
- Jassem, A.M.; Dhumad, A.M. Synthesis, Antimicrobial Activity, Anti-HIV Activity, and Molecular Docking of Novel 5-, 6- and 7-Membered Ring (1 H -Pyrrol-2-yl)aminolactams. ChemistrySelect 2021, 6, 2641–2647. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
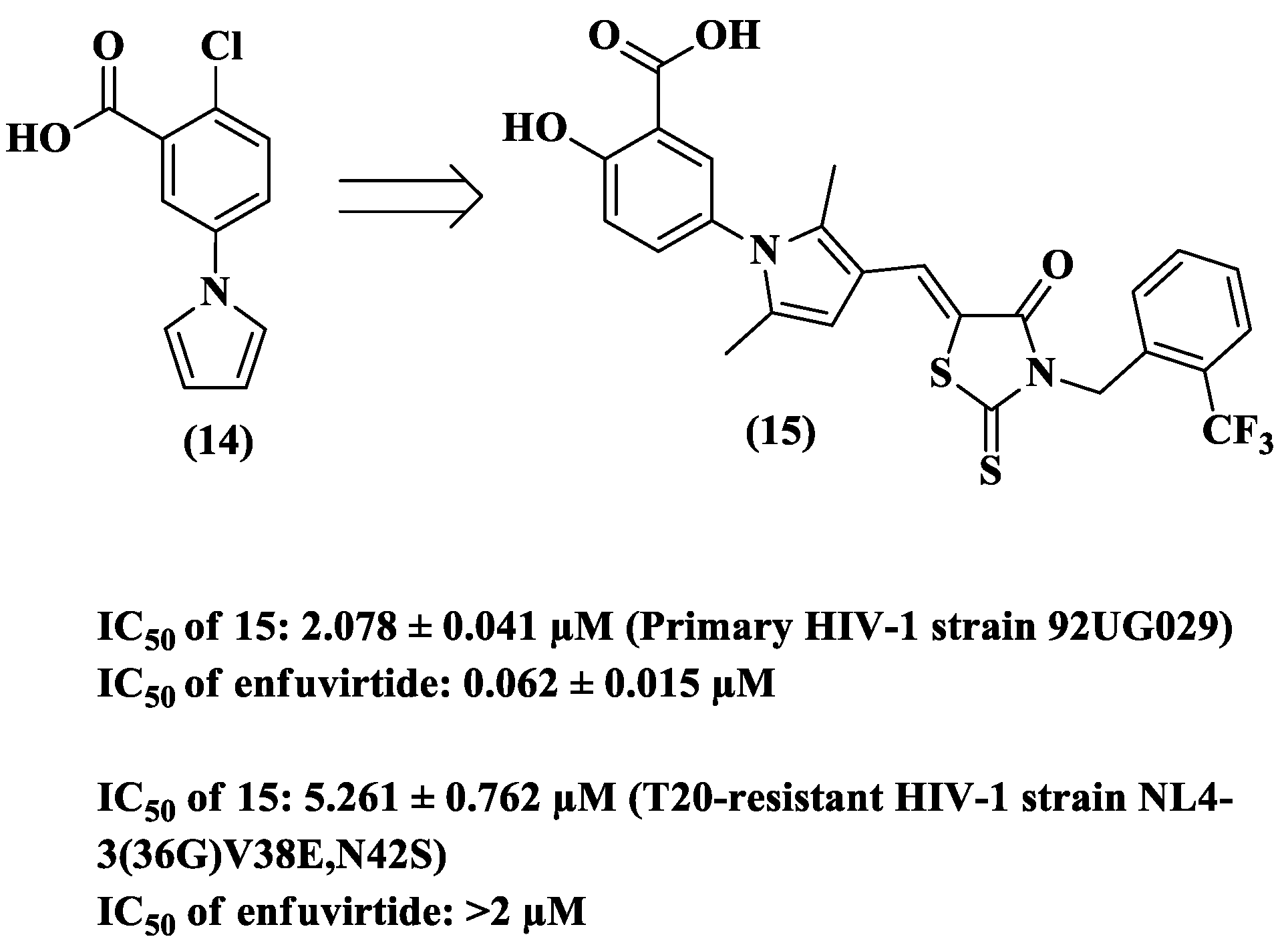{kind=link}
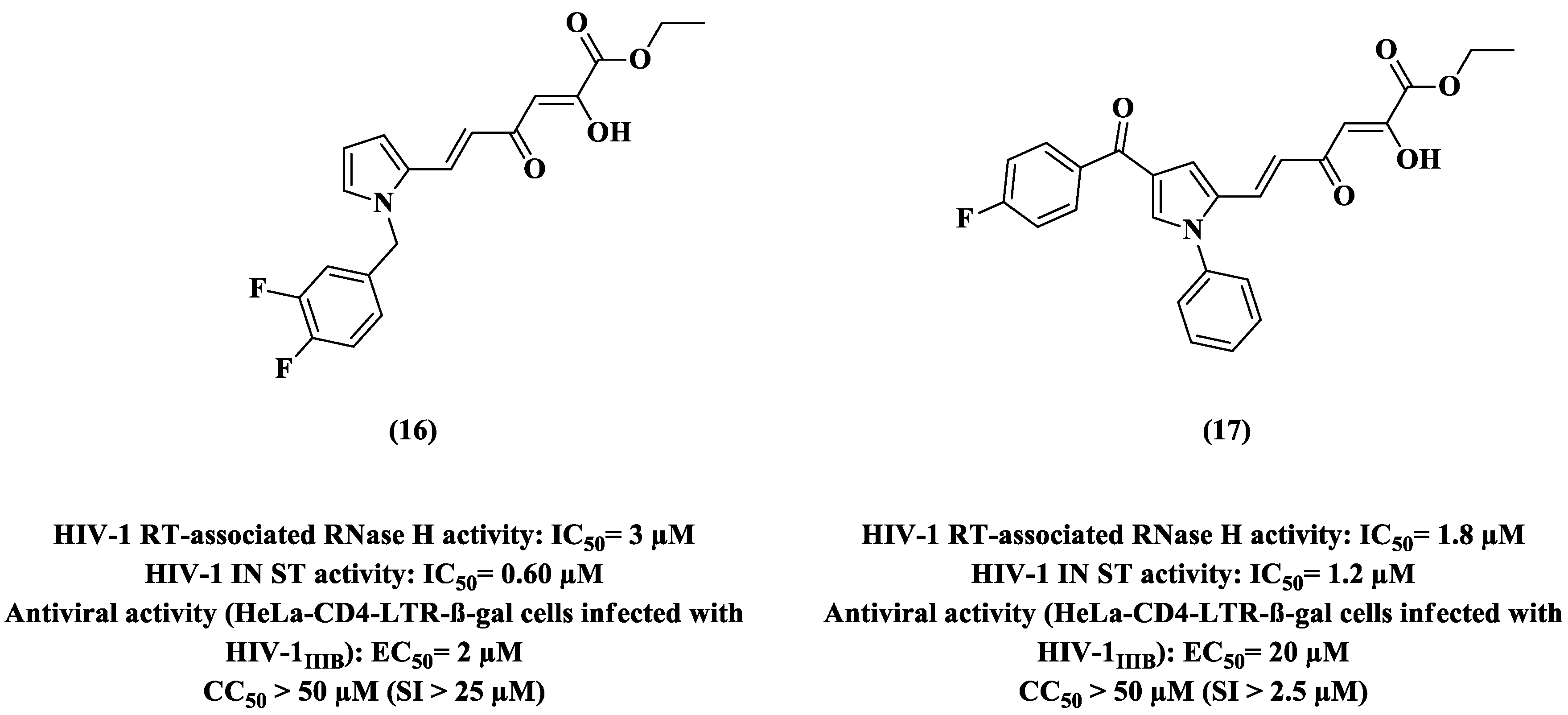{kind=link}
{kind=link}
{kind=link}
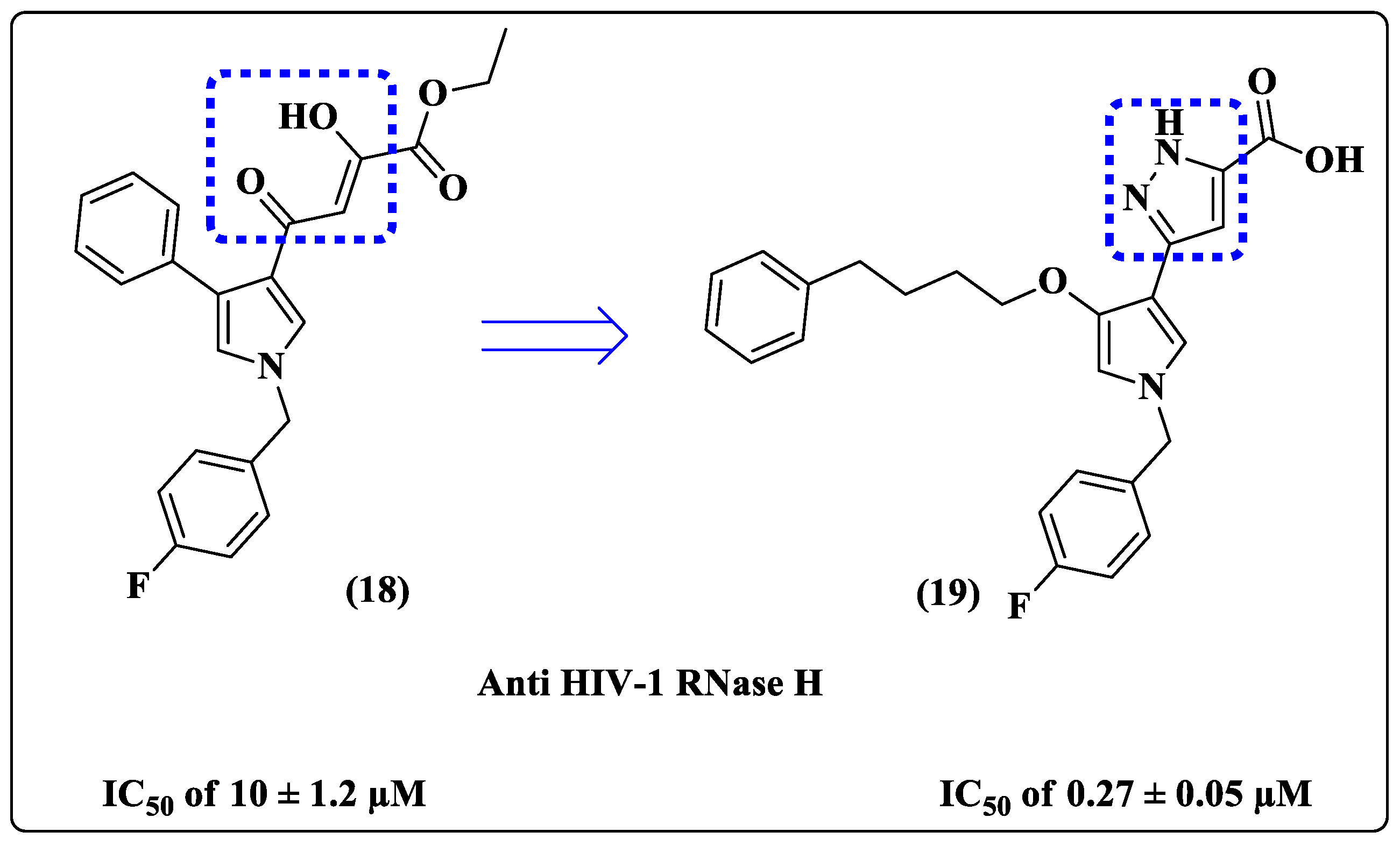
{kind=link}
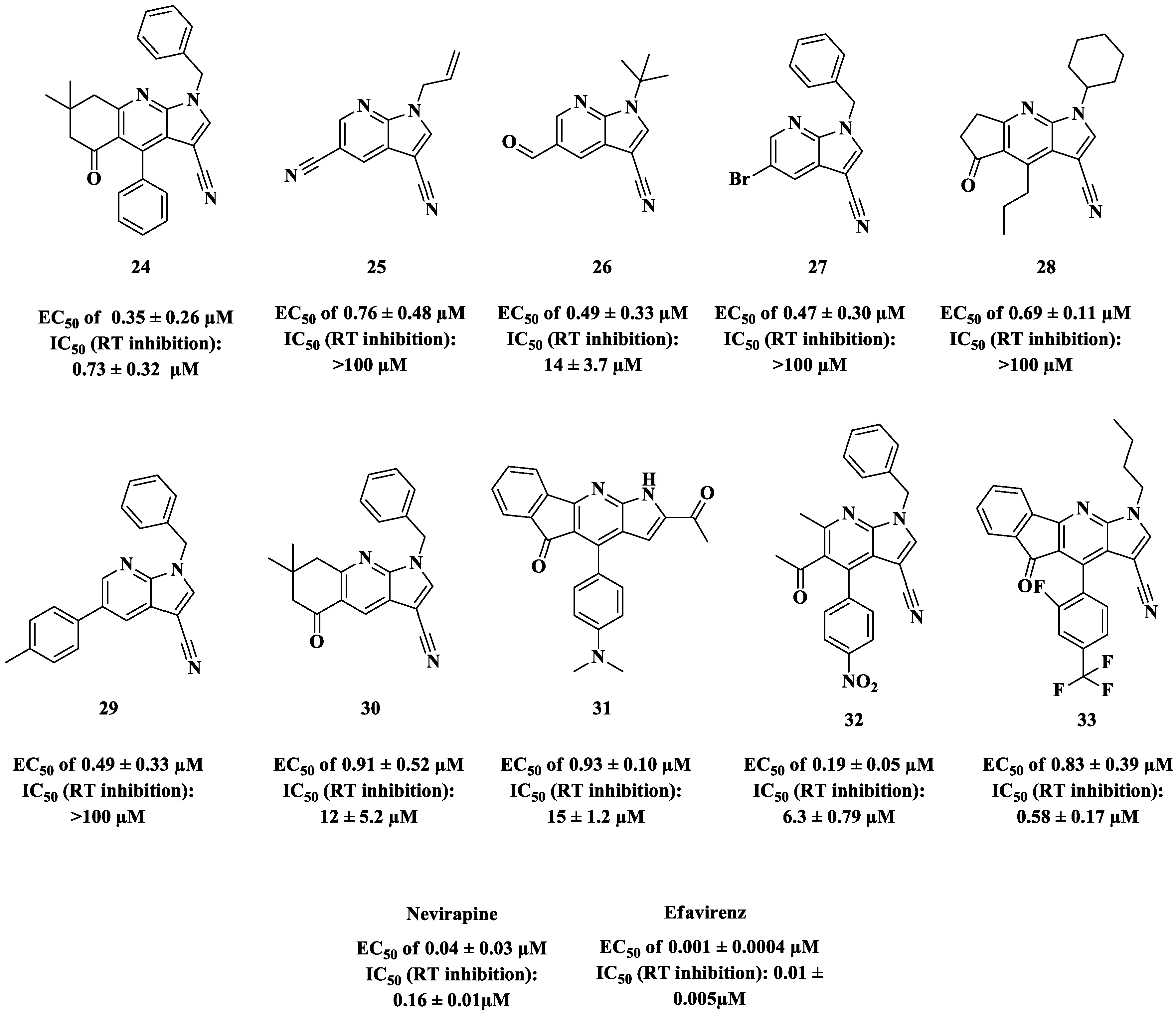{kind=link}
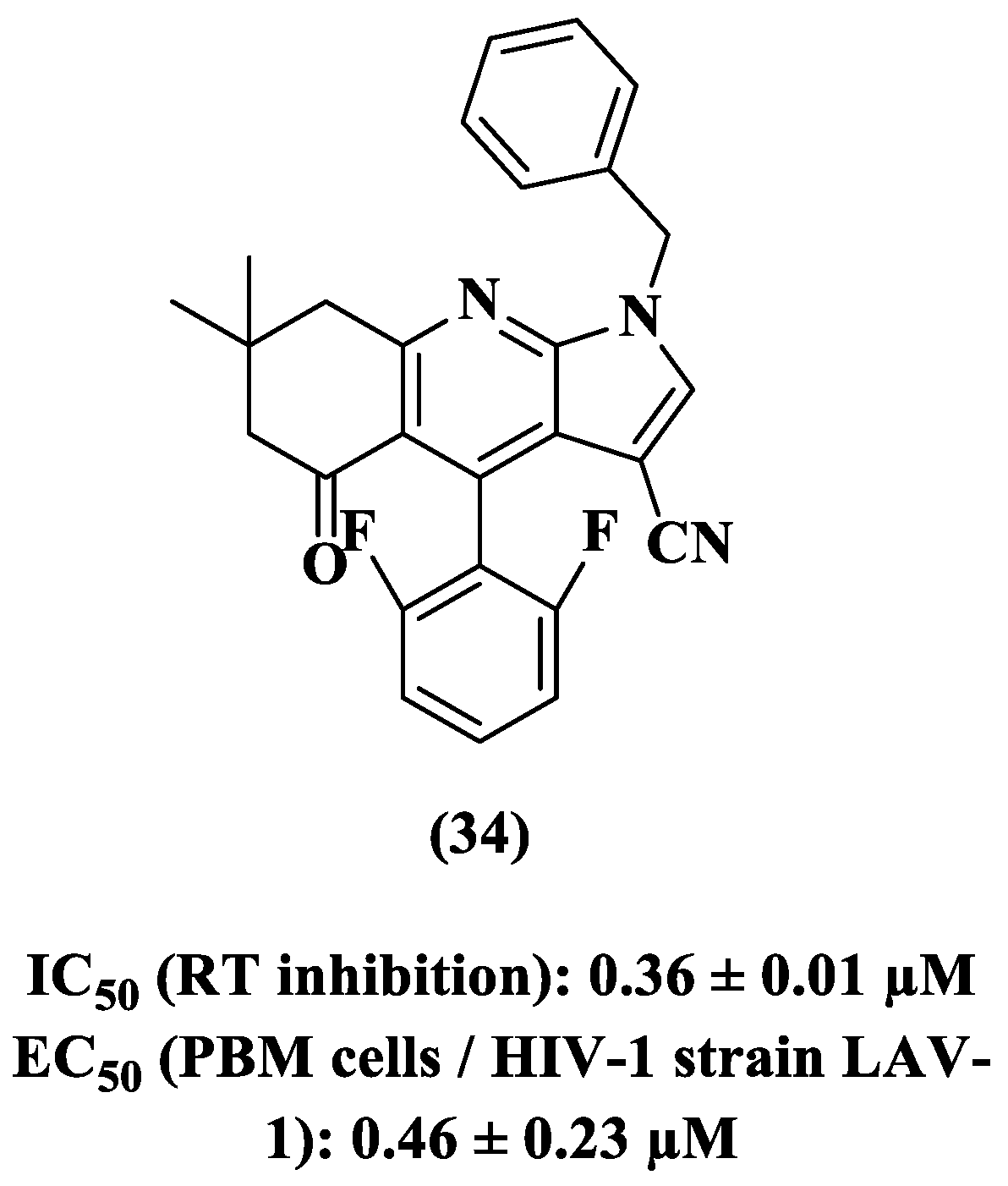{kind=link}
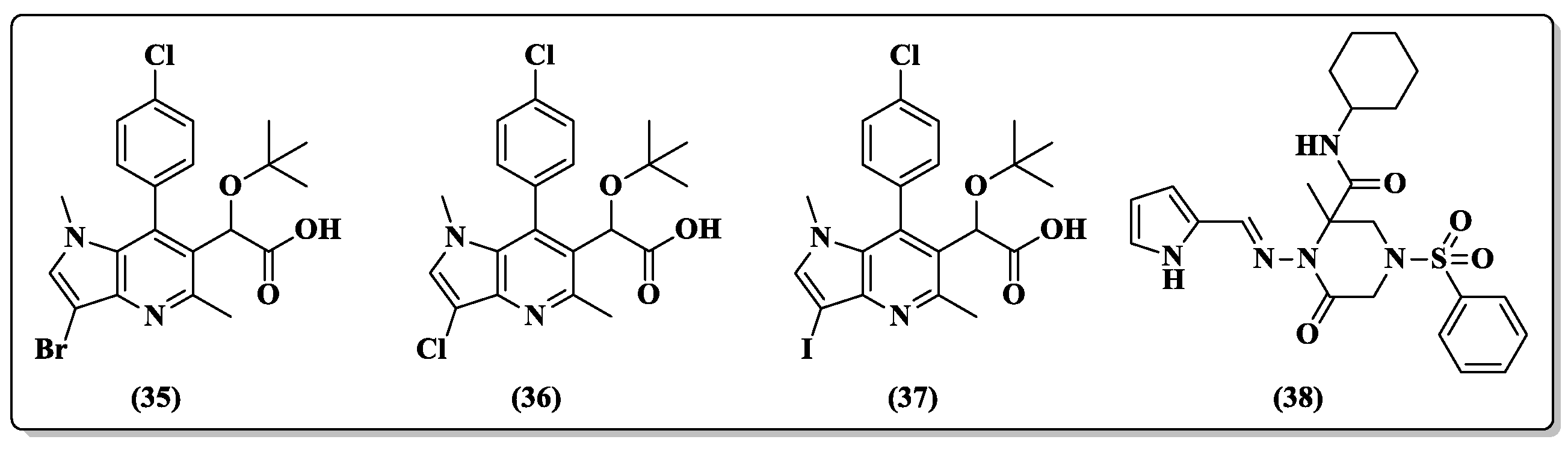{kind=link}
| NBD- Analog | TZM-bl Cells (Mean ± SD) a | MT-2 Cells (Mean ± SD) a | SI b | Inhibition of HIV-1 RT | ||
|---|---|---|---|---|---|---|
| IC50 (μM) | CC50 (μM) | IC50 (μM) | CC50 (μM) | IC50 (μM) | ||
| 1 | 2.2 ± 0.2 d/2.5 ± 0.2 f | 23.6 ± 1.1 d/~28 f | 0.85 ± 0.06 d/1.5 ± 0.06 f | 24.8 ± 2.3 d/~28 f | 10.7 d/~11.2 f | 47 d/43.4 f |
| 2 f | 4.7 ± 1.1 | 23.7 ± 1.1 | 4.7 ± 0.6 | >108 | 5.04 | - |
| S-(3) | 2.1 ± 0.2 | 33.6 ± 0.8 | 2.7 ± 0.33 | 25.8 ± 8.8 | - | - |
| S-(4) | 0.59 ± 0.06 | 40.5 ± 1.3 | 0.52 ± 0.05 | 30.5 ± 0.6 | - | - |
| 5 | 0.45 ± 0.05 | 38.8 ± 0.8 | 0.76 ± 0.3 | 38 ± 1 | 86.2 | 7.2 |
| 6 | 0.64 ± 0.06 | 39.5 ± 2.3 | 0.96 ± 0.1 | 37 ± 1.5 | 61.7 | 8.4 |
| 7 | 0.089 ± 0.001 | 21.9 ± 0.5 | 0.18 ± 0.001 | 22.1 ± 1 | 246 | NT c |
| 8 | 0.16 ± 0.004 | 109.3 ± 2 | NT c | NT c | 683.1 | NT c |
| 9 | 2.3 ± 0.1 | 145.6 ± 7.6 | 3.8 ± 1.4 | 193 ± 6.3 | 63.3 | 30.6 ± 2.7 |
| 10 | 4.3 ± 0.8 | 142.3 ± 2.6 | 4.5 ± 0.9 | 191.5 ± 2.6 | 33.1 | 39.5 ± 2.8 |
| 11 | 2.7 ± 0.3 | 95.8 ± 1.4 | 2.98 ± 0.1 | 180 ± 2 | 35.5 | 28.1 ± 7.9 |
| 12 g [48] | 10.8 ± 0.6 μM | 145.9 ± 8.9 μM | NT c | NT c | - | NT c |
| 13 g | 21.3 ± 5.0 μM | - | NT c | NT c | - | NT c |
| Temsavir | <1 nM e | >100 e | NT c | NT c | - | NT c |
| Compound | % of Inhibition (Tested Concentration) | % of Cell Viability | |||||
|---|---|---|---|---|---|---|---|
| 10 μM | 50 μM | 100 μM | 150 μM | ||||
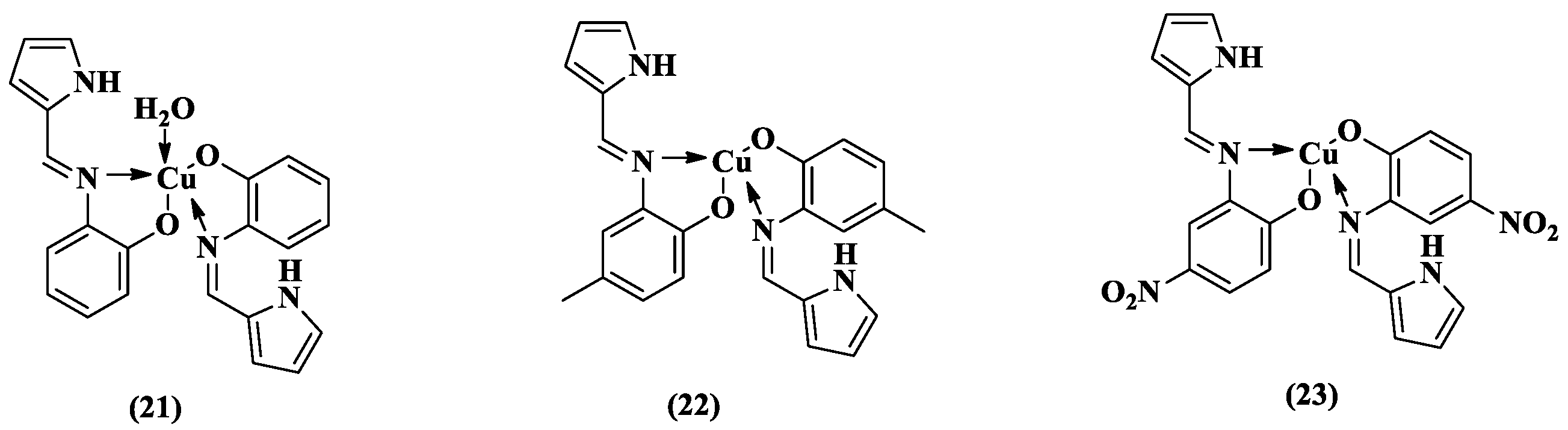
| 21 | 2.11 (22.16 μM) | 46.54 (110.84 μM) | 70.18 (221.69 μM) | 38 | 32 | 25 | 20 |
| 22 | 10.82 (21.68 μM) | 46.54 (108.43 μM) | 67.54 (216.87 μM) | 100 | 100 | 100 | 100 |
| 23 | 6.34 (19.11 μM) | 35.17 (95.59 μM) | 48.44 (191.18 μM) | 100 | 100 | 100 | 100 |
| Compound | Antiviral Activity (EC50) (μM) | CC50 (μM) b | |||
|---|---|---|---|---|---|
| HTLV-IIIB a | HIV-1, IIIB b | HIV-2, ROD b | HIV-1, IIIB b | HIV-2, ROD b | |
| 35 | 5.02 | NT c | NT c | NT c | NT c |
| 36 | 5.03 | NT c | NT c | NT c | NT c |
| 37 | 5.07 | NT c | NT c | NT c | NT c |
| 38 | NT c | 2.74 ± 1.08 | >17.13 | 18.93 ± 4.0 | >18.95 |
| AZT | 0.0063 | NT c | NT c | NT c | NT c |
| Raltegravir | 0.0063 | NT c | NT c | NT c | NT c |
| Efavirenz | NT c | 0.00441 | NT c | >6.327 | NT c |
| Nevirapine | NT c | 0.198 | NT c | >14.02 | NT c |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianco, M.d.C.A.D.; Marinho, D.I.L.F.; Hoelz, L.V.B.; Bastos, M.M.; Boechat, N. Pyrroles as Privileged Scaffolds in the Search for New Potential HIV Inhibitors. Pharmaceuticals 2021, 14, 893. https://doi.org/10.3390/ph14090893
Bianco MdCAD, Marinho DILF, Hoelz LVB, Bastos MM, Boechat N. Pyrroles as Privileged Scaffolds in the Search for New Potential HIV Inhibitors. Pharmaceuticals. 2021; 14(9):893. https://doi.org/10.3390/ph14090893
Chicago/Turabian StyleBianco, Maria da Conceição Avelino Dias, Debora Inacio Leite Firmino Marinho, Lucas Villas Boas Hoelz, Monica Macedo Bastos, and Nubia Boechat. 2021. "Pyrroles as Privileged Scaffolds in the Search for New Potential HIV Inhibitors" Pharmaceuticals 14, no. 9: 893. https://doi.org/10.3390/ph14090893
APA StyleBianco, M. d. C. A. D., Marinho, D. I. L. F., Hoelz, L. V. B., Bastos, M. M., & Boechat, N. (2021). Pyrroles as Privileged Scaffolds in the Search for New Potential HIV Inhibitors. Pharmaceuticals, 14(9), 893. https://doi.org/10.3390/ph14090893