
TGF-β Inhibitors for Therapeutic Management of Kidney Fibrosis


Abstract
:1. Introduction

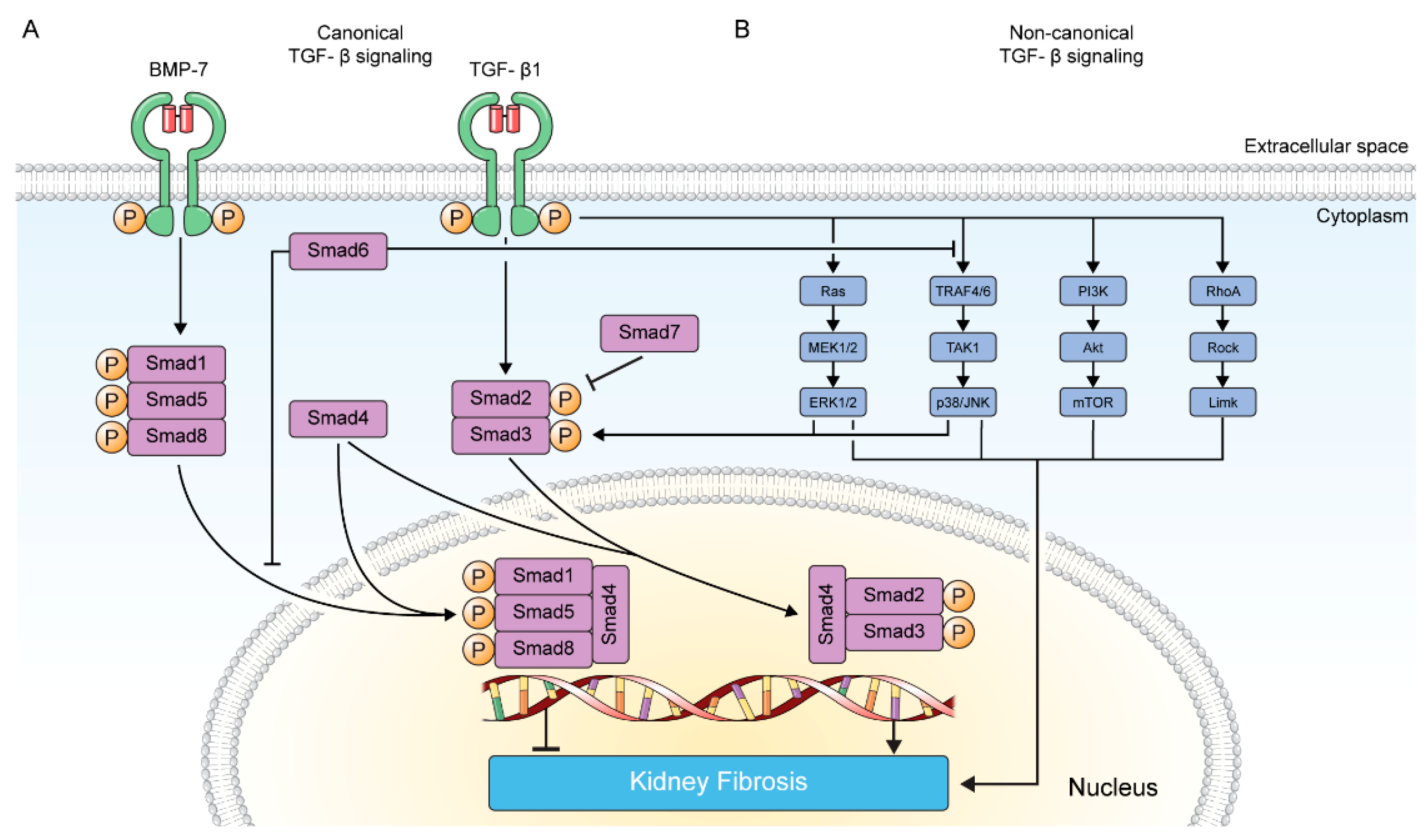
2. TGF-β Signaling in Kidney Fibrosis
2.1. Canonical TGF-β Signaling Pathway
2.2. Non-Canonical TGF-β Signaling Pathway
2.2.1. MAP Kinase Pathway
2.2.2. p38/JNK Pathway
2.2.3. PI3K/Akt Pathway
2.2.4. RhoA GTPase Pathway
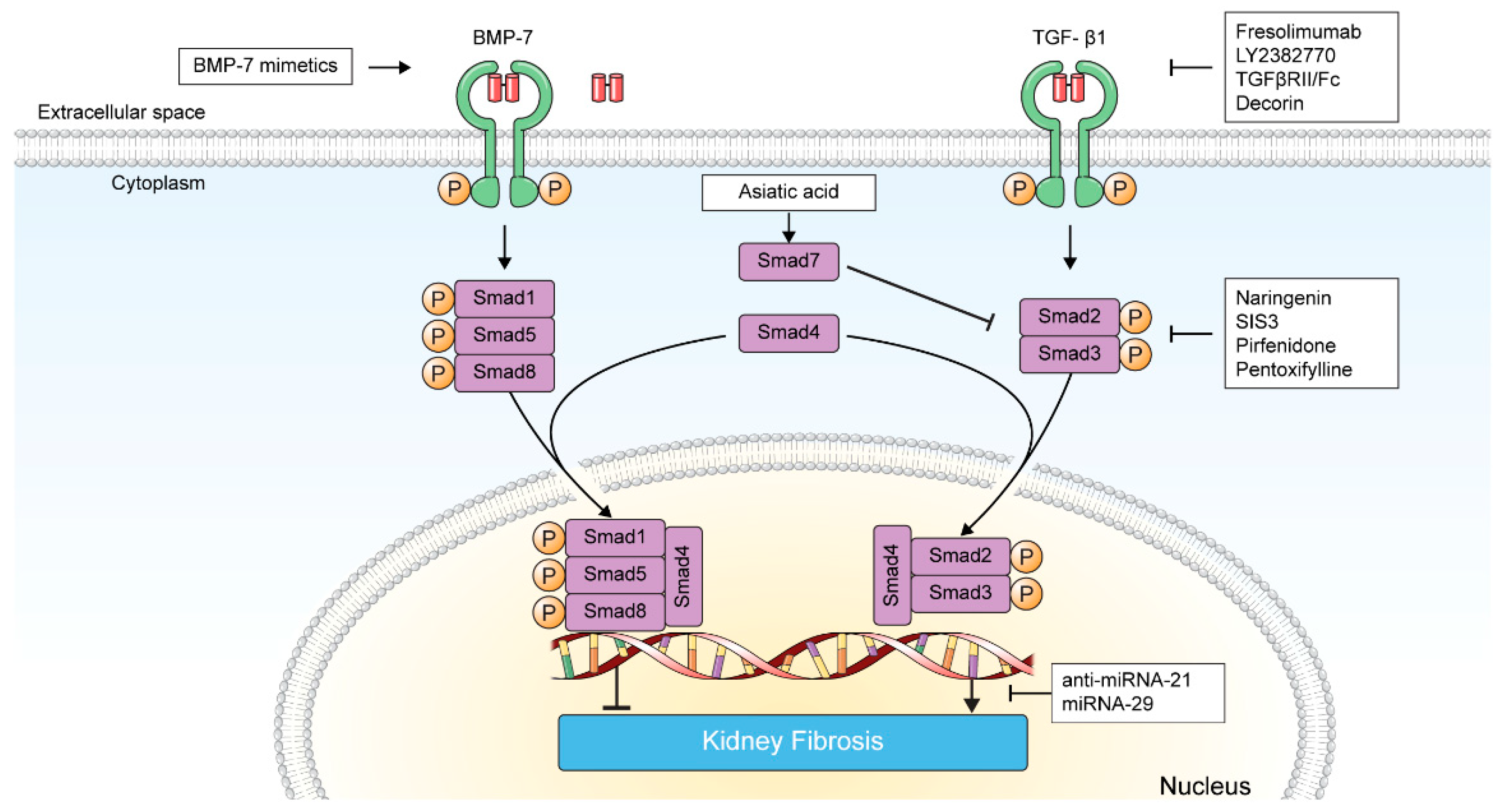
3. Targeting TGF-β in Kidney Fibrosis
3.1. Direct Inhibition of TGF-β-TGF-β Receptor Interaction
3.1.1. Fresolimumab

3.1.2. LY2382770

{kind=link}
{kind=link}
{kind=link}
| Agent | ClinicalTrial.gov Identifier | Notes | Reference |
|---|---|---|---|
| Fresolimumab | NCT00464321 | Treatment-resistant primary FSGS 1 N = 16 Fresolimumab was safe and well tolerated | [55] |
| NCT01665391 | Steroid resistant primary FSGS N = 36 Non-significant but greater eGFR 2 decline in the placebo group than in either of the fresolimumab-treated groups | [16] | |
| LY2382770 | NCT01113801 | Diabetic nephropathy N = 258 LY2382770 did not slow progression of diabetic nephropathy | [15] |
| VPI-2690B | NCT02251067 | Diabetic nephropathy N = 165 VPI-2690B failed to improve change in serum creatinine level | [56] |
| THR-184 | NCT01830920 | Cardiac surgery requiring CPB 3 N = 401 Administration of perioperative THR-184 failed to demonstrate beneficial effects on kidney function | [57] |
| Pirfenidone | NCT00001959 | FSGS N = 18 The decline in eGFR improved after pirfenidone treatment | [17] |
| NCT00063583 | Diabetic nephropathy N = 77 eGFR change was not statistically different between the placebo and pirfenidone groups | [18] | |
| NCT04258397 | CKD 4 (eGFR ≥ 20 mL/min/1.73 m2) N = 200 (Recruiting) | [58] | |
| Pentoxifylline | NCT00285298 | CKD with proteinuria (≥1 g/24 h) N = 40 Pentoxifylline group showed significantly slower eGFR decline compared with the placebo group | [59] |
| - | Diabetic nephropathy N = 169 Addition of pentoxifylline to RAS 6 inhibitors resulted in a smaller decrease in eGFR and a greater reduction in residual albuminuria | [60] | |
| - | CKD (eGFR < 60 mL/min/1.73 m2) N = 91 Pentoxifylline decreased inflammatory markers in CKD and stabilized renal function | [61] | |
| NCT03625648 | Diabetic nephropathy N = 2510 (recruiting) Primary outcome: Time to KFRT 5 or death | [62] | |
| NCT05487755 | Diabetic nephropathy N = 90 (planned) Primary outcome: Change in serum creatinine and urine albumin-to-creatinine ratio | [63] |
3.1.3. TGFβRII/Fc
3.1.4. Decorin
3.1.5. VPI-2690B
3.2. Inhibition of TGF-β Signaling
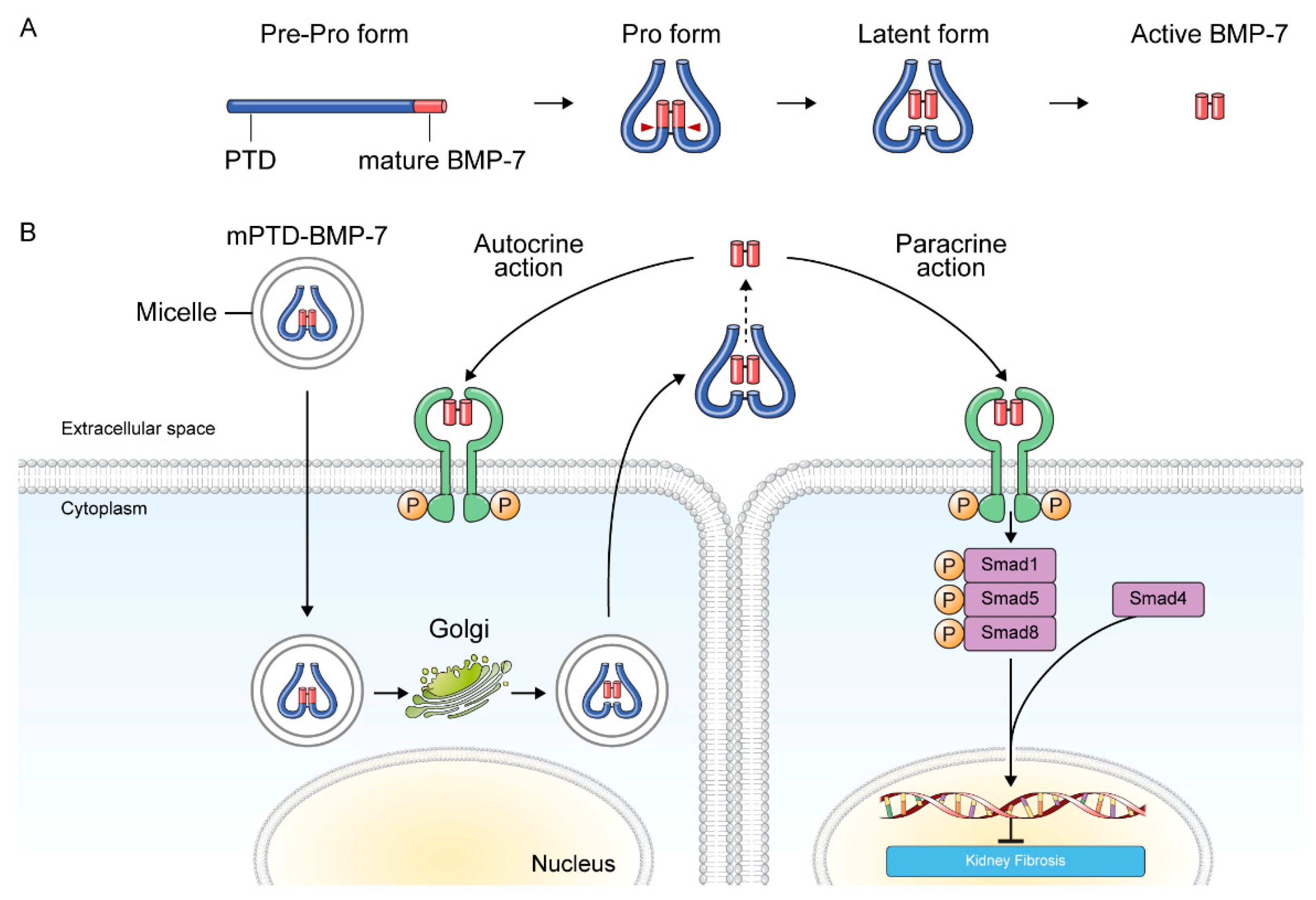
3.2.1. BMP-7 and BMP-7 Agonists

3.2.2. Smad Agonists/Inhibitors
3.2.3. Pirfenidone
3.2.4. Pentoxifylline
3.3. Inhibition of TGF-β-Induced Transcription Product
MicroRNAs
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xie, Y.; Bowe, B.; Mokdad, A.H.; Xian, H.; Yan, Y.; Li, T.; Maddukuri, G.; Tsai, C.Y.; Floyd, T.; Al-Aly, Z. Analysis of the global burden of disease study highlights the global, regional, and national trends of chronic kidney disease epidemiology from 1990 to 2016. Kidney Int. 2018, 94, 567–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- United States Renal Data System. 2021 USRDS Annual Data Report: Epidemiology of Kidney Disease in the United States; National Institutes of Health: Bethesda, MD, USA; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2021.
- Boenink, R.; Astley, M.E.; Huijben, J.A.; Stel, V.S.; Kerschbaum, J.; Ots-Rosenberg, M.; Asberg, A.A.; Lopot, F.; Golan, E.; Castro de la Nuez, P.; et al. The era registry annual report 2019: Summary and age comparisons. Clin. Kidney J. 2022, 15, 452–472. [Google Scholar] [CrossRef] [PubMed]
- Risdon, R.A.; Sloper, J.C.; De Wardener, H.E. Relationship between renal function and histological changes found in renal-biopsy specimens from patients with persistent glomerular nephritis. Lancet 1968, 2, 363–366. [Google Scholar] [CrossRef]
- Bohle, A.; Grund, K.E.; Mackensen, S.; Tolon, M. Correlations between renal interstitium and level of serum creatinine. Morphometric investigations of biopsies in perimembranous glomerulonephritis. Virchows Arch. A Pathol. Anat. Histol. 1977, 373, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Eddy, A.A.; Neilson, E.G. Chronic kidney disease progression. J. Am. Soc. Nephrol. 2006, 17, 2964–2966. [Google Scholar] [CrossRef] [Green Version]
- Mutsaers, H.A.M.; Norregaard, R. Prostaglandin E2 receptors as therapeutic targets in renal fibrosis. Kidney Res. Clin. Pract. 2022, 41, 4–13. [Google Scholar] [CrossRef]
- Bhatia, D.; Capili, A.; Choi, M.E. Mitochondrial dysfunction in kidney injury, inflammation, and disease: Potential therapeutic approaches. Kidney Res. Clin. Pract. 2020, 39, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, M.; Kalluri, R. Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. Am. J. Physiol. Cell Physiol. 2013, 304, C216–C225. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Muragaki, Y.; Saika, S.; Roberts, A.B.; Ooshima, A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 2003, 112, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Russo, L.M.; del Re, E.; Brown, D.; Lin, H.Y. Evidence for a role of transforming growth factor (TGF)-beta1 in the induction of postglomerular albuminuria in diabetic nephropathy: Amelioration by soluble TGF-beta type II receptor. Diabetes 2007, 56, 380–388. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Voelker, J.; Berg, P.H.; Sheetz, M.; Duffin, K.; Shen, T.; Moser, B.; Greene, T.; Blumenthal, S.S.; Rychlik, I.; Yagil, Y.; et al. Anti-TGF-beta1 antibody therapy in patients with diabetic nephropathy. J. Am. Soc. Nephrol. 2017, 28, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Vincenti, F.; Fervenza, F.C.; Campbell, K.N.; Diaz, M.; Gesualdo, L.; Nelson, P.; Praga, M.; Radhakrishnan, J.; Sellin, L.; Singh, A.; et al. A phase 2, double-blind, placebo-controlled, randomized study of fresolimumab in patients with steroid-resistant primary focal segmental glomerulosclerosis. Kidney Int. Rep. 2017, 2, 800–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, M.E.; Smith, D.C.; Branton, M.H.; Penzak, S.R.; Kopp, J.B. Pirfenidone slows renal function decline in patients with focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2007, 2, 906–913. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; Ix, J.H.; Mathew, A.V.; Cho, M.; Pflueger, A.; Dunn, S.R.; Francos, B.; Sharma, S.; Falkner, B.; McGowan, T.A.; et al. Pirfenidone for diabetic nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1144–1151. [Google Scholar] [CrossRef] [Green Version]
- Li, M.O.; Sanjabi, S.; Flavell, R.A. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 2006, 25, 455–471. [Google Scholar] [CrossRef] [Green Version]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Bottinger, E.P.; Bitzer, M. TGF-beta signaling in renal disease. J. Am. Soc. Nephrol. 2002, 13, 2600–2610. [Google Scholar] [CrossRef]
- Meng, X.M.; Chung, A.C.; Lan, H.Y. Role of the TGF-beta/BMP-7/Smad pathways in renal diseases. Clin. Sci. 2013, 124, 243–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basile, D.P.; Ullah, M.M.; Collet, J.A.; Mehrotra, P. T helper 17 cells in the pathophysiology of acute and chronic kidney disease. Kidney Res. Clin. Pract. 2021, 40, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Huminiecki, L.; Goldovsky, L.; Freilich, S.; Moustakas, A.; Ouzounis, C.; Heldin, C.H. Emergence, development and diversification of the TGF-beta signalling pathway within the animal kingdom. BMC Evol. Biol. 2009, 9, 28. [Google Scholar] [CrossRef] [Green Version]
- Wrana, J.L. Signaling by the TGF-beta superfamily. Cold Spring Harb. Perspect. Biol. 2013, 5, a011197. [Google Scholar] [CrossRef] [Green Version]
- Hinck, A.P. Structural studies of the tgf-betas and their receptors—Insights into evolution of the TGF-beta superfamily. FEBS Lett. 2012, 586, 1860–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates erk map kinase signalling through direct phosphorylation of shca. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Ning, X.; Li, R.; Yang, Z.; Yang, X.; Sun, S.; Qian, Q. Signalling pathways involved in hypoxia-induced renal fibrosis. J. Cell. Mol. Med. 2017, 21, 1248–1259. [Google Scholar] [CrossRef] [Green Version]
- ten Dijke, P.; Arthur, H.M. Extracellular control of TGF-beta signalling in vascular development and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Tzavlaki, K.; Moustakas, A. TGF-β signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef] [Green Version]
- Abdollah, S.; Macias-Silva, M.; Tsukazaki, T.; Hayashi, H.; Attisano, L.; Wrana, J.L. TbetaRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J. Biol. Chem. 1997, 272, 27678–27685. [Google Scholar] [CrossRef]
- Souchelnytskyi, S.; Tamaki, K.; Engstrom, U.; Wernstedt, C.; ten Dijke, P.; Heldin, C.H. Phosphorylation of Ser465 and Ser467 in the C terminus of Smad2 mediates interaction with Smad4 and is required for transforming growth factor-beta signaling. J. Biol. Chem. 1997, 272, 28107–28115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Sun, Y.; Constantinescu, S.N.; Karam, E.; Weinberg, R.A.; Lodish, H.F. Transforming growth factor beta-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10669–10674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, A.; Lagna, G.; Massague, J.; Hemmati-Brivanlou, A. Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev. 1998, 12, 186–197. [Google Scholar] [CrossRef] [Green Version]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 2001, 276, 12477–12480. [Google Scholar] [CrossRef] [Green Version]
- Funaba, M.; Zimmerman, C.M.; Mathews, L.S. Modulation of Smad2-mediated signaling by extracellular signal-regulated kinase. J. Biol. Chem. 2002, 277, 41361–41368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretzschmar, M.; Doody, J.; Timokhina, I.; Massague, J. A mechanism of repression of TGF-beta/Smad signaling by oncogenic ras. Genes Dev. 1999, 13, 804–816. [Google Scholar] [CrossRef]
- Seong, H.A.; Jung, H.; Ha, H. Murine protein serine/threonine kinase 38 stimulates TGF-beta signaling in a kinase-dependent manner via direct phosphorylation of smad proteins. J. Biol. Chem. 2010, 285, 30959–30970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, J.Y.; Shin, I.; Arteaga, C.L. Type I transforming growth factor beta receptor binds to and activates phosphatidylinositol 3-kinase. J. Biol. Chem. 2005, 280, 10870–10876. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef] [PubMed]
- National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: Evaluation, classification, and stratification. Am. J. Kidney Dis. 2002, 39, S1–S266. [Google Scholar]
- Burton, C.; Harris, K.P. The role of proteinuria in the progression of chronic renal failure. Am. J. Kidney Dis. 1996, 27, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Ruggenenti, P.; Perna, A.; Mosconi, L.; Pisoni, R.; Remuzzi, G. Urinary protein excretion rate is the best independent predictor of ESRF in non-diabetic proteinuric chronic nephropathies. “Gruppo Italiano di Studi Epidemiologici in Nefrologia” (gisen). Kidney Int. 1998, 53, 1209–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddy, A.A. Proteinuria and interstitial injury. Nephrol. Dial. Transplant. 2004, 19, 277–281. [Google Scholar] [CrossRef] [Green Version]
- Akhurst, R.J. TGF-beta antagonists: Why suppress a tumor suppressor? J. Clin. Investig. 2002, 109, 1533–1536. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. Companies waver in efforts to target transforming growth factor beta in cancer. J. Natl. Cancer Inst. 2009, 101, 1664–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaswen, L.; Kulkarni, A.B.; Fredrickson, T.; Mittleman, B.; Schiffman, R.; Payne, S.; Longenecker, G.; Mozes, E.; Karlsson, S. Autoimmune manifestations in the transforming growth factor-beta 1 knockout mouse. Blood 1996, 87, 1439–1445. [Google Scholar] [CrossRef] [Green Version]
- Li, M.O.; Flavell, R.A. TGF-beta: A master of all T cell trades. Cell 2008, 134, 392–404. [Google Scholar] [CrossRef] [Green Version]
- Ling, H.; Li, X.; Jha, S.; Wang, W.; Karetskaya, L.; Pratt, B.; Ledbetter, S. Therapeutic role of TGF-beta-neutralizing antibody in mouse cyclosporin a nephropathy: Morphologic improvement associated with functional preservation. J. Am. Soc. Nephrol. 2003, 14, 377–388. [Google Scholar] [CrossRef]
- Dahly-Vernon, A.J.; Sharma, M.; McCarthy, E.T.; Savin, V.J.; Ledbetter, S.R.; Roman, R.J. Transforming growth factor-beta, 20-hete interaction, and glomerular injury in dahl salt-sensitive rats. Hypertension 2005, 45, 643–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benigni, A.; Zoja, C.; Campana, M.; Corna, D.; Sangalli, F.; Rottoli, D.; Gagliardini, E.; Conti, S.; Ledbetter, S.; Remuzzi, G. Beneficial effect of TGF-beta antagonism in treating diabetic nephropathy depends on when treatment is started. Nephron. Exp Nephrol. 2006, 104, e158–e168. [Google Scholar] [CrossRef]
- Trachtman, H.; Fervenza, F.C.; Gipson, D.S.; Heering, P.; Jayne, D.R.; Peters, H.; Rota, S.; Remuzzi, G.; Rump, L.C.; Sellin, L.K.; et al. A phase 1, single-dose study of fresolimumab, an anti-TGF-beta antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney Int. 2011, 79, 1236–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US National Library of Medicine. 2014. Available online: https://clinicaltrials.gov/ct2/show/NCT02251067 (accessed on 26 October 2022).
- Himmelfarb, J.; Chertow, G.M.; McCullough, P.A.; Mesana, T.; Shaw, A.D.; Sundt, T.M.; Brown, C.; Cortville, D.; Dagenais, F.; de Varennes, B.; et al. Perioperative thr-184 and aki after cardiac surgery. J. Am. Soc. Nephrol. 2018, 29, 670–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US National Library of Medicine. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04258397 (accessed on 26 October 2022).
- Perkins, R.M.; Aboudara, M.C.; Uy, A.L.; Olson, S.W.; Cushner, H.M.; Yuan, C.M. Effect of pentoxifylline on GFR decline in ckd: A pilot, double-blind, randomized, placebo-controlled trial. Am. J. Kidney Dis. 2009, 53, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Gonzalez, J.F.; Mora-Fernandez, C.; Muros de Fuentes, M.; Chahin, J.; Mendez, M.L.; Gallego, E.; Macia, M.; del Castillo, N.; Rivero, A.; Getino, M.A.; et al. Effect of pentoxifylline on renal function and urinary albumin excretion in patients with diabetic kidney disease: The predian trial. J. Am. Soc. Nephrol. 2015, 26, 220–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goicoechea, M.; Garcia de Vinuesa, S.; Quiroga, B.; Verdalles, U.; Barraca, D.; Yuste, C.; Panizo, N.; Verde, E.; Munoz, M.A.; Luno, J. Effects of pentoxifylline on inflammatory parameters in chronic kidney disease patients: A randomized trial. J. Nephrol. 2012, 25, 969–975. [Google Scholar] [CrossRef]
- Leehey, D.J.; Carlson, K.; Reda, D.J.; Craig, I.; Clise, C.; Conner, T.A.; Agarwal, R.; Kaufman, J.S.; Anderson, R.J.; Lammie, D.; et al. Pentoxifylline in diabetic kidney disease (VA PTXRx): Protocol for a pragmatic randomised controlled trial. BMJ Open 2021, 11, e053019. [Google Scholar] [CrossRef] [PubMed]
- US National Library of Medicine. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05487755 (accessed on 26 October 2022).
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Lin, H.Y.; Moustakas, A.; Knaus, P.; Wells, R.G.; Henis, Y.I.; Lodish, H.F. The soluble exoplasmic domain of the type II transforming growth factor (TGF)-beta receptor. A heterogeneously glycosylated protein with high affinity and selectivity for TGF-beta ligands. J. Biol. Chem. 1995, 270, 2747–2754. [Google Scholar] [CrossRef]
- Isaka, Y.; Akagi, Y.; Ando, Y.; Tsujie, M.; Sudo, T.; Ohno, N.; Border, W.A.; Noble, N.A.; Kaneda, Y.; Hori, M.; et al. Gene therapy by transforming growth factor-beta receptor-IgG Fc chimera suppressed extracellular matrix accumulation in experimental glomerulonephritis. Kidney Int. 1999, 55, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Mann, D.M.; Ruoslahti, E. Negative regulation of transforming growth factor-beta by the proteoglycan decorin. Nature 1990, 346, 281–284. [Google Scholar] [CrossRef]
- Williams, K.J.; Qiu, G.; Usui, H.K.; Dunn, S.R.; McCue, P.; Bottinger, E.; Iozzo, R.V.; Sharma, K. Decorin deficiency enhances progressive nephropathy in diabetic mice. Am. J. Pathol. 2007, 171, 1441–1450. [Google Scholar] [CrossRef] [Green Version]
- Merline, R.; Lazaroski, S.; Babelova, A.; Tsalastra-Greul, W.; Pfeilschifter, J.; Schluter, K.D.; Gunther, A.; Iozzo, R.V.; Schaefer, R.M.; Schaefer, L. Decorin deficiency in diabetic mice: Aggravation of nephropathy due to overexpression of profibrotic factors, enhanced apoptosis and mononuclear cell infiltration. J. Physiol. Pharmacol. 2009, 60 (Suppl. 4), 5–13. [Google Scholar]
- Border, W.A.; Noble, N.A.; Yamamoto, T.; Harper, J.R.; Yamaguchi, Y.; Pierschbacher, M.D.; Ruoslahti, E. Natural inhibitor of transforming growth factor-beta protects against scarring in experimental kidney disease. Nature 1992, 360, 361–364. [Google Scholar] [CrossRef]
- Isaka, Y.; Brees, D.K.; Ikegaya, K.; Kaneda, Y.; Imai, E.; Noble, N.A.; Border, W.A. Gene therapy by skeletal muscle expression of decorin prevents fibrotic disease in rat kidney. Nat. Med. 1996, 2, 418–423. [Google Scholar] [CrossRef]
- Worthington, J.J.; Klementowicz, J.E.; Travis, M.A. TGF-beta: A sleeping giant awoken by integrins. Trends Biochem. Sci. 2011, 36, 47–54. [Google Scholar] [CrossRef]
- Sarrazy, V.; Koehler, A.; Chow, M.L.; Zimina, E.; Li, C.X.; Kato, H.; Caldarone, C.A.; Hinz, B. Integrins αvβ5 and αvβ3 promote latent TGF-beta1 activation by human cardiac fibroblast contraction. Cardiovasc. Res. 2014, 102, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Breyer, M.D.; Susztak, K. The next generation of therapeutics for chronic kidney disease. Nat. Rev. Drug Discov. 2016, 15, 568–588. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Q.; Tang, B.; Zhang, C. Signaling pathways of chronic kidney diseases, implications for therapeutics. Signal Transduct. Target. Ther. 2022, 7, 182. [Google Scholar] [CrossRef] [PubMed]
- Bon, H.; Hales, P.; Lumb, S.; Holdsworth, G.; Johnson, T.; Qureshi, O.; Twomey, B.M. Spontaneous extracellular matrix accumulation in a human in vitro model of renal fibrosis is mediated by alphav integrins. Nephron 2019, 142, 328–350. [Google Scholar] [CrossRef] [PubMed]
- Li, R.X.; Yiu, W.H.; Tang, S.C. Role of bone morphogenetic protein-7 in renal fibrosis. Front. Physiol. 2015, 6, 114. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Hanai, J.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef]
- Wang, S.N.; Lapage, J.; Hirschberg, R. Loss of tubular bone morphogenetic protein-7 in diabetic nephropathy. J. Am. Soc. Nephrol. 2001, 12, 2392–2399. [Google Scholar] [CrossRef]
- Motazed, R.; Colville-Nash, P.; Kwan, J.T.; Dockrell, M.E. BMP-7 and proximal tubule epithelial cells: Activation of multiple signaling pathways reveals a novel anti-fibrotic mechanism. Pharm. Res. 2008, 25, 2440–2446. [Google Scholar] [CrossRef]
- Wang, S.; Hirschberg, R. Bone morphogenetic protein-7 signals opposing transforming growth factor beta in mesangial cells. J. Biol. Chem. 2004, 279, 23200–23206. [Google Scholar] [CrossRef] [Green Version]
- Vukicevic, S.; Basic, V.; Rogic, D.; Basic, N.; Shih, M.S.; Shepard, A.; Jin, D.; Dattatreyamurty, B.; Jones, W.; Dorai, H.; et al. Osteogenic protein-1 (bone morphogenetic protein-7) reduces severity of injury after ischemic acute renal failure in rat. J. Clin. Investig. 1998, 102, 202–214. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Shah, A.A.; Kalluri, R. Bone morphogenic protein-7 induces mesenchymal to epithelial transition in adult renal fibroblasts and facilitates regeneration of injured kidney. J. Biol. Chem. 2005, 280, 8094–8100. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef] [Green Version]
- Veerasamy, M.; Nguyen, T.Q.; Motazed, R.; Pearson, A.L.; Goldschmeding, R.; Dockrell, M.E. Differential regulation of E-cadherin and alpha-smooth muscle actin by BMP 7 in human renal proximal tubule epithelial cells and its implication in renal fibrosis. Am. J. Physiol. Ren. Physiol. 2009, 297, F1238–F1248. [Google Scholar] [CrossRef] [PubMed]
- Mitu, G.; Hirschberg, R. Bone morphogenetic protein-7 (BMP7) in chronic kidney disease. Front. Biosci. 2008, 13, 4726–4739. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Surendran, K.; Zawaideh, M.A.; Mathew, S.; Hruska, K.A. Bone morphogenetic protein 7: A novel treatment for chronic renal and bone disease. Curr. Opin. Nephrol. Hypertens. 2004, 13, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Bottiglio, C.; Kumar, N.; Maeshima, Y.; Strutz, F.; Muller, G.A.; Kalluri, R. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am. J. Physiol. Ren. Physiol. 2003, 285, F1060–F1067. [Google Scholar] [CrossRef] [Green Version]
- Morrissey, J.; Hruska, K.; Guo, G.; Wang, S.; Chen, Q.; Klahr, S. Bone morphogenetic protein-7 improves renal fibrosis and accelerates the return of renal function. J. Am. Soc. Nephrol. 2002, 13 (Suppl. 1), S14–S21. [Google Scholar] [CrossRef]
- Peng, W.; Zhou, X.; Xu, T.; Mao, Y.; Zhang, X.; Liu, H.; Liang, L.; Liu, L.; Liu, L.; Xiao, Y.; et al. BMP-7 ameliorates partial epithelial-mesenchymal transition by restoring snon protein level via Smad1/5 pathway in diabetic kidney disease. Cell Death Dis. 2022, 13, 254. [Google Scholar] [CrossRef]
- Lo, K.W.; Ulery, B.D.; Ashe, K.M.; Laurencin, C.T. Studies of bone morphogenetic protein-based surgical repair. Adv. Drug Deliv. Rev. 2012, 64, 1277–1291. [Google Scholar] [CrossRef] [Green Version]
- Swencki-Underwood, B.; Mills, J.K.; Vennarini, J.; Boakye, K.; Luo, J.; Pomerantz, S.; Cunningham, M.R.; Farrell, F.X.; Naso, M.F.; Amegadzie, B. Expression and characterization of a human BMP-7 variant with improved biochemical properties. Protein Expr. Purif. 2008, 57, 312–319. [Google Scholar] [CrossRef]
- Sugimoto, H.; LeBleu, V.S.; Bosukonda, D.; Keck, P.; Taduri, G.; Bechtel, W.; Okada, H.; Carlson, W., Jr.; Bey, P.; Rusckowski, M.; et al. Activin-like kinase 3 is important for kidney regeneration and reversal of fibrosis. Nat. Med. 2012, 18, 396–404. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Jeong, C.H.; Song, S.H.; Um, J.E.; Kim, H.S.; Yun, J.S.; Han, D.; Cho, E.S.; Nam, B.Y.; Yook, J.I.; et al. Micellized protein transduction domain-bone morphogenetic protein-7 efficiently blocks renal fibrosis via inhibition of transforming growth factor-beta-mediated epithelial-mesenchymal transition. Front. Pharmacol. 2020, 11, 591275. [Google Scholar] [CrossRef]
- Kim, N.H.; Cha, Y.H.; Kim, H.S.; Lee, S.E.; Huh, J.K.; Kim, J.K.; Kim, J.M.; Ryu, J.K.; Kim, H.J.; Lee, Y.; et al. A platform technique for growth factor delivery with novel mode of action. Biomaterials 2014, 35, 9888–9896. [Google Scholar] [CrossRef] [PubMed]
- Vendeville, A.; Rayne, F.; Bonhoure, A.; Bettache, N.; Montcourrier, P.; Beaumelle, B. HIV-1 Tat enters T cells using coated pits before translocating from acidified endosomes and eliciting biological responses. Mol. Biol. Cell 2004, 15, 2347–2360. [Google Scholar] [CrossRef] [PubMed]
- Wadia, J.S.; Stan, R.V.; Dowdy, S.F. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med. 2004, 10, 310–315. [Google Scholar] [CrossRef]
- Midgley, A.C.; Wei, Y.; Zhu, D.; Gao, F.; Yan, H.; Khalique, A.; Luo, W.; Jiang, H.; Liu, X.; Guo, J.; et al. Multifunctional natural polymer nanoparticles as antifibrotic gene carriers for CKD therapy. J. Am. Soc. Nephrol. 2020, 31, 2292–2311. [Google Scholar] [CrossRef]
- Wang, S.; Hirschberg, R. BMP7 antagonizes TGF-beta -dependent fibrogenesis in mesangial cells. Am. J. Physiol. Ren. Physiol. 2003, 284, F1006–F1013. [Google Scholar] [CrossRef] [Green Version]
- Cook, S.D.; Wolfe, M.W.; Salkeld, S.L.; Rueger, D.C. Effect of recombinant human osteogenic protein-1 on healing of segmental defects in non-human primates. J. Bone Jt. Surg. Am. 1995, 77, 734–750. [Google Scholar] [CrossRef]
- Magin, M.N.; Delling, G. Improved lumbar vertebral interbody fusion using rhOP-1: A comparison of autogenous bone graft, bovine hydroxylapatite (Bio-Sss), and BMP-7 (rhOP-1) in sheep. Spine 2001, 26, 469–478. [Google Scholar] [CrossRef]
- Ripamonti, U.; Crooks, J.; Rueger, D.C. Induction of bone formation by recombinant human osteogenic protein-1 and sintered porous hydroxyapatite in adult primates. Plast. Reconstr. Surg. 2001, 107, 977–988. [Google Scholar] [CrossRef]
- Blattert, T.R.; Delling, G.; Dalal, P.S.; Toth, C.A.; Balling, H.; Weckbach, A. Successful transpedicular lumbar interbody fusion by means of a composite of osteogenic protein-1 (rhBMP-7) and hydroxyapatite carrier: A comparison with autograft and hydroxyapatite in the sheep spine. Spine 2002, 27, 2697–2705. [Google Scholar] [CrossRef]
- Lu, H.H.; Kofron, M.D.; El-Amin, S.F.; Attawia, M.A.; Laurencin, C.T. In vitro bone formation using muscle-derived cells: A new paradigm for bone tissue engineering using polymer-bone morphogenetic protein matrices. Biochem. Biophys. Res. Commun. 2003, 305, 882–889. [Google Scholar] [CrossRef]
- Kim, H.J.; Han, M.A.; Shin, J.Y.; Jeon, J.H.; Lee, S.J.; Yoon, M.Y.; Kim, H.J.; Choi, E.J.; Do, S.H.; Yang, V.C.; et al. Intra-articular delivery of synovium-resident mesenchymal stem cells via BMP-7-loaded fibrous PLGA scaffolds for cartilage repair. J. Control. Release 2019, 302, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Pluhar, G.E.; Turner, A.S.; Pierce, A.R.; Toth, C.A.; Wheeler, D.L. A comparison of two biomaterial carriers for osteogenic protein-1 (BMP-7) in an ovine critical defect model. J Bone Jt. Surg. Br. 2006, 88, 960–966. [Google Scholar] [CrossRef]
- Neuerburg, C.; Mittlmeier, L.M.; Keppler, A.M.; Westphal, I.; Glass, A.; Saller, M.M.; Herlyn, P.K.E.; Richter, H.; Bocker, W.; Schieker, M.; et al. Growth factor-mediated augmentation of long bones: Evaluation of a BMP-7 loaded thermoresponsive hydrogel in a murine femoral intramedullary injection model. J. Orthop. Surg. Res. 2019, 14, 297. [Google Scholar] [CrossRef] [Green Version]
- Mantripragada, V.P.; Jayasuriya, A.C. Bone regeneration using injectable BMP-7 loaded chitosan microparticles in rat femoral defect. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 63, 596–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Chen, Y.G. Smad7: Not only a regulator, but also a cross-talk mediator of TGF-beta signalling. Biochem. J. 2011, 434, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Huang, X.R.; Li, A.G.; Liu, F.; Li, J.H.; Truong, L.D.; Wang, X.J.; Lan, H.Y. Signaling mechanism of TGF-beta1 in prevention of renal inflammation: Role of Smad7. J. Am. Soc. Nephrol. 2005, 16, 1371–1383. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y. Smad7 as a therapeutic agent for chronic kidney diseases. Front. Biosci. 2008, 13, 4984–4992. [Google Scholar] [CrossRef]
- Schiffer, M.; Bitzer, M.; Roberts, I.S.; Kopp, J.B.; ten Dijke, P.; Mundel, P.; Bottinger, E.P. Apoptosis in podocytes induced by TGF-beta and Smad7. J. Clin. Investig. 2001, 108, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y. Smads as therapeutic targets for chronic kidney disease. Kidney Res. Clin. Pract. 2012, 31, 4–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Qu, X.; Yao, J.; Caruana, G.; Ricardo, S.D.; Yamamoto, Y.; Yamamoto, H.; Bertram, J.F. Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes 2010, 59, 2612–2624. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Meng, X.M.; Huang, X.R.; Lan, H.Y. The preventive and therapeutic implication for renal fibrosis by targetting TGF-beta/smad3 signaling. Clin. Sci. 2018, 132, 1403–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.M.; Zhang, Y.; Huang, X.R.; Ren, G.L.; Li, J.; Lan, H.Y. Treatment of renal fibrosis by rebalancing TGF-beta/Smad signaling with the combination of asiatic acid and naringenin. Oncotarget 2015, 6, 36984–36997. [Google Scholar] [CrossRef]
- Taniguchi, H.; Ebina, M.; Kondoh, Y.; Ogura, T.; Azuma, A.; Suga, M.; Taguchi, Y.; Takahashi, H.; Nakata, K.; Sato, A.; et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur. Respir. J. 2010, 35, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.L.; Rojas, M.; Pardo, A.; Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 2017, 16, 810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tampe, D.; Zeisberg, M. Potential approaches to reverse or repair renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 226–237. [Google Scholar] [CrossRef]
- Ruwanpura, S.M.; Thomas, B.J.; Bardin, P.G. Pirfenidone: Molecular mechanisms and potential clinical applications in lung disease. Am. J. Respir. Cell Mol. Biol. 2020, 62, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Shihab, F.S.; Bennett, W.M.; Yi, H.; Andoh, T.F. Pirfenidone treatment decreases transforming growth factor-beta1 and matrix proteins and ameliorates fibrosis in chronic cyclosporine nephrotoxicity. Am. J. Transplant. 2002, 2, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Fukagawa, M.; Kuroda, T.; Hata, S.; Iwasaki, Y.; Nemoto, M.; Shirai, K.; Yamauchi, S.; Margolin, S.B.; Shimizu, F.; et al. Pirfenidone prevents collagen accumulation in the remnant kidney in rats with partial nephrectomy. Kidney Int. Suppl. 1997, 63, S239–S243. [Google Scholar] [PubMed]
- Al-Bayati, M.A.; Xie, Y.; Mohr, F.C.; Margolin, S.B.; Giri, S.N. Effect of pirfenidone against vanadate-induced kidney fibrosis in rats. Biochem. Pharmacol. 2002, 64, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Giri, S.N.; Al-Bayati, M.A.; Du, X.; Schelegle, E.; Mohr, F.C.; Margolin, S.B. Amelioration of doxorubicin-induced cardiac and renal toxicity by pirfenidone in rats. Cancer Chemother. Pharmacol. 2004, 53, 141–150. [Google Scholar]
- Miric, G.; Dallemagne, C.; Endre, Z.; Margolin, S.; Taylor, S.M.; Brown, L. Reversal of cardiac and renal fibrosis by pirfenidone and spironolactone in streptozotocin-diabetic rats. Br. J. Pharmacol. 2001, 133, 687–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Kuroda, T.; Hata, S.; Fukagawa, M.; Margolin, S.B.; Kurokawa, K. Pirfenidone improves renal function and fibrosis in the post-obstructed kidney. Kidney Int. 1998, 54, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salhiyyah, K.; Forster, R.; Senanayake, E.; Abdel-Hadi, M.; Booth, A.; Michaels, J.A. Pentoxifylline for intermittent claudication. Cochrane Database Syst. Rev. 2015, 9, CD005262. [Google Scholar] [CrossRef]
- Subramaniyan, V.; Chakravarthi, S.; Jegasothy, R.; Seng, W.Y.; Fuloria, N.K.; Fuloria, S.; Hazarika, I.; Das, A. Alcohol-associated liver disease: A review on its pathophysiology, diagnosis and drug therapy. Toxicol. Rep. 2021, 8, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Strutz, F.; Heeg, M.; Kochsiek, T.; Siemers, G.; Zeisberg, M.; Muller, G.A. Effects of pentoxifylline, pentifylline and gamma-interferon on proliferation, differentiation, and matrix synthesis of human renal fibroblasts. Nephrol. Dial. Transplant. 2000, 15, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.L.; Chen, Y.M.; Chien, C.T.; Chiang, W.C.; Tsai, C.C.; Tsai, T.J. Pentoxifylline attenuated the renal disease progression in rats with remnant kidney. J. Am. Soc. Nephrol. 2002, 13, 2916–2929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Meng, F.; Song, J.; Zhang, L.; Wang, J.; Li, D.; Li, L.; Dong, P.; Yang, B.; Chen, Y. Pentoxifylline ameliorates cardiac fibrosis, pathological hypertrophy, and cardiac dysfunction in angiotensin ii-induced hypertensive rats. J. Cardiovasc. Pharmacol. 2016, 67, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hua, J.; Guo, C.X.; Wang, W.X.; Wang, B.J.; Yang, D.L.; Wei, P.; Lu, Y.P. Pentoxifylline inhibits liver fibrosis via hedgehog signaling pathway. J. Huazhong Univ. Sci. Technol. Med. Sci. 2016, 36, 372–376. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, Z.; Zhou, B.; Ma, K.; Jiang, M. Pentoxifylline inhibits pulmonary fibrosis by regulating cellular senescence in mice. Front. Pharmacol. 2022, 13, 848263. [Google Scholar] [CrossRef]
- Chen, Y.M.; Chiang, W.C.; Yang, Y.; Lai, C.F.; Wu, K.D.; Lin, S.L. Pentoxifylline attenuates proteinuria in anti-Thy1 glomerulonephritis via downregulation of nuclear factor-kappaB and Smad2/3 signaling. Mol. Med. 2015, 21, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Maruno, S.; Tanaka, T.; Nangaku, M. Exploring molecular targets in diabetic kidney disease. Kidney Res. Clin. Pract. 2022, 41, S33–S45. [Google Scholar] [CrossRef] [PubMed]
- de Morales, A.M.; Goicoechea, M.; Verde, E.; Carbayo, J.; Barbieri, D.; Delgado, A.; Verdalles, U.; de Jose, A.P.; Luno, J. Pentoxifylline, progression of chronic kidney disease (CKD) and cardiovascular mortality: Long-term follow-up of a randomized clinical trial. J. Nephrol. 2019, 32, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.L. The whereabouts of microrna actions: Cytoplasm and beyond. Trends Cell Biol. 2015, 25, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.M.; Mott, J.L. Overview of microrna biology. Semin. Liver Dis. 2015, 35, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.A.; Ganepola, G.A.P.; Rutledge, J.R.; Chang, D.H. The potential role of dysregulated mirnas in Alzheimer’s disease pathogenesis and progression. J. Alzheimers Dis. 2019, 67, 1123–1145. [Google Scholar] [CrossRef]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Treguer, K.; Carmona, G.; Bonauer, A.; et al. MicroRNA-34a regulates cardiac ageing and function. Nature 2013, 495, 107–110. [Google Scholar] [CrossRef]
- Jin, Z.Q. Microrna targets and biomarker validation for diabetes-associated cardiac fibrosis. Pharmacol. Res. 2021, 174, 105941. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Choi, C.; Yoo, T.H. Extracellular vesicles in kidneys and their clinical potential in renal diseases. Kidney Res. Clin. Pract. 2021, 40, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, W.; Liao, J.; Tang, F.; Gao, G.; Peng, J.; Fu, X.; Zhan, Y.; Chen, Z.; Xu, W.; et al. MicroRNA-21: A critical pathogenic factor of diabetic nephropathy. Front. Endocrinol. 2022, 13, 895010. [Google Scholar] [CrossRef] [PubMed]
- Zarjou, A.; Yang, S.; Abraham, E.; Agarwal, A.; Liu, G. Identification of a microrna signature in renal fibrosis: Role of miR-21. Am. J. Physiol. Ren. Physiol. 2011, 301, F793–F801. [Google Scholar] [CrossRef] [Green Version]
- McClelland, A.D.; Herman-Edelstein, M.; Komers, R.; Jha, J.C.; Winbanks, C.E.; Hagiwara, S.; Gregorevic, P.; Kantharidis, P.; Cooper, M.E. MiR-21 promotes renal fibrosis in diabetic nephropathy by targeting pten and Smad7. Clin. Sci. 2015, 129, 1237–1249. [Google Scholar] [CrossRef]
- Chung, A.C.; Lan, H.Y. MicroRNAs in renal fibrosis. Front. Physiol. 2015, 6, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Li, W.; Yu, W.; Rao, T.; Li, H.; Ruan, Y.; Yuan, R.; Li, C.; Ning, J.; Li, S.; et al. Exosomal miR-21 from tubular cells contributes to renal fibrosis by activating fibroblasts via targeting pten in obstructed kidneys. Theranostics 2021, 11, 8660–8673. [Google Scholar] [CrossRef] [PubMed]
- Chau, B.N.; Xin, C.; Hartner, J.; Ren, S.; Castano, A.P.; Linn, G.; Li, J.; Tran, P.T.; Kaimal, V.; Huang, X.; et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci. Transl. Med. 2012, 4, 121ra118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Chung, A.C.; Chen, H.Y.; Meng, X.M.; Lan, H.Y. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J. Am. Soc. Nephrol. 2011, 22, 1668–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Chung, A.C.; Chen, H.Y.; Dong, Y.; Meng, X.M.; Li, R.; Yang, W.; Hou, F.F.; Lan, H.Y. miR-21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia 2013, 56, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Chung, A.C.; Huang, X.R.; Meng, X.M.; Hui, D.S.; Yu, C.M.; Sung, J.J.; Lan, H.Y. TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol. 2011, 22, 1462–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Komers, R.; Carew, R.; Winbanks, C.E.; Xu, B.; Herman-Edelstein, M.; Koh, P.; Thomas, M.; Jandeleit-Dahm, K.; Gregorevic, P.; et al. Suppression of microRNA-29 expression by TGF-beta1 promotes collagen expression and renal fibrosis. J. Am. Soc. Nephrol. 2012, 23, 252–265. [Google Scholar] [CrossRef] [PubMed]
| Carrier Type | Preclinical Model | Reference |
|---|---|---|
| Collagen | Bone defects in non-human primates | [100] |
| Vertebral interbody fusion in sheep | [101] | |
| Hydroxyapatite | Orthotopic skull defects in baboons | [102] |
| Spinal fusion in sheep | [103] | |
| Poly(D,L-lactide-co-glycolide) | Bone formation from rabbit muscle cells | [104] |
| Osteochondral defect in rabbit knee | [105] | |
| CMC 1-Collagen | Tibial bone defects in sheep | [106] |
| 1,4 Butane-diisocyanate-hydrogel | Femoral intramedullary injection in mice | [107] |
| Micelle | Unilateral ureteral obstruction in mice/pig | [94] |
| Chitosan nanoparticle | Unilateral ureteral obstruction in mice | [98] |
| Femoral bone defect in rat | [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, C.H.; Yoo, T.-H. TGF-β Inhibitors for Therapeutic Management of Kidney Fibrosis. Pharmaceuticals 2022, 15, 1485. https://doi.org/10.3390/ph15121485
Park CH, Yoo T-H. TGF-β Inhibitors for Therapeutic Management of Kidney Fibrosis. Pharmaceuticals. 2022; 15(12):1485. https://doi.org/10.3390/ph15121485
Chicago/Turabian StylePark, Cheol Ho, and Tae-Hyun Yoo. 2022. "TGF-β Inhibitors for Therapeutic Management of Kidney Fibrosis" Pharmaceuticals 15, no. 12: 1485. https://doi.org/10.3390/ph15121485
APA StylePark, C. H., & Yoo, T. -H. (2022). TGF-β Inhibitors for Therapeutic Management of Kidney Fibrosis. Pharmaceuticals, 15(12), 1485. https://doi.org/10.3390/ph15121485