
Over 40 Years of Fosmidomycin Drug Research: A Comprehensive Review and Future Opportunities
 , , ,
, , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
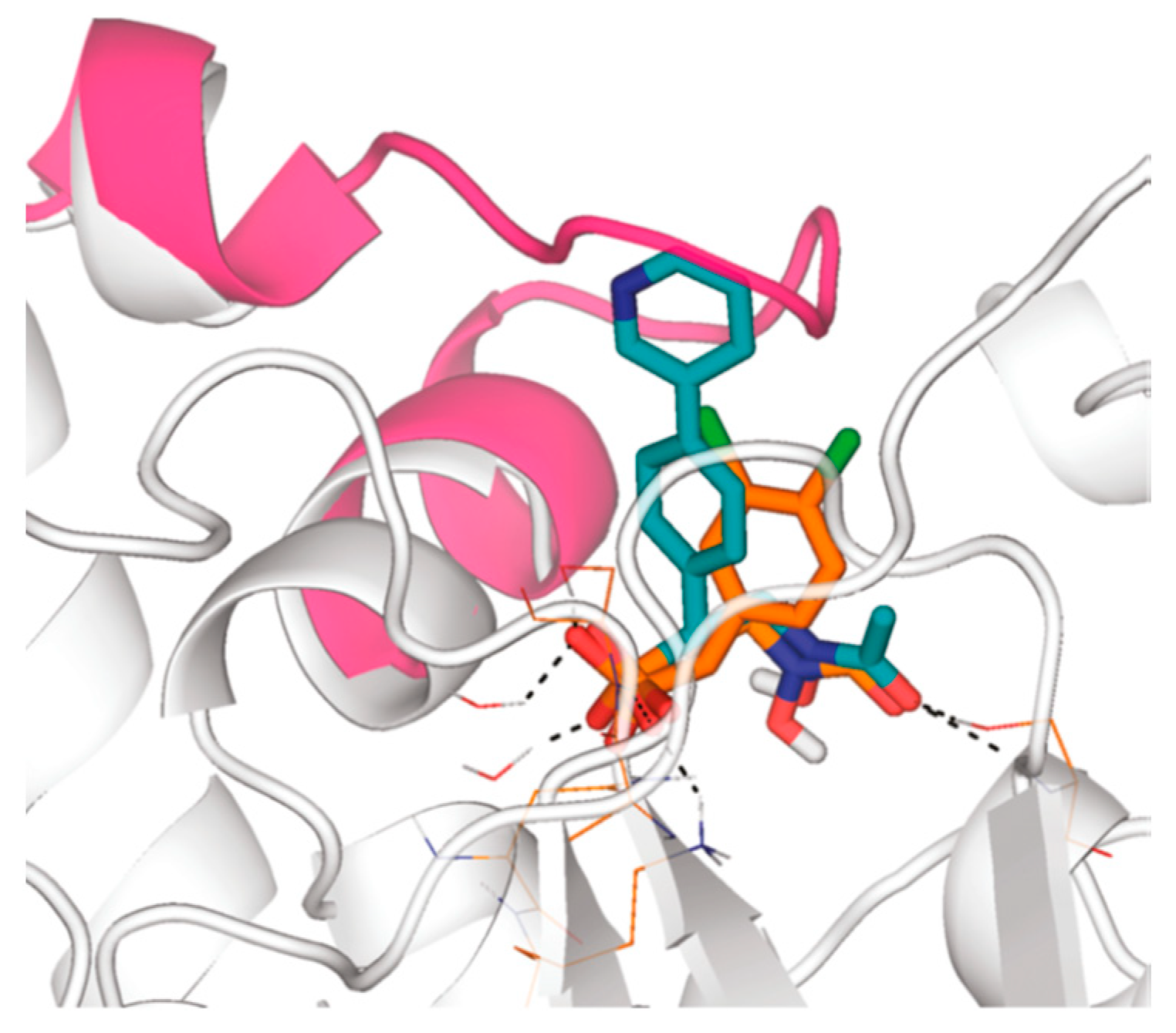{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
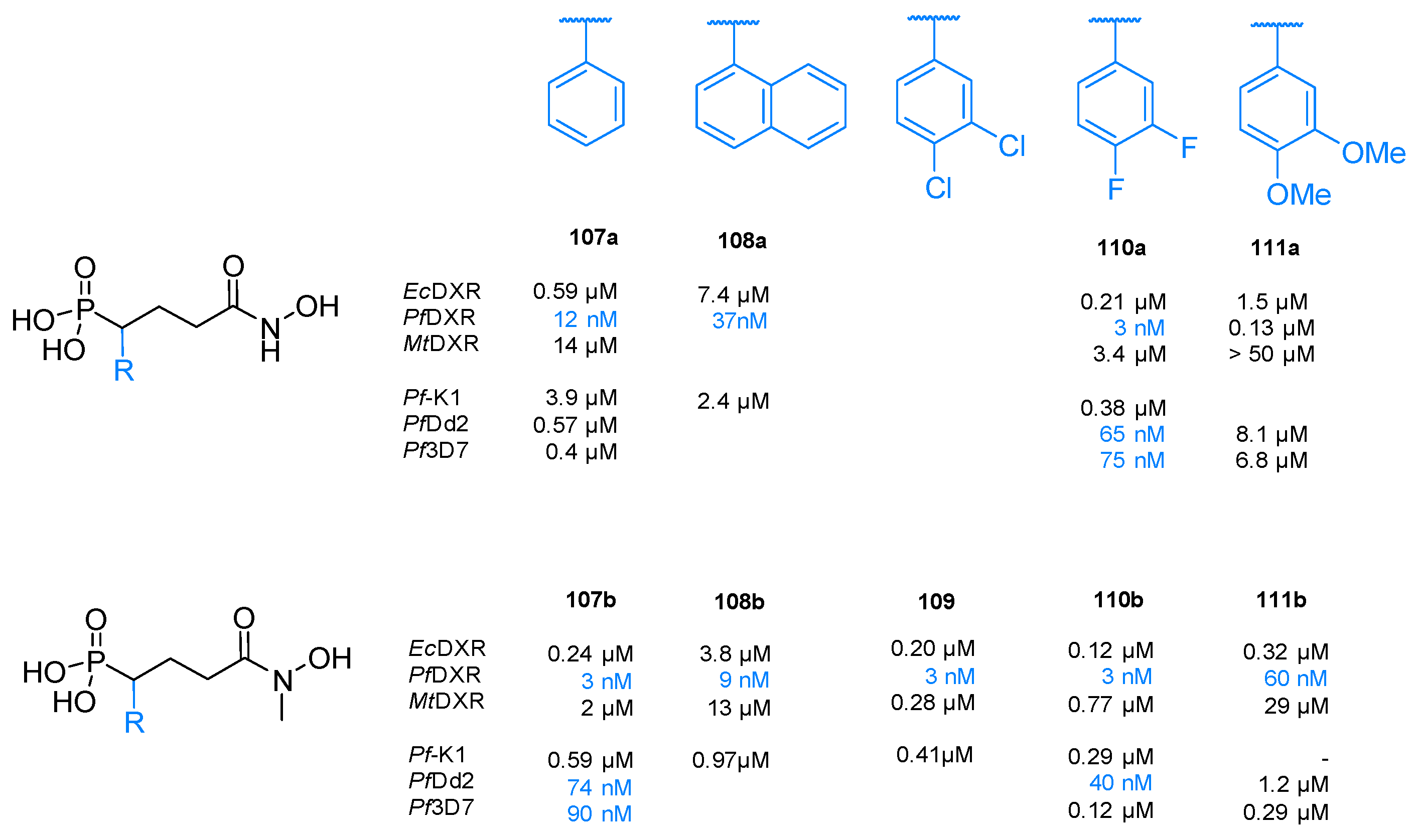{kind=link}
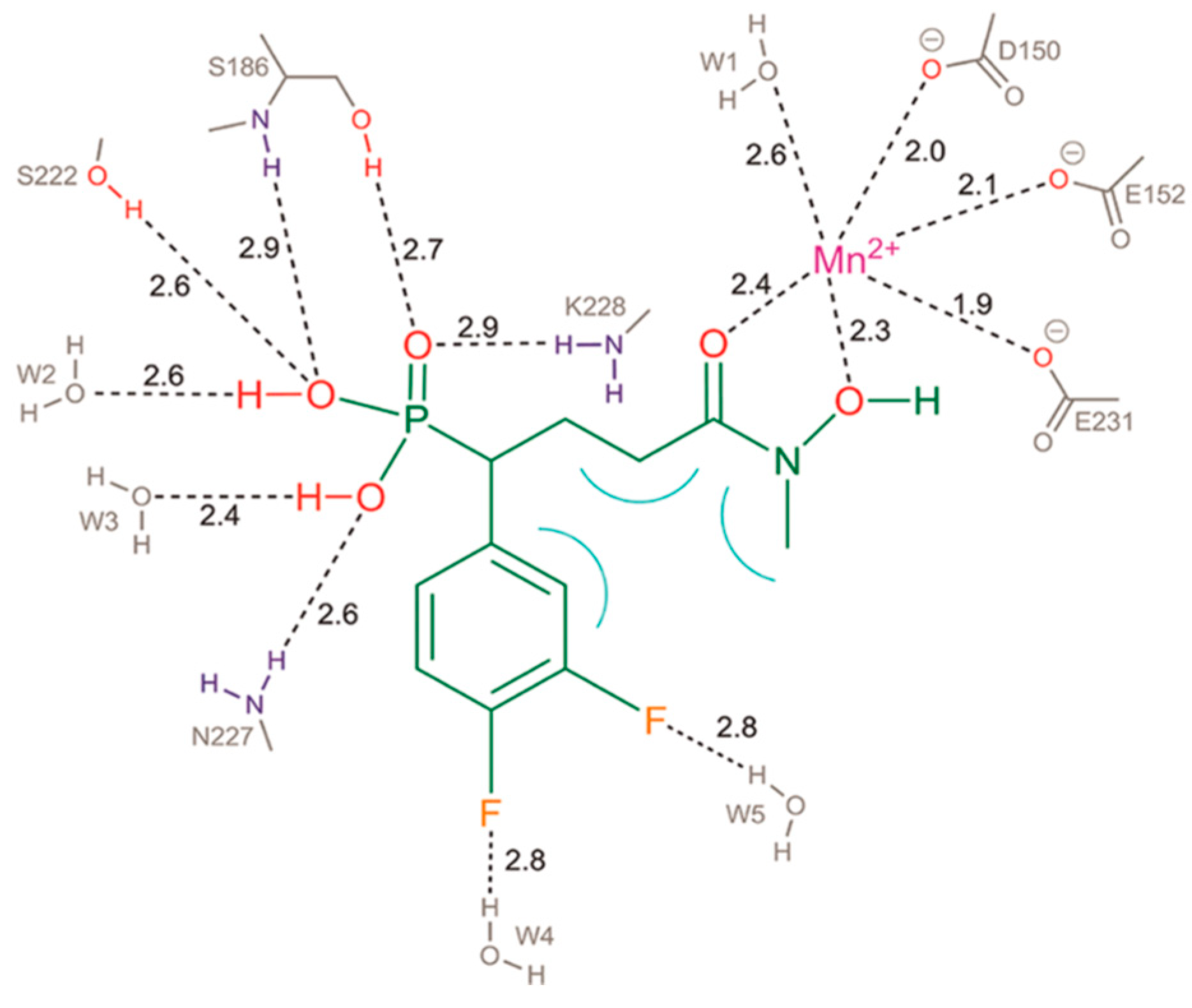{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
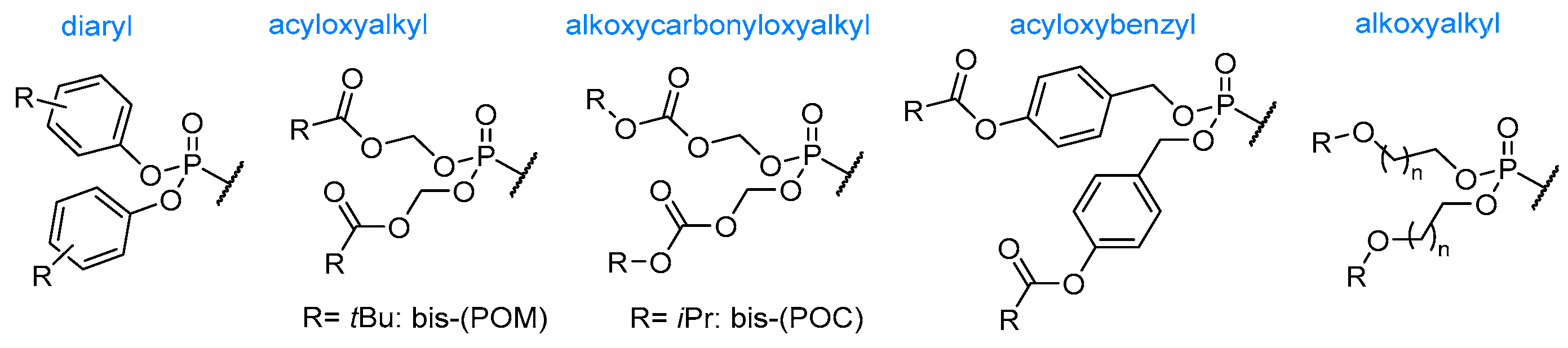{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Discovery and Evaluation of Fosmidomycin (1) and Related Natural Products
2.1. Anti-Infective Activity of Fosmidomycin
2.1.1. Parasites
2.1.2. Gram-Positive Bacteria
2.1.3. Gram-Negative Bacteria
2.2. Pharmacokinetic Profile of Fosmidomycin
2.3. Clinical Trials from 1985 to 2018
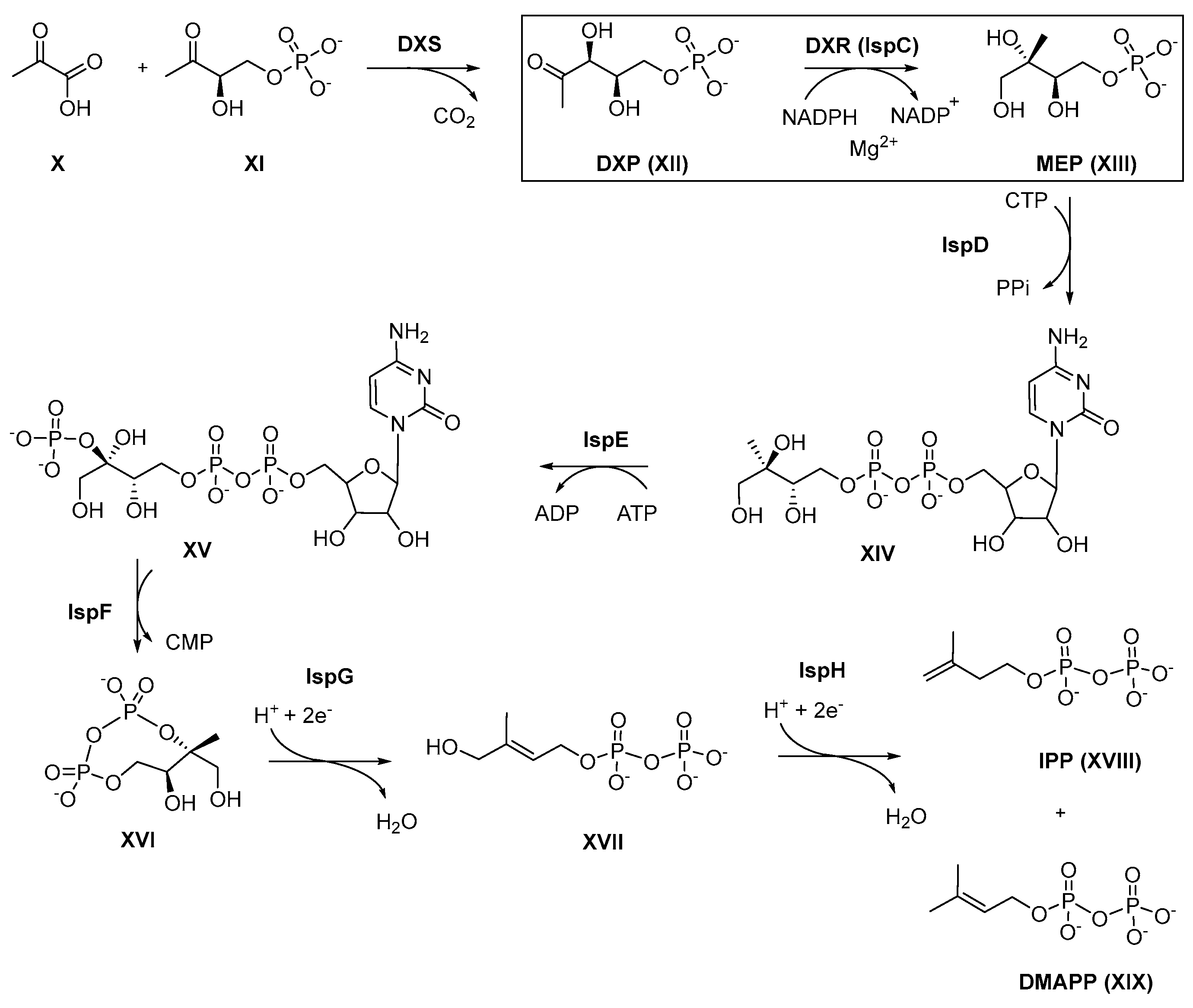
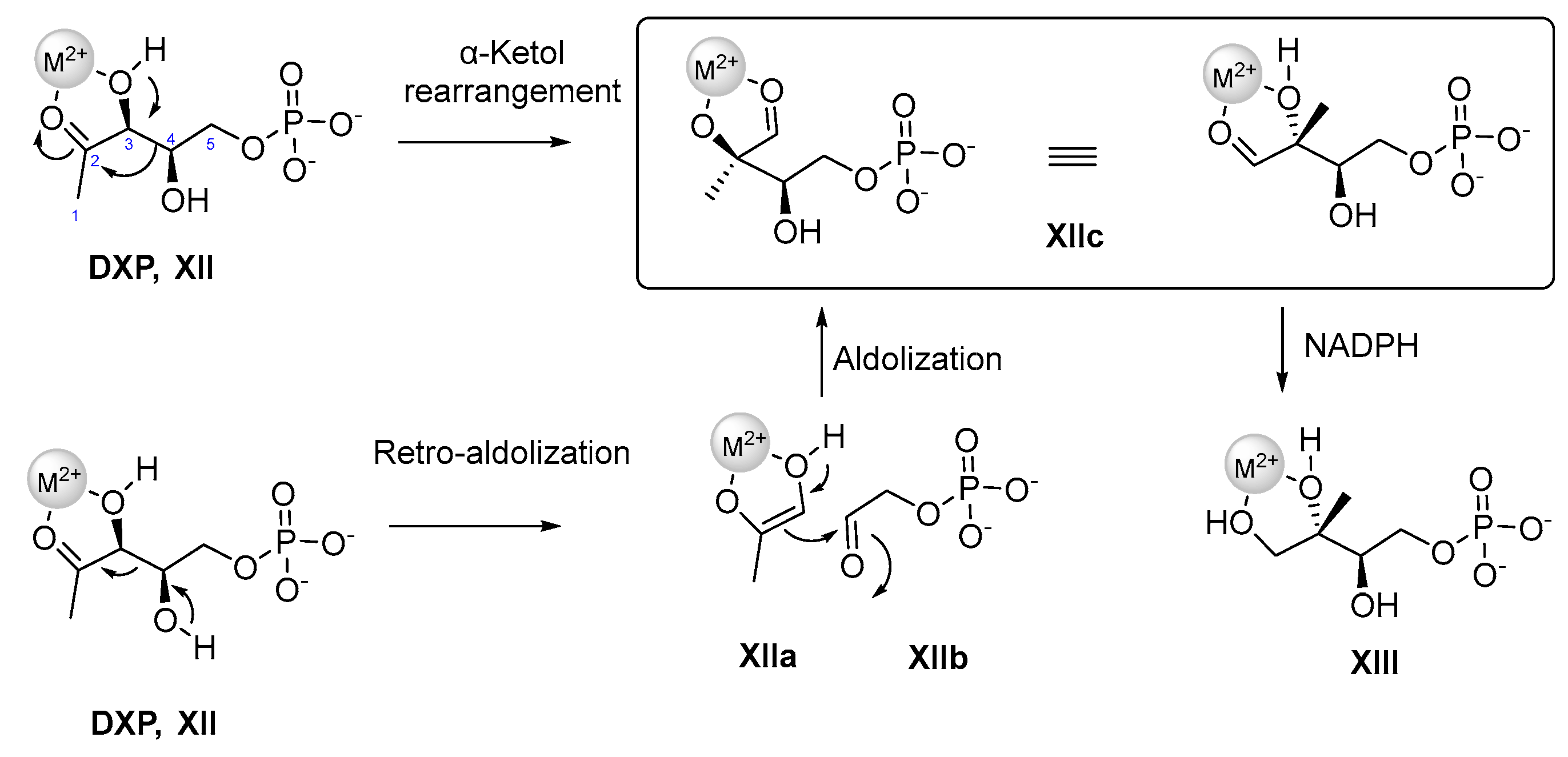
3. 1 Targeting the Deoxy-D-xylulose-5-phosphate Reductoisomerase (DXR)

3.1. Crystal Structures of DXR
Active Site
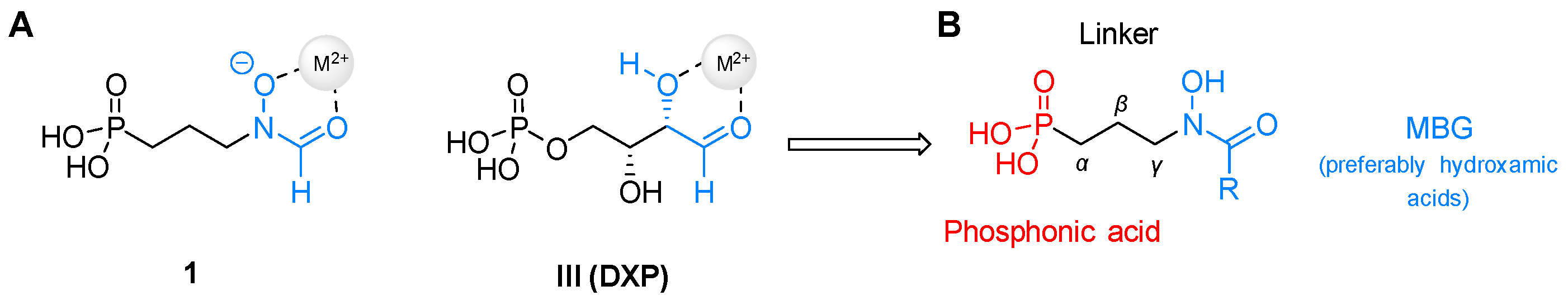
4. Structural Modifications of Fosmidomycin and FR900098
4.1. Modifications of the Retro-Hydroxamate Moiety
4.1.1. Inversion of the Retro-Hydroxamate Moiety
4.1.2. Alteration of the Acyl Moiety and Replacement of the Hydroxamic Acid Moiety
4.1.3. Development of Bisubstrate Inhibitors
4.2. Modifications of the Propyl Linker
4.2.1. Linker Length Variation
4.2.2. α,β-Unsaturated Propenyl Linker
4.2.3. Oxa Analogs
4.2.4. Conformationally Restricted Analogs
4.3. α-, β- and γ-Substituted Fosmidomycin Analogs
4.3.1. α-Phenyl and α-Biaryl-Substituted Analogs
4.3.2. α-Halogenated Phosphonic Acid Derivatives
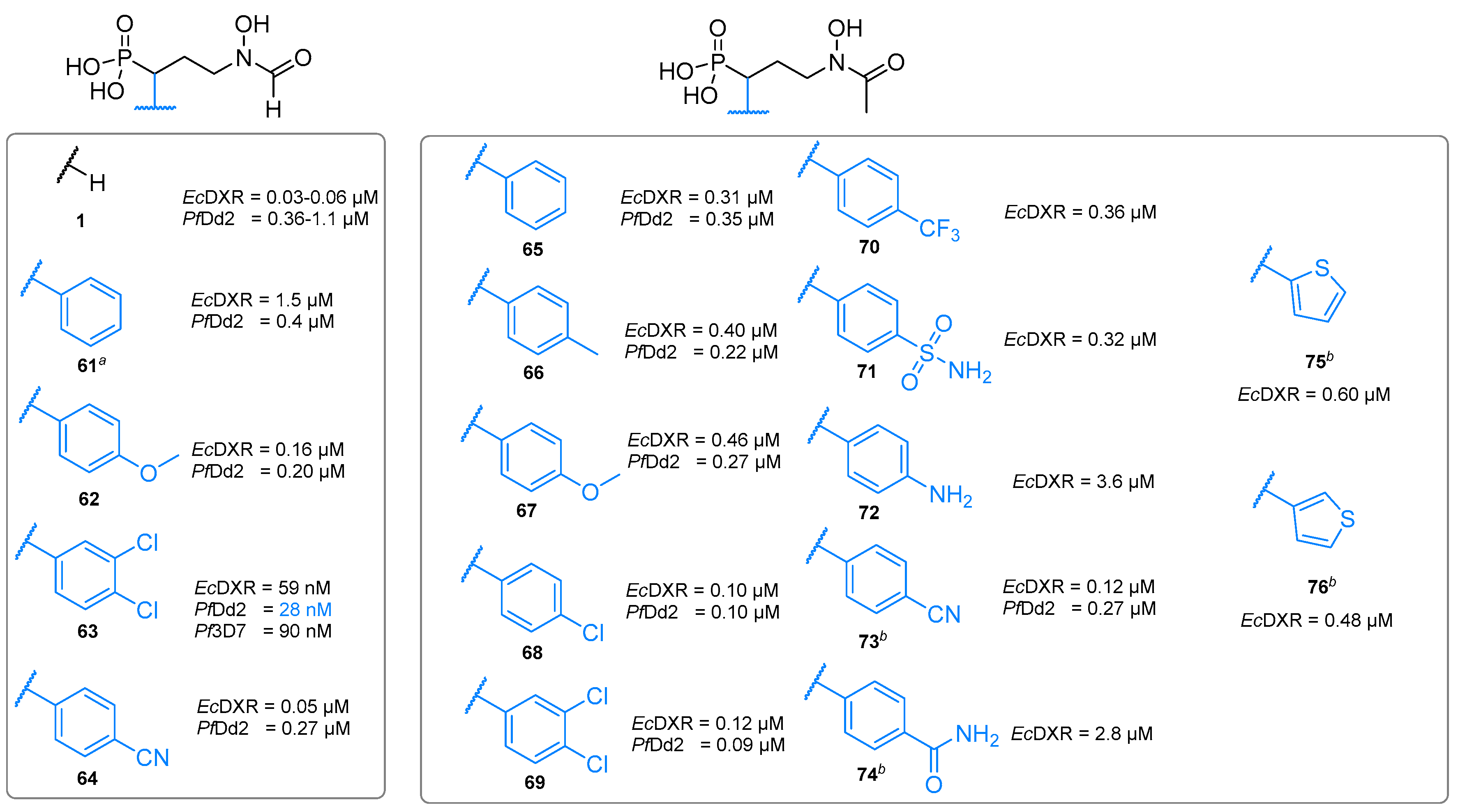
4.3.3. Structurally Diverse Substituents in the α-Position
4.3.4. α-Substituted Reverse Carba Analogs
4.3.5. Reverse α-Substituted Oxa, Thia and Aza Analogs
4.3.6. β- and γ-Substituted Analogs
5. Phosphonic Acid Isosteres and Bioisosteres
6. Conclusions Regarding Structure–Activity Relationship
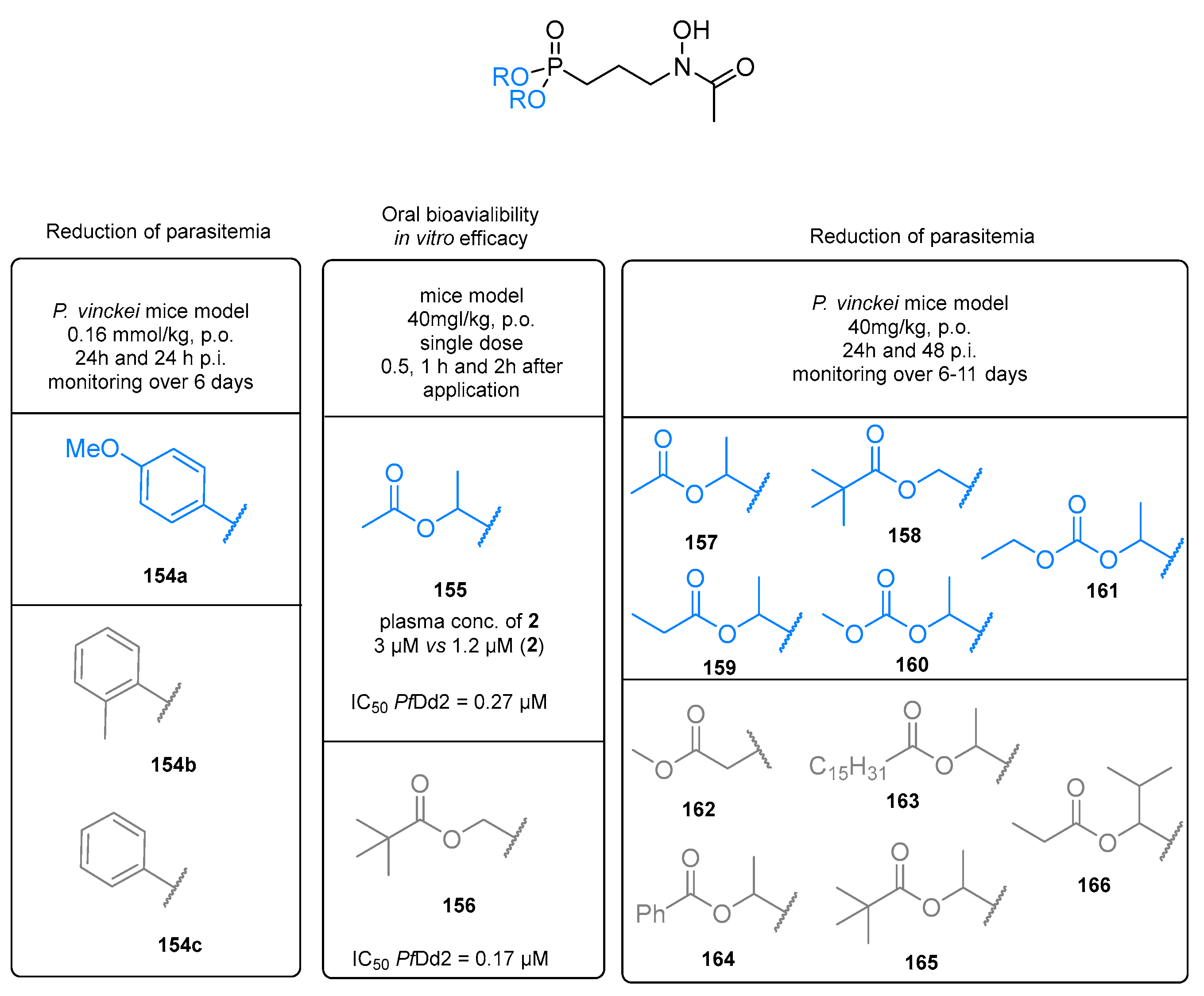
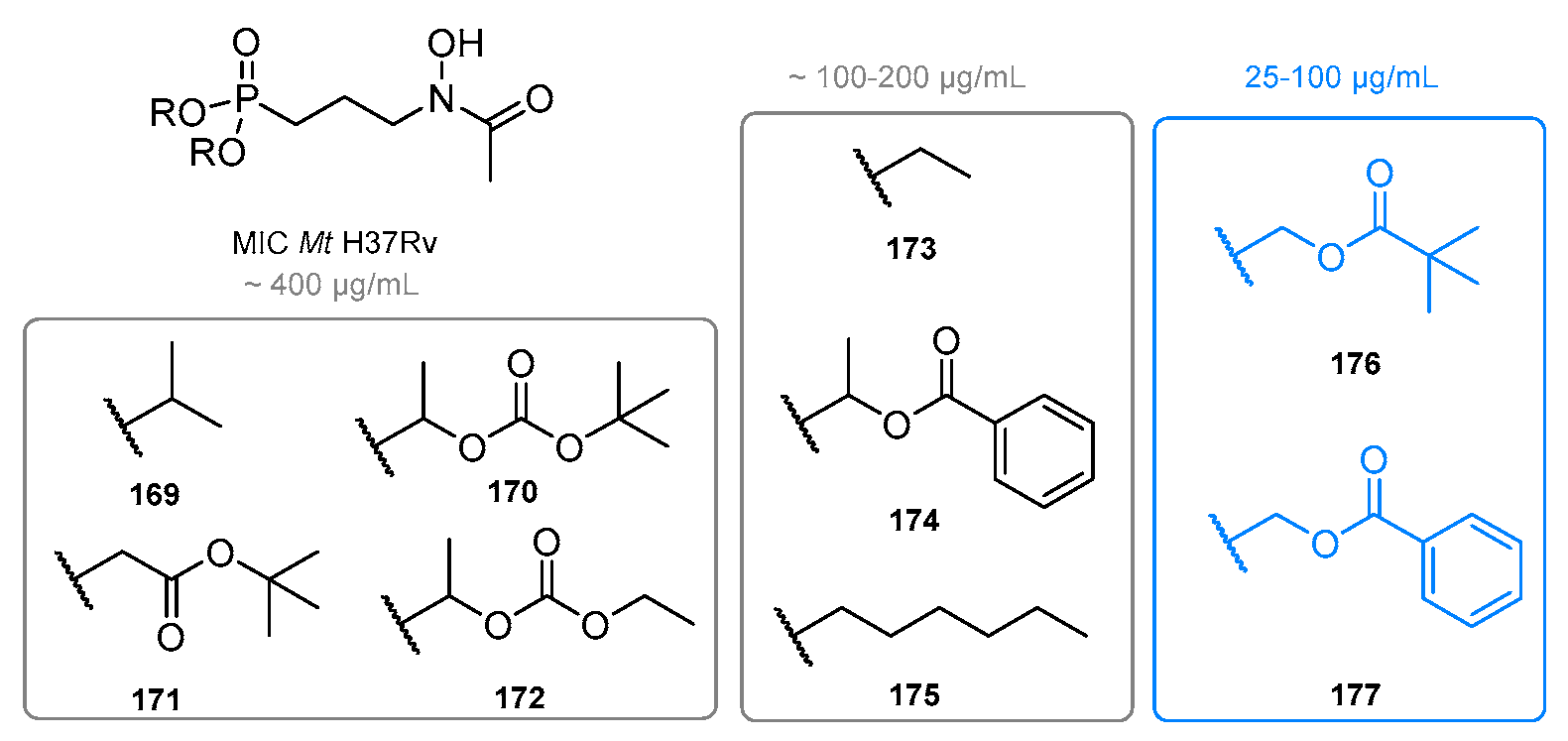
7. Prodrugs of Fosmidomycin and Its Analogs
7.1. Lipophilic Phosphonic Acid Esters
7.1.1. Ester Prodrugs of Fosmidomycin and FR900098
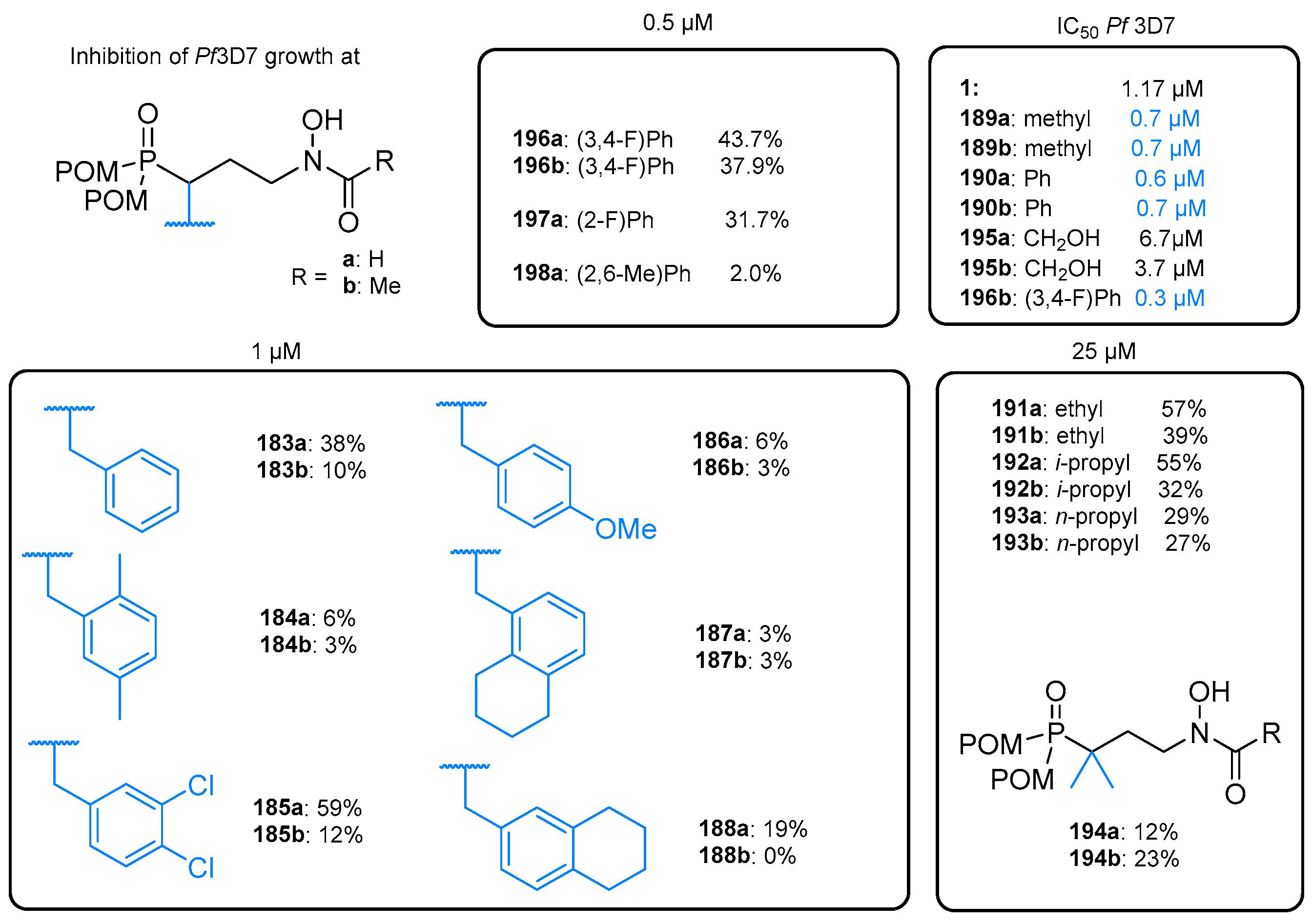
7.1.2. Ester Prodrugs of Fosmidomycin Analogs
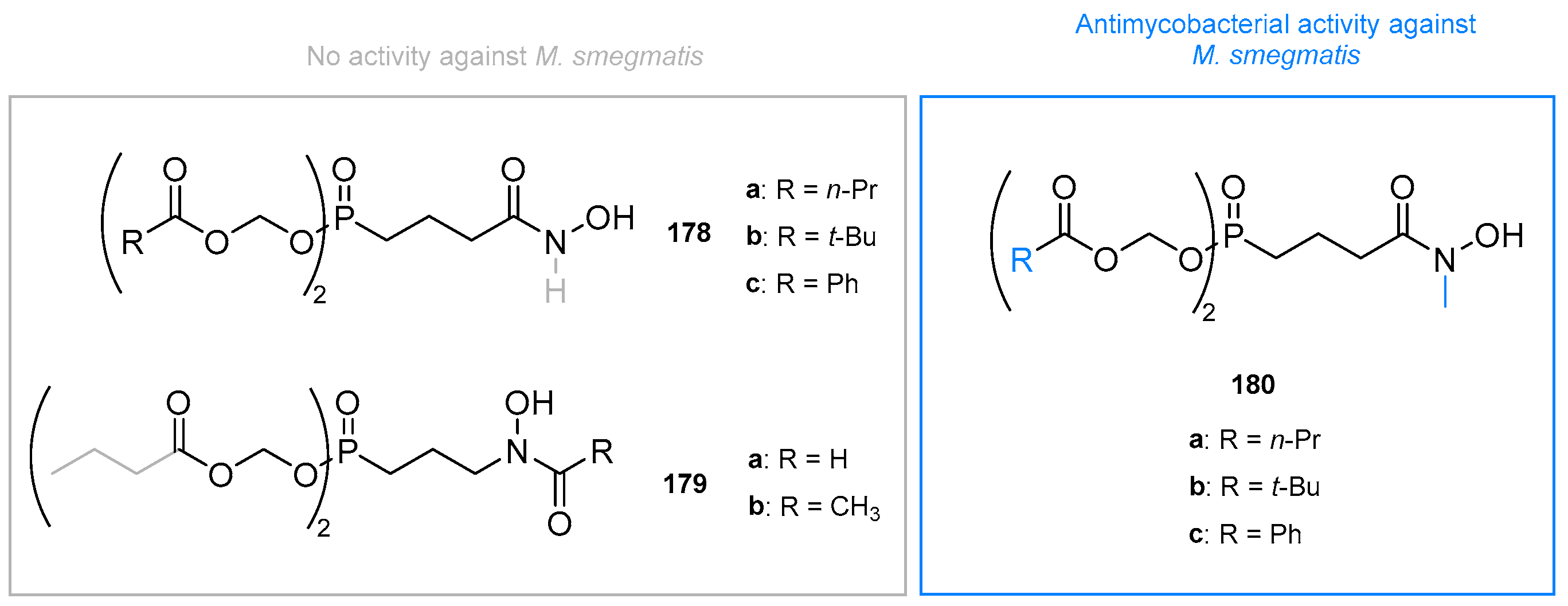
7.2. Double Prodrugs
7.3. Amino Acid Esters and Phosphonamidate Prodrugs
8. Fosmidomycin Conjugates and Hybrids
9. DXR-Inhibitors Not Based on Fosmidomycin
10. Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Rohmer, M.; Knani, M.; Simonin, P.; Sutter, B.; Sahm, H. Isoprenoid Biosynthesis in Bacteria: A Novel Pathway for the Early Steps Leading to Isopentenyl Diphosphate. Biochem. J. 1993, 295, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disch, A.; Schwender, J.; Müller, C.; Lichtenthaler, H.K.; Rohmer, M. Distribution of the Mevalonate and Glyceraldehyde Phosphate/Pyruvate Pathways for Isoprenoid Biosynthesis in Unicellular Algae and the Cyanobacterium Synechocystis PCC 6714. Biochem. J. 1998, 333, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Arigoni, D.; Sagner, S.; Latzel, C.; Eisenreich, W.; Bacher, A.; Zenk, M.H. Terpenoid Biosynthesis from 1-Deoxy-D-xylulose in Higher Plants by Intramolecular Skeletal Rearrangement. Proc. Natl. Acad. Sci. USA 1997, 94, 10600–10605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwender, J.; Zeidler, J.; Gröner, R.; Müller, C.; Focke, M.; Braun, S.; Lichtenthaler, F.W.; Lichtenthaler, H.K. Incorporation of 1-Deoxy-D-Xylulose into Isoprene and Phytol by Higher Plants and Algae. FEBS Lett. 1997, 414, 129–134. [Google Scholar] [CrossRef] [Green Version]
- Cvejić, J.H.; Rohmer, M. CO2 as Main Carbon Source for Isoprenoid Biosynthesis via the Mevalonate-independent Methylerythritol 4-Phosphate Route in the Marine Diatoms Phaeodactylum tricornutum and Nitzschia ovalis. Phytochemistry 2000, 53, 21–28. [Google Scholar] [CrossRef]
- Eberl, M.; Hintz, M.; Reichenberg, A.; Kollas, A.-K.; Wiesner, J.; Jomaa, H. Microbial Isoprenoid Biosynthesis and Human γδ T Cell Activation. FEBS Lett. 2003, 544, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Coppens, I. Targeting Lipid Biosynthesis and Salvage in Apicomplexan Parasites for Improved Chemotherapies. Nat. Rev. Microbiol. 2013, 11, 823–835. [Google Scholar] [CrossRef]
- Hunter, W.N. The Non-Mevalonate Pathway of Isoprenoid Precursor Biosynthesis. J. Biol. Chem. 2007, 282, 21573–21577. [Google Scholar] [CrossRef] [Green Version]
- Okuhara, M.; Kuroda, Y.; Goto, T.; Okamoto, M.; Terano, H.; Kohsaka, M.; Aoki, H.; Imanaka, H. Studies on New Phosphonic Acid Antibiotics. I. FR-900098, Isolation and Characterization. J. Antibiot. 1980, 33, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, T.; Hashimoto, M.; Hemmi, K.; Takeno, H. Hydroxyaminohydrocarbonphosphonic Acids. U.S. Patent US4206156A, 3 June 1980. [Google Scholar]
- Patterson, D.R. Herbicidal Hydroxyamino Phosphonic Acids and Derivatives. U.S. Patent US4693742A, 15 September 1987. [Google Scholar]
- Kuzuyama, T.; Shimizu, T.; Takahashi, S.; Seto, H. Fosmidomycin, a Specific Inhibitor of 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase in the Nonmevalonate Pathway for Terpenoid Biosynthesis. Tetrahedron Lett. 1998, 39, 7913–7916. [Google Scholar] [CrossRef]
- Jomaa, H.; Wiesner, J.; Sanderbrand, S.; Altincicek, B.; Weidemeyer, C.; Hintz, M.; Türbachova, I.; Eberl, M.; Zeidler, J.; Lichtenthaler, H.K.; et al. Inhibitors of the Nonmevalonate Pathway of Isoprenoid Biosynthesis as Antimalarial Drugs. Science 1999, 285, 1573–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeidler, J.; Schwender, J.; Müller, C.; Wiesner, J.; Weidemeyer, C.; Beck, E.; Jomaa, H.; Lichtenthaler, H. Inhibition of the Non-Mevalonate 1-Deoxy-D-Xylulose-5-Phosphate Pathway of Plant Isoprenoid Biosynthesis by Fosmidomycin. Z. Für Nat. C 1998, 53, 980–986. [Google Scholar] [CrossRef]
- Baumeister, S.; Wiesner, J.; Reichenberg, A.; Hintz, M.; Bietz, S.; Harb, O.S.; Roos, D.S.; Kordes, M.; Friesen, J.; Matuschewski, K.; et al. Fosmidomycin Uptake into Plasmodium and Babesia-Infected Erythrocytes Is Facilitated by Parasite-Induced New Permeability Pathways. PLoS ONE 2011, 6, e19334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chofor, R.; Risseeuw, M.D.P.; Pouyez, J.; Johny, C.; Wouters, J.; Dowd, C.S.; Couch, R.D.; Van Calenbergh, S. Synthetic Fosmidomycin Analogues with Altered Chelating Moieties Do Not Inhibit 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase or Plasmodium falciparum Growth in Vitro. Molecules 2014, 19, 2571–2587. [Google Scholar] [CrossRef] [Green Version]
- Iguchi, E.; Okuhara, M.; Kohsaka, M.; Aoki, H.; Imanaka, H. Studies on New Phosphonic Acid Antibiotics. II. Taxonomic Studies on Producing Organisms of the Phosphonic Acid and Related Compounds. J. Antibiot. 1980, 33, 19–23. [Google Scholar] [CrossRef] [Green Version]
- Parkinson, E.I.; Erb, A.; Eliot, A.C.; Ju, K.-S.; Metcalf, W.W. Fosmidomycin Biosynthesis Diverges from Related Phosphonate Natural Products. Nat. Chem. Biol. 2019, 15, 1049–1056. [Google Scholar] [CrossRef]
- Eliot, A.C.; Griffin, B.M.; Thomas, P.M.; Johannes, T.W.; Kelleher, N.L.; Zhao, H.; Metcalf, W.W. Cloning, Expression, and Biochemical Characterization of Streptomyces rubellomurinus Genes Required for Biosynthesis of Antimalarial Compound FR900098. Chem. Biol. 2008, 15, 765–770. [Google Scholar] [CrossRef] [Green Version]
- Johannes, T.W.; DeSieno, M.A.; Griffin, B.M.; Thomas, P.M.; Kelleher, N.L.; Metcalf, W.W.; Zhao, H. Deciphering the Late Biosynthetic Steps of Antimalarial Compound FR-900098. Chem. Biol. 2010, 17, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, Y.; Okuhara, M.; Goto, T.; Okamoto, M.; Terano, H.; Kohsaka, M.; Aoki, H.; Imanaka, H. Studies on New Phosphonic Acid Antibiotics. IV. Structure Determination of FR-33289, FR-31564 and FR-32863. J. Antibiot. 1980, 33, 29–35. [Google Scholar] [CrossRef] [Green Version]
- Shigi, Y. Inhibition of Bacterial Isoprenoid Synthesis by Fosmidomycin, a Phosphonic Acid-containing Antibiotic. J. Antimicrob. Chemother. 1989, 24, 131–145. [Google Scholar] [CrossRef]
- Missinou, M.A.; Borrmann, S.; Schindler, A.; Issifou, S.; Adegnika, A.A.; Matsiegui, P.B.; Binder, R.; Lell, B.; Wiesner, J.; Baranek, T.; et al. Fosmidomycin for Malaria. Lancet 2002, 360, 1941–1942. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.C.; Brooks, C.F.; Goodman, C.D.; Sturm, A.; McFadden, G.I.; Sundriyal, S.; Anglin, J.L.; Song, Y.; Moreno, S.N.; Striepen, B. Apicoplast Isoprenoid Precursor Synthesis and the Molecular Basis of Fosmidomycin Resistance in Toxoplasma gondii. J. Exp. Med. 2011, 208, 1547–1559. [Google Scholar] [CrossRef]
- Armstrong, C.M.; Meyers, D.J.; Imlay, L.S.; Freel Meyers, C.; Odom, A.R. Resistance to the Antimicrobial Agent Fosmidomycin and an FR900098 Prodrug through Mutations in the Deoxyxylulose Phosphate Reductoisomerase Gene (dxr). Antimicrob. Agents Chemother. 2015, 59, 5511–5519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ball, H.S.; Girma, M.B.; Zainab, M.; Soojhawon, I.; Couch, R.D.; Noble, S.M. Characterization and Inhibition of 1-Deoxy-d-Xylulose 5-Phosphate Reductoisomerase: A Promising Drug Target in Acinetobacter baumannii and Klebsiella pneumoniae. ACS Infect. Dis. 2021, 7, 2987–2998. [Google Scholar] [CrossRef] [PubMed]
- Altincicek, B.; Hintz, M.; Sanderbrand, S.; Wiesner, J.; Beck, E.; Jomaa, H. Tools for Discovery of Inhibitors of the 1-Deoxy-D-Xylulose 5-Phosphate (DXP) Synthase and DXP Reductoisomerase: An Approach with Enzymes from the Pathogenic Bacterium Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2000, 190, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Misic, A.M.; Cain, C.L.; Morris, D.O.; Rankin, S.C.; Beiting, D.P.; Fey, P.D. Divergent Isoprenoid Biosynthesis Pathways in Staphylococcus Species Constitute a Drug Target for Treating Infections in Companion Animals. mSphere 2016, 1, e00258-16. [Google Scholar] [CrossRef] [Green Version]
- Lange, B.M.; Rujan, T.; Martin, W.; Croteau, R. Isoprenoid Biosynthesis: The Evolution of Two Ancient and Distinct Pathways Across Genomes. Proc. Natl. Acad. Sci. USA 2000, 97, 13172–13177. [Google Scholar] [CrossRef] [Green Version]
- Van Laar, T.A.; Lin, Y.-H.; Miller, C.L.; Karna, S.L.R.; Chambers, J.P.; Seshu, J. Effect of Levels of Acetate on the Mevalonate Pathway of Borrelia burgdorferi. PLoS ONE 2012, 7, e38171. [Google Scholar] [CrossRef] [Green Version]
- Reuter, K.; Sanderbrand, S.; Jomaa, H.; Wiesner, J.; Steinbrecher, I.; Beck, E.; Hintz, M.; Klebe, G.; Stubbs, M.T. Crystal Structure of 1-Deoxy-D-xylulose-5-phosphate Reductoisomerase, a Crucial Enzyme in the Non-mevalonate Pathway of Isoprenoid Biosynthesis. J. Biol. Chem. 2002, 277, 5378–5384. [Google Scholar] [CrossRef] [Green Version]
- Mac Sweeney, A.; Lange, R.; Fernandes, R.P.M.; Schulz, H.; Dale, G.E.; Douangamath, A.; Proteau, P.J.; Oefner, C. The Crystal Structure of E.coli 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase in a Ternary Complex with the Antimalarial Compound Fosmidomycin and NADPH Reveals a Tight-Binding Closed Enzyme Conformation. J. Mol. Biol. 2005, 345, 115–127. [Google Scholar] [CrossRef]
- Brown, A.C.; Parish, T. Dxr is Essential in Mycobacterium tuberculosis and Fosmidomycin Resistance is Due to a Lack of Uptake. BMC Microbiol. 2008, 8, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, G.; Deng, L.; Xue, J.; Moreno, S.N.J.; Striepen, B.; Song, Y. Expression, Characterization and Inhibition of Toxoplasma gondii 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase. Bioorg. Med. Chem. Lett. 2013, 23, 2158–2161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howe, R.; Kelly, M.; Jimah, J.; Hodge, D.; Odom, A.R. Isoprenoid Biosynthesis Inhibition Disrupts Rab5 Localization and Food Vacuolar Integrity in Plasmodium falciparum. Eukaryot. Cell 2013, 12, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botté, C.Y.; Dubar, F.; McFadden, G.I.; Maréchal, E.; Biot, C. Plasmodium falciparum Apicoplast Drugs: Targets or Off-Targets? Chem. Rev. 2012, 112, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Brücher, K.; Gräwert, T.; Konzuch, S.; Held, J.; Lienau, C.; Behrendt, C.; Illarionov, B.; Maes, L.; Bacher, A.; Wittlin, S.; et al. Prodrugs of Reverse Fosmidomycin Analogues. J. Med. Chem. 2015, 58, 2025–2035. [Google Scholar] [CrossRef]
- He, L.; He, P.; Luo, X.; Li, M.; Yu, L.; Guo, J.; Zhan, X.; Zhu, G.; Zhao, J. The MEP Pathway in Babesia orientalis Apicoplast, a Potential Target for Anti-Babesiosis Drug Development. Parasites Vectors 2018, 11, 452. [Google Scholar] [CrossRef]
- Sivakumar, T.; Aboulaila, M.; Khukhuu, A.; Iseki, H.; Alhassan, A.; Yokoyama, N.; Igarashi, I. In Vitro Inhibitory Effect of Fosmidomycin on the Asexual Growth of Babesia bovis and Babesia bigemina. J. Protozool. Res. 2008, 18, 71–78. [Google Scholar]
- Clastre, M.; Goubard, A.; Prel, A.; Mincheva, Z.; Viaud-Massuart, M.C.; Bout, D.; Rideau, M.; Velge-Roussel, F.; Laurent, F. The Methylerythritol Phosphate Pathway for Isoprenoid Biosynthesis in Coccidia: Presence and Sensitivity to Fosmidomycin. Exp. Parasitol. 2007, 116, 375–384. [Google Scholar] [CrossRef]
- San Jose, G.; Jackson, E.R.; Haymond, A.; Johny, C.; Edwards, R.L.; Wang, X.; Brothers, R.C.; Edelstein, E.K.; Odom, A.R.; Boshoff, H.I.; et al. Structure–Activity Relationships of the MEPicides: N-Acyl and O-Linked Analogs of FR900098 as Inhibitors of Dxr from Mycobacterium tuberculosis and Yersinia pestis. ACS Infect. Dis. 2016, 2, 923–935. [Google Scholar] [CrossRef] [Green Version]
- Uh, E.; Jackson, E.R.; San Jose, G.; Maddox, M.; Lee, R.E.; Lee, R.E.; Boshoff, H.I.; Dowd, C.S. Antibacterial and Antitubercular Activity of Fosmidomycin, FR900098, and Their Lipophilic Analogs. Bioorg. Med. Chem. Lett. 2011, 21, 6973–6976. [Google Scholar] [CrossRef] [Green Version]
- Edwards, R.L.; Heueck, I.; Lee, S.G.; Shah, I.T.; Miller, J.J.; Jezewski, A.J.; Mikati, M.O.; Wang, X.; Brothers, R.C.; Heidel, K.M.; et al. Potent, Specific MEPicides for Treatment of Zoonotic Staphylococci. PLoS Pathog. 2020, 16, e1007806. [Google Scholar] [CrossRef] [PubMed]
- Ropponen, H.-K.; Richter, R.; Hirsch, A.K.H.; Lehr, C.-M. Mastering the Gram-negative Bacterial Barrier—Chemical Approaches to Increase Bacterial Bioavailability of Antibiotics. Adv. Drug Deliv. Rev. 2021, 172, 339–360. [Google Scholar] [CrossRef] [PubMed]
- Haemers, T.; Wiesner, J.; Poecke, S.V.; Goeman, J.; Henschker, D.; Beck, E.; Jomaa, H.; Calenbergh, S.V. Synthesis of α-Substituted Fosmidomycin Analogues as Highly Potent Plasmodium falciparum Growth Inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 1888–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisaki, S.; Ohnuma, S.; Horiuchi, T.; Takahashi, I.; Tsukui, S.; Nishimura, Y.; Nishino, T.; Kitabatake, M.; Inokuchi, H. Cloning of a Gene from Escherichia coli that Confers Resistance to Fosmidomycin as a Consequence of Amplification. Gene 1996, 175, 83–87. [Google Scholar] [CrossRef]
- Nishino, K.; Yamaguchi, A. Analysis of a Complete Library of Putative Drug Transporter Genes in Escherichia coli. J. Bacteriol. 2001, 183, 5803–5812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauret-Güeto, S.; Urós, E.M.; Ibáñez, E.; Boronat, A.; Rodríguez-Concepción, M. A Mutant Pyruvate Dehydrogenase E1 Subunit Allows Survival of Escherichia coli Strains Defective in 1-Deoxy-D-Xylulose 5-Phosphate Synthase. FEBS Lett. 2006, 580, 736–740. [Google Scholar] [CrossRef] [Green Version]
- Perez-Gil, J.; Uros, E.M.; Sauret-Güeto, S.; Lois, L.M.; Kirby, J.; Nishimoto, M.; Baidoo, E.E.; Keasling, J.D.; Boronat, A.; Rodriguez-Concepcion, M. Mutations in Escherichia coli aceE and ribB Genes Allow Survival of Strains Defective in the First Step of the Isoprenoid Biosynthesis Pathway. PLoS ONE 2012, 7, e43775. [Google Scholar] [CrossRef] [Green Version]
- Messiaen, A.S.; Verbrugghen, T.; Declerck, C.; Ortmann, R.; Schlitzer, M.; Nelis, H.; Van Calenbergh, S.; Coenye, T. Resistance of the Burkholderia cepacia Complex to Fosmidomycin and Fosmidomycin Derivatives. Int. J. Antimicrob. Agents 2011, 38, 261–264. [Google Scholar] [CrossRef] [Green Version]
- Malott, R.J.; Wu, C.-H.; Lee, T.D.; Hird, T.J.; Dalleska, N.F.; Zlosnik, J.E.A.; Newman, D.K.; Speert, D.P. Fosmidomycin Decreases Membrane Hopanoids and Potentiates the Effects of Colistin on Burkholderia multivorans Clinical Isolates. Antimicrob. Agents Chemother. 2014, 58, 5211–5219. [Google Scholar] [CrossRef] [Green Version]
- Mackie, R.; McKenney, E.; van Hoek, M. Resistance of Francisella Novicida to Fosmidomycin Associated with Mutations in the Glycerol-3-Phosphate Transporter. Front. Microbiol. 2012, 3, 226. [Google Scholar] [CrossRef] [Green Version]
- Jawaid, S.; Seidle, H.; Zhou, W.; Abdirahman, H.; Abadeer, M.; Hix, J.H.; van Hoek, M.L.; Couch, R.D. Kinetic Characterization and Phosphoregulation of the Francisella tularensis 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase (MEP Synthase). PLoS ONE 2009, 4, e8288. [Google Scholar] [CrossRef] [PubMed]
- Biological and Chemical Terrorism: Strategic Plan for Preparedness and Response. Recommendations of the CDC Strategic Planning Workgroup. MMWR Recomm. Rep. 2000, 49, 1–14.
- Nguyen, V.K.; Parra-Rojas, C.; Hernandez-Vargas, E.A. The 2017 Plague Outbreak in Madagascar: Data Descriptions and Epidemic Modelling. Epidemics 2018, 25, 20–25. [Google Scholar] [CrossRef]
- Haymond, A.; Johny, C.; Dowdy, T.; Schweibenz, B.; Villarroel, K.; Young, R.; Mantooth, C.J.; Patel, T.; Bases, J.; San Jose, G.; et al. Kinetic Characterization and Allosteric Inhibition of the Yersinia pestis 1-Deoxy-D-xylulose 5-phosphate Reductoisomerase (MEP Synthase). PLoS ONE 2014, 9, e106243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ball, H.S.; Girma, M.; Zainab, M.; Riley, H.; Behrendt, C.T.; Lienau, C.; Konzuch, S.; Avelar, L.A.A.; Lungerich, B.; Soojhawon, I.; et al. Inhibition of the Yersinia pestis Methylerythritol Phosphate Pathway of Isoprenoid Biosynthesis by α-Phenyl-Substituted Reverse Fosmidomycin Analogues. ACS Omega 2020, 5, 5170–5175. [Google Scholar] [CrossRef] [Green Version]
- Michalopoulos, A.; Falagas, M.E. Treatment of Acinetobacter Infections. Expert Opin. Pharmacother. 2010, 11, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Tzouvelekis, L.S.; Markogiannakis, A.; Psichogiou, M.; Tassios, P.T.; Daikos, G.L. Carbapenemases in Klebsiella pneumoniae and Other Enterobacteriaceae: An Evolving Crisis of Global Dimensions. Clin. Microbiol. Rev. 2012, 25, 682–707. [Google Scholar] [CrossRef] [Green Version]
- Głowińska, A.; Trochimczuk, A.W. Polymer-Supported Phosphoric, Phosphonic and Phosphinic Acids—From Synthesis to Properties and Applications in Separation Processes. Molecules 2020, 25, 4236. [Google Scholar] [CrossRef]
- Kuemmerle, H.P.; Murakawa, T.; De Santis, F. Pharmacokinetic Evaluation of Fosmidomycin, a New Phosphonic Acid Antibiotic. Chemioterapia 1987, 6, 113–119. [Google Scholar]
- Murakawa, T.; Sakamoto, H.; Fukada, S.; Konishi, T.; Nishida, M. Pharmacokinetics of Fosmidomycin, a New Phosphonic Acid Antibiotic. Antimicrob. Agents Chemother. 1982, 21, 224–230. [Google Scholar] [CrossRef] [Green Version]
- Wiesner, J.; Borrmann, S.; Jomaa, H. Fosmidomycin for the Treatment of Malaria. Parasitol. Res. 2003, 90, S71–S76. [Google Scholar] [CrossRef] [PubMed]
- Ruangweerayut, R.; Looareesuwan, S.; Hutchinson, D.; Chauemung, A.; Banmairuroi, V.; Na-Bangchang, K. Assessment of the Pharmacokinetics and Dynamics of Two Combination Regimens of Fosmidomycin-Clindamycin in Patients with Acute Uncomplicated Falciparum Malaria. Malar. J. 2008, 7, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, T.; Ishibashi, K.; Terakawa, M.; Nishiyama, M.; Itoh, N.; Noguchi, H. Pharmacokinetics and Metabolism of Fosmidomycin, a New Phosphonic Acid, in Rats and Dogs. Eur. J. Drug Metab. Pharmacokinet. 1982, 7, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, J.; Henschker, D.; Hutchinson, D.B.; Beck, E.; Jomaa, H. In Vitro and In Vivo Synergy of Fosmidomycin, a Novel Antimalarial Drug, with Clindamycin. Antimicrob. Agents Chemother. 2002, 46, 2889–2894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na-Bangchang, K.; Ruengweerayut, R.; Karbwang, J.; Chauemung, A.; Hutchinson, D. Pharmacokinetics and Pharmacodynamics of Fosmidomycin Monotherapy and Combination Therapy with Clindamycin in the Treatment of Multidrug Resistant Falciparum Malaria. Malar. J. 2007, 6, 70. [Google Scholar] [CrossRef] [Green Version]
- Wiesner, J.; Ziemann, C.; Hintz, M.; Reichenberg, A.; Ortmann, R.; Schlitzer, M.; Fuhst, R.; Timmesfeld, N.; Vilcinskas, A.; Jomaa, H. FR-900098, an Antimalarial Development Candidate that Inhibits the Non-Mevalonate Isoprenoid Biosynthesis Pathway, Shows no Evidence of Acute Toxicity and Genotoxicity. Virulence 2016, 7, 718–728. [Google Scholar] [CrossRef] [Green Version]
- Uppala, R.; Arthanareeswari, M. Determination of Hydroxylamine Genotoxic Impurity by Derivatization in Penicillamine Drug Substance by GCHS-MS. Mater. Today Proc. 2021, 34, 506–509. [Google Scholar] [CrossRef]
- Kuemmerle, H.P.; Murakawa, T.; Soneoka, K.; Konishi, T. Fosmidomycin: A New Phosphonic Acid Antibiotic. Part I: Phase I Tolerance Studies. Int. J. Clin. Pharmacol. Ther. Toxicol. 1985, 23, 515–520. [Google Scholar]
- Wiesner, J.; Reichenberg, A.; Hintz, M.; Ortmann, R.; Schlitzer, M.; Van Calenbergh, S.; Borrmann, S.; Lell, B.; Kremsner, P.G.; Hutchinson, D.; et al. Fosmidomycin as an Antimalarial Agent. In Isoprenoid Synthesis in Plants and Microorganisms; Bach, T., Rohmer, M., Eds.; Springer: New York, NY, USA, 2012. [Google Scholar]
- World Health, O. Guidelines for the Treatment of Malaria, 3rd ed.; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Fernandes, J.F.; Lell, B.; Agnandji, S.T.; Obiang, R.M.; Bassat, Q.; Kremsner, P.G.; Mordmüller, B.; Grobusch, M.P. Fosmidomycin as an Antimalarial Drug: A Meta-Analysis of Clinical Trials. Future Microbiol. 2015, 10, 1375–1390. [Google Scholar] [CrossRef]
- Borrmann, S.; Lundgren, I.S.; Oyakhirome, S.; Impouma, B.; Matsiegui, P.; Adegnika, A.; Issifou, S.; Kun, J.; Hutchinson, D.; Wiesner, J.; et al. Fosmidomycin plus Clindamycin for Treatment of Pediatric Patients Aged 1 to 14 Years with Plasmodium falciparum Malaria. Antimicrob. Agents Chemother. 2006, 50, 2713–2718. [Google Scholar] [CrossRef] [Green Version]
- Lanaspa, M.; Moraleda, C.; Machevo, S.; González, R.; Serrano, B.; Macete, E.; Cisteró, P.; Mayor, A.; Hutchinson, D.; Kremsner, P.G.; et al. Inadequate Efficacy of a New Formulation of Fosmidomycin-Clindamycin Combination in Mozambican Children Less than Three Years Old with Uncomplicated Plasmodium falciparum Malaria. Antimicrob. Agents Chemother. 2012, 56, 2923–2928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mombo-Ngoma, G.; Remppis, J.; Sievers, M.; Zoleko Manego, R.; Endamne, L.; Kabwende, L.; Veletzky, L.; Nguyen, T.T.; Groger, M.; Lötsch, F.; et al. Efficacy and Safety of Fosmidomycin–Piperaquine as Nonartemisinin-Based Combination Therapy for Uncomplicated Falciparum Malaria: A Single-Arm, Age De-Escalation Proof-of-Concept Study in Gabon. Clin. Infect. Dis. 2017, 66, 1823–1830. [Google Scholar] [CrossRef] [PubMed]
- Eberl, M.; Oldfield, E.; Herrmann, T. Immuno-Antibiotics: Targeting Microbial Metabolic Pathways Sensed by Unconventional T Cells. Immunother. Adv. 2021, 1, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Deutsche Malaria Gesellschaft GmbH. DMG Receives Financing from European Union Malaria Fund. Available online: https://www.dmg-deutschemalaria.com/news/eu-malaria-fund-financing/ (accessed on 14 September 2021).
- Zhao, L.; Chang, W.C.; Xiao, Y.; Liu, H.W.; Liu, P. Methylerythritol Phosphate Pathway of Isoprenoid Biosynthesis. Annu. Rev. Biochem. 2013, 82, 497–530. [Google Scholar] [CrossRef] [PubMed]
- Kuzuyama, T.; Takahashi, S.; Watanabe, H.; Seto, H. Direct Formation of 2-C-Methyl-D-Erythritol 4-Phosphate from 1-Deoxy-D-Xylulose 5-Phosphate by 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase, a New Enzyme in the Non-Mevalonate Pathway to Isopentenyl Diphosphate. Tetrahedron Lett. 1998, 39, 4509–4512. [Google Scholar] [CrossRef]
- Chellapandi, P.; Prathiviraj, R.; Prisilla, A. Deciphering Structure, Function and Mechanism of Plasmodium IspD Homologs from Their Evolutionary Imprints. J. Comput. Aided Mol. Des. 2019, 33, 419–436. [Google Scholar] [CrossRef]
- Hoeffler, J.-F.; Tritsch, D.; Grosdemange-Billiard, C.; Rohmer, M. Isoprenoid Biosynthesis via the Methylerythritol Phosphate Pathway. Mechanistic Investigations of the 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase. Eur. J. Biochem. 2002, 269, 4446–4457. [Google Scholar] [CrossRef]
- Wong, U.; Cox, R. The Chemical Mechanism of D-1-Deoxyxylulose-5-Phosphate Reductoisomerase from Escherichia coli. Angew. Chem. 2007, 46, 4926–4929. [Google Scholar] [CrossRef]
- Koppisch, A.T.; Fox, D.T.; Blagg, B.S.J.; Poulter, C.D.E. coli MEP Synthase: Steady-State Kinetic Analysis and Substrate Binding. Biochemistry 2002, 41, 236–243. [Google Scholar] [CrossRef]
- Proteau, P.J. 1-Deoxy-D-xylulose 5-Phosphate Reductoisomerase: An Overview. Bioorg. Chem. 2004, 32, 483–493. [Google Scholar] [CrossRef]
- Argyrou, A.; Blanchard, J.S. Kinetic and Chemical Mechanism of Mycobacterium tuberculosis 1-Deoxy-D-xylulose-5-phosphate Isomeroreductase. Biochemistry 2004, 43, 4375–4384. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Cheve, G.; Avery, M.; McCurdy, C. Targeting the Methyl Erythritol Phosphate (MEP) Pathway for Novel Antimalarial, Antibacterial and Herbicidal Drug Discovery: Inhibition of 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase (DXR) Enzyme. Curr. Pharm. Des. 2007, 13, 1161–1177. [Google Scholar] [CrossRef] [PubMed]
- The UniProt, C. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A Better Web Interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umeda, T.; Tanaka, N.; Kusakabe, Y.; Nakanishi, M.; Kitade, Y.; Nakamura, K.T. Molecular Basis of Fosmidomycin’s Action on the Human Malaria Parasite Plasmodium falciparum. Sci. Rep. 2011, 1, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriksson, L.M.; Unge, T.; Carlsson, J.; Aqvist, J.; Mowbray, S.L.; Jones, T.A. Structures of Mycobacterium tuberculosis 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase Provide New Insights into Catalysis. J. Biol. Chem. 2007, 282, 19905–19916. [Google Scholar] [CrossRef] [Green Version]
- Konzuch, S.; Umeda, T.; Held, J.; Hähn, S.; Brücher, K.; Lienau, C.; Behrendt, C.T.; Gräwert, T.; Bacher, A.; Illarionov, B.; et al. Binding Modes of Reverse Fosmidomycin Analogs Toward the Antimalarial Target IspC. J. Med. Chem. 2014, 57, 8827–8838. [Google Scholar] [CrossRef]
- Yajima, S.; Nonaka, T.; Kuzuyama, T.; Seto, H.; Ohsawa, K. Crystal Structure of 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase Complexed With Cofactors: Implications of a Flexible Loop Movement Upon Substrate Binding. J. Biochem. 2002, 131, 313–317. [Google Scholar] [CrossRef]
- Henriksson, L.M.; Björkelid, C.; Mowbray, S.L.; Unge, T. The 1.9 A Resolution Structure of Mycobacterium tuberculosis 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase, a Potential Drug Target. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 807–813. [Google Scholar] [CrossRef] [Green Version]
- Codd, R. Traversing the coordination chemistry and chemical biology of hydroxamic acids. Coord. Chem. Rev. 2008, 252, 1387–1408. [Google Scholar] [CrossRef]
- Kuntz, L.; Tritsch, D.; Grosdemange-Billiard, C.; Hemmerlin, A.; Willem, A.; Bach, T.J.; Rohmer, M. Isoprenoid Biosynthesis as a Target for Antibacterial and Antiparasitic Drugs: Phosphonohydroxamic Acids as Inhibitors of Deoxyxylulose Phosphate Reducto-Isomerase. Biochem. J. 2005, 386, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.-H.; Fernandes, R.P.M.; Proteau, P.J. Evaluation of Fosmidomycin Analogs as Inhibitors of the Synechocystis sp. PCC6803 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase. Bioorg. Med. Chem. 2006, 14, 2375–2385. [Google Scholar] [CrossRef]
- Zinglé, C.; Kuntz, L.; Tritsch, D.; Grosdemange-Billiard, C.; Rohmer, M. Isoprenoid Biosynthesis via the Methylerythritol Phosphate Pathway: Structural Variations around Phosphonate Anchor and Spacer of Fosmidomycin, a Potent Inhibitor of Deoxyxylulose Phosphate Reductoisomerase. J. Org. Chem. 2010, 75, 3203–3207. [Google Scholar] [CrossRef]
- Behrendt, C.T.; Kunfermann, A.; Illarionova, V.; Matheeussen, A.; Pein, M.K.; Gräwert, T.; Kaiser, J.; Bacher, A.; Eisenreich, W.; Illarionov, B.; et al. Reverse Fosmidomycin Derivatives Against the Antimalarial Drug Target IspC (Dxr). J. Med. Chem. 2011, 54, 6796–6802. [Google Scholar] [CrossRef]
- Behrendt, C.T.; Kunfermann, A.; Illarionova, V.; Matheeussen, A.; Gräwert, T.; Groll, M.; Rohdich, F.; Bacher, A.; Eisenreich, W.; Fischer, M.; et al. Synthesis and Antiplasmodial Activity of Highly Active Reverse Analogues of the Antimalarial Drug Candidate Fosmidomycin. ChemMedChem 2010, 5, 1673–1676. [Google Scholar] [CrossRef] [PubMed]
- Giessmann, D.; Heidler, P.; Haemers, T.; Van Calenbergh, S.; Reichenberg, A.; Jomaa, H.; Weidemeyer, C.; Sanderbrand, S.; Wiesner, J.; Link, A. Towards New Antimalarial Drugs: Synthesis of Non-Hydrolyzable Phosphate Mimics as Feed for a Predictive QSAR Study on 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase Inhibitors. Chem. Biodivers. 2008, 5, 643–656. [Google Scholar] [CrossRef]
- Ortmann, R.; Wiesner, J.; Silber, K.; Klebe, G.; Jomaa, H.; Schlitzer, M. Novel Deoxyxylulosephosphate-Reductoisomerase Inhibitors: Fosmidomycin Derivatives with Spacious Acyl Residues. Arch. Der Pharm. 2007, 340, 483–490. [Google Scholar] [CrossRef]
- Andaloussi, M.; Lindh, M.; Björkelid, C.; Suresh, S.; Wieckowska, A.; Iyer, H.; Karlén, A.; Larhed, M. Substitution of the Phosphonic Acid and Hydroxamic Acid Functionalities of the DXR Inhibitor FR900098: An attempt to Improve the Activity Against Mycobacterium Tuberc. Bioorg. Med. Chem. Lett. 2011, 21, 5403–5407. [Google Scholar] [CrossRef]
- Mercklé, L.; Andrés-Gómez, A.D.; Dick, B.; Cox, R.; Godfrey, C. A Fragment-Based Approach to Understanding Inhibition of 1-Desoxy-D-Xylulose-5-Phosphate Reductoisomerase. ChemBioChem 2005, 6, 1866–1874. [Google Scholar] [CrossRef]
- Adeyemi, C.M.; Faridoon; Isaacs, M.; Mnkandhla, D.; Hoppe, H.C.; Krause, R.W.M.; Kaye, P.T. Synthesis and Antimalarial Activity of N-Benzylated (N-Arylcarbamoyl)alkylphosphonic Acid Derivatives. Bioorg. Med. Chem. 2016, 24, 6131–6138. [Google Scholar] [CrossRef] [PubMed]
- Bodill, T.; Conibear, A.C.; Blatch, G.L.; Lobb, K.A.; Kaye, P.T. Synthesis and Evaluation of Phosphonated N-Heteroarylcarboxamides as DOXP-Reductoisomerase (DXR) Inhibitors. Bioorg. Med. Chem. 2011, 19, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Bodill, T.; Conibear, A.C.; Mutorwa, M.K.; Goble, J.L.; Blatch, G.L.; Lobb, K.A.; Klein, R.; Kaye, P.T. Exploring DOXP-Reductoisomerase Binding Limits Using Phosphonated N-Aryl and N-Heteroarylcarboxamides as DXR Inhibitors. Bioorg. Med. Chem. 2013, 21, 4332–4341. [Google Scholar] [CrossRef]
- Kurz, T.; Geffken, D.; Wackendorff, C. Hydroxyurea analogues of Fosmidomycin. Z. Für Nat. 2003, 58, 106–110. [Google Scholar] [CrossRef]
- Zinglé, C.; Kuntz, L.; Tritsch, D.; Grosdemange-Billiard, C.; Rohmer, M. Modifications Around the Hydroxamic Acid Chelating Group of Fosmidomycin, an Inhibitor of the Metalloenzyme 1-Deoxyxylulose 5-Phosphate Reductoisomerase (DXR). Bioorg. Med. Chem. Lett. 2012, 22, 6563–6567. [Google Scholar] [CrossRef] [PubMed]
- Mancini, G.; Bouda, M.; Gamrat, J.M.; Tomsho, J.W. Synthesis and Antimicrobial Evaluation of γ-Borono Phosphonate Compounds in Escherichia coli and Mycobacterium smegmatis. ACS Omega 2019, 4, 14551–14559. [Google Scholar] [CrossRef] [Green Version]
- Montel, S.; Midrier, C.; Volle, J.-N.; Braun, R.; Haaf, K.; Willms, L.; Pirat, J.-L.; Virieux, D. Functionalized Phosphanyl-Phosphonic Acids as Unusual Complexing Units as Analogues of Fosmidomycin. Eur. J. Org. Chem. 2012, 2012, 3237–3248. [Google Scholar] [CrossRef]
- Deng, L.; Sundriyal, S.; Rubio, V.; Shi, Z.-Z.; Song, Y. Coordination Chemistry Based Approach to Lipophilic Inhibitors of 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase. J. Med. Chem. 2009, 52, 6539–6542. [Google Scholar] [CrossRef]
- Masini, T.; Kroezen, B.S.; Hirsch, A.K.H. Druggability of the Enzymes of the Non-Mevalonate-Pathway. Drug Discov. Today 2013, 18, 1256–1262. [Google Scholar] [CrossRef]
- San Jose, G.; Jackson, E.R.; Uh, E.; Johny, C.; Haymond, A.; Lundberg, L.; Pinkham, C.; Kehn-Hall, K.; Boshoff, H.I.; Couch, R.D.; et al. Design of Potential Bisubstrate Inhibitors Against Mycobacterium tuberculosis (Mtb) 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase (Dxr)—Evidence of a Novel Binding Mode. MedChemComm 2013, 4, 1099–1104. [Google Scholar] [CrossRef] [Green Version]
- Girma, M.B.; Ball, H.S.; Wang, X.; Brothers, R.C.; Jackson, E.R.; Meyers, M.J.; Dowd, C.S.; Couch, R.D. Mechanism of Action of N-Acyl and N-Alkoxy Fosmidomycin Analogs: Mono- and Bisubstrate Inhibition of IspC from Plasmodium falciparum, a Causative Agent of Malaria. ACS Omega 2021, 6, 27630–27639. [Google Scholar] [CrossRef] [PubMed]
- Jansson, A.M.; Więckowska, A.; Björkelid, C.; Yahiaoui, S.; Sooriyaarachchi, S.; Lindh, M.; Bergfors, T.; Dharavath, S.; Desroses, M.; Suresh, S.; et al. DXR Inhibition by Potent Mono- and Disubstituted Fosmidomycin Analogues. J. Med. Chem. 2013, 56, 6190–6199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmi, K.; Takeno, K.; Hashimoto, M.; Kamiya, T. Studies on Phosphonic Acid Antibiotics. IV. Synthesis and Antibacterial Activity of Analogs of 3-(N-Acetyl-N-hydroxyamino)-propylphosphonic Acid (FR-900098). Chem. Pharm. Bull. 1982, 30, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, E.R.; San Jose, G.; Brothers, R.C.; Edelstein, E.K.; Sheldon, Z.; Haymond, A.; Johny, C.; Boshoff, H.I.; Couch, R.D.; Dowd, C.S. The Effect of Chain Length and Unsaturation on Mtb Dxr Inhibition and Antitubercular Killing Activity of FR900098 Analogs. Bioorg. Med. Chem. Lett. 2014, 24, 649–653. [Google Scholar] [CrossRef]
- Devreux, V.; Wiesner, J.; Jomaa, H.; Van der Eycken, J.; Van Calenbergh, S. Synthesis and Evaluation of α,β-Unsaturated α-Aryl-substituted Fosmidomycin Analogues as DXR Inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 4920–4923. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Edwards, R.L.; Ball, H.; Johnson, C.; Haymond, A.; Girma, M.; Manikkam, M.; Brothers, R.C.; McKay, K.T.; Arnett, S.D.; et al. MEPicides: α,β-Unsaturated Fosmidomycin Analogues as DXR Inhibitors against Malaria. J. Med. Chem. 2018, 61, 8847–8858. [Google Scholar] [CrossRef]
- Katayama, N.; Tsubotani, S.; Nozaki, Y.; Harada, S.; Ono, H. Fosfadecin and Fosfocytocin, New Nucleotide Antibiotics Produced by Bacteria. J. Antibiot. 1990, 43, 238–246. [Google Scholar] [CrossRef] [Green Version]
- Haemers, T.; Wiesner, J.; Giessmann, D.; Verbrugghen, T.; Hillaert, U.; Ortmann, R.; Jomaa, H.; Link, A.; Schlitzer, M.; Van Calenbergh, S. Synthesis of β- and γ-Oxa Isosteres of Fosmidomycin and FR900098 as Antimalarial Candidates. Bioorg. Med. Chem. 2008, 16, 3361–3371. [Google Scholar] [CrossRef] [Green Version]
- Devreux, V.; Wiesner, J.; Goeman, J.L.; Van der Eycken, J.; Jomaa, H.; Van Calenbergh, S. Synthesis and Biological Evaluation of Cyclopropyl Analogues of Fosmidomycin as Potent Plasmodium falciparum Growth Inhibitors. J. Med. Chem. 2006, 49, 2656–2660. [Google Scholar] [CrossRef] [Green Version]
- Haemers, T.; Wiesner, J.; Busson, R.; Jomaa, H.; Van Calenbergh, S. Synthesis of α-Aryl-Substituted and Conformationally Restricted Fosmidomycin Analogues as Promising Antimalarials. Eur. J. Org. Chem. 2006, 2006, 3856–3863. [Google Scholar] [CrossRef] [Green Version]
- Kurz, T.; Schlueter, K.; Pein, M.; Behrendt, C.T.; Bergmann, B.; Walter, R.D. Conformationally Restrained Aromatic Analogues of Fosmidomycin and FR900098. Arch. Der Pharm. 2007, 340, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Verbrugghen, T.; Cos, P.; Maes, L.; Van Calenbergh, S. Synthesis and Evaluation of α-Halogenated Analogues of 3-(Acetylhydroxyamino)propylphosphonic Acid (FR900098) as Antimalarials. J. Med. Chem. 2010, 53, 5342–5346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andaloussi, M.; Henriksson, L.M.; Wiȩckowska, A.; Lindh, M.; Björkelid, C.; Larsson, A.M.; Suresh, S.; Iyer, H.; Srinivasa, B.R.; Bergfors, T.; et al. Design, Synthesis, and X-ray Crystallographic Studies of α-Aryl Substituted Fosmidomycin Analogues as Inhibitors of Mycobacterium tuberculosis 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase. J. Med. Chem. 2011, 54, 4964–4976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordqvist, A.; Björkelid, C.; Andaloussi, M.; Jansson, A.M.; Mowbray, S.L.; Karlén, A.; Larhed, M. Synthesis of Functionalized Cinnamaldehyde Derivatives by an Oxidative Heck Reaction and their Use as Starting Materials for Preparation of Mycobacterium tuberculosis 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase Inhibitors. J. Org. Chem. 2011, 76, 8986–8998. [Google Scholar] [CrossRef] [PubMed]
- Devreux, V.; Wiesner, J.; Jomaa, H.; Rozenski, J.; Van der Eycken, J.; Van Calenbergh, S. Divergent Strategy for the Synthesis of α-Aryl-Substituted Fosmidomycin Analogues. J. Org. Chem. 2007, 72, 3783–3789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlüter, K.; Walter, R.D.; Bergmann, B.; Kurz, T. Arylmethyl Substituted Derivatives of Fosmidomycin: Synthesis and Antimalarial Activity. Eur. J. Med. Chem. 2006, 41, 1385–1397. [Google Scholar] [CrossRef] [PubMed]
- Kurz, T.; Geffken, D.; Kaula, U. Phosphororganische Verbindungen und deren Verwendung. DE10356410A1, 23 June 2005. [Google Scholar]
- Kurz, T.; Schlüter, K.; Kaula, U.; Bergmann, B.; Walter, R.D.; Geffken, D. Synthesis and Antimalarial Activity of Chain Substituted Pivaloyloxymethyl Ester Analogues of Fosmidomycin and FR900098. Bioorg. Med. Chem. 2006, 14, 5121–5135. [Google Scholar] [CrossRef] [PubMed]
- Perruchon, J.; Ortmann, R.; Altenkämper, M.; Silber, K.; Wiesner, J.; Jomaa, H.; Klebe, G.; Schlitzer, M. Studies Addressing the Importance of Charge in the Binding of Fosmidomycin-like Molecules to Deoxyxylulosephosphate Reductoisomerase. ChemMedChem 2008, 3, 1232–1241. [Google Scholar] [CrossRef]
- Dreneau, A.; Krebs, F.S.; Munier, M.; Ngov, C.; Tritsch, D.; Lièvremont, D.; Rohmer, M.; Grosdemange-Billiard, C. α,α-Difluorophosphonohydroxamic Acid Derivatives Among the Best Antibacterial Fosmidomycin Analogues. Molecules 2021, 26, 5111. [Google Scholar] [CrossRef] [PubMed]
- Chekan, J.R.; Cogan, D.P.; Nair, S.K. Molecular Basis for Resistance Against Phosphonate Antibiotics and Herbicides. MedChemComm 2016, 7, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Verbrugghen, T.; Vandurm, P.; Pouyez, J.; Maes, L.; Wouters, J.; Van Calenbergh, S. Alpha-Heteroatom Derivatized Analogues of 3-(Acetylhydroxyamino)propyl Phosphonic Acid (FR900098) as Antimalarials. J. Med. Chem. 2013, 56, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Kunfermann, A.; Lienau, C.; Illarionov, B.; Held, J.; Gräwert, T.; Behrendt, C.T.; Werner, P.; Hähn, S.; Eisenreich, W.; Riederer, U.; et al. IspC as Target for Antiinfective Drug Discovery: Synthesis, Enantiomeric Separation, and Structural Biology of Fosmidomycin Thia Isosters. J. Med. Chem. 2013, 56, 8151–8162. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Diao, J.; Cai, G.; Deng, L.; Zheng, B.; Yao, Y.; Song, Y. Antimalarial and Structural Studies of Pyridine-Containing Inhibitors of 1-Deoxyxylulose-5-phosphate Reductoisomerase. ACS Med. Chem. Lett. 2013, 4, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Steinbacher, S.; Kaiser, J.; Eisenreich, W.; Huber, R.; Bacher, A.; Rohdich, F. Structural Basis of Fosmidomycin Action Revealed by the Complex with 2-C-Methyl-D-Erythritol 4-Phosphate Synthase (IspC). Implications for the Catalytic Mechanism and Anti-Malaria Drug Development. J. Biol. Chem. 2003, 278, 18401–18407. [Google Scholar] [CrossRef]
- Yajima, S.; Hara, K.; Iino, D.; Sasaki, Y.; Kuzuyama, T.; Ohsawa, K.; Seto, H. Structure of 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase in a Quaternary Complex with a Magnesium Ion, NADPH and the Antimalarial Drug Gosmidomycin. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2007, 63, 466–470. [Google Scholar] [CrossRef] [Green Version]
- Brücher, K.; Illarionov, B.; Held, J.; Tschan, S.; Kunfermann, A.; Pein, M.K.; Bacher, A.; Gräwert, T.; Maes, L.; Mordmüller, B.; et al. α-Substituted β-Oxa Isosteres of Fosmidomycin: Synthesis and Biological Evaluation. J. Med. Chem. 2012, 55, 6566–6575. [Google Scholar] [CrossRef]
- Lienau, C.; Gräwert, T.; Alves Avelar, L.A.; Illarionov, B.; Held, J.; Knaab, T.C.; Lungerich, B.; van Geelen, L.; Meier, D.; Geissler, S.; et al. Novel Reverse Thia-Analogs of Fosmidomycin: Synthesis and Antiplasmodial Activity. Eur. J. Med. Chem. 2019, 181, 111555. [Google Scholar] [CrossRef]
- Adeyemi, C.M.; Hoppe, H.C.; Isaacs, M.; Klein, R.; Lobb, K.A.; Kaye, P.T. Synthesis of N-Substituted Phosphoramidic Acid Esters as “Reverse” Fosmidomycin Analogues. Tetrahedron 2019, 75, 2371–2378. [Google Scholar] [CrossRef]
- Chofor, R.; Sooriyaarachchi, S.; Risseeuw, M.D.P.; Bergfors, T.; Pouyez, J.; Johny, C.; Haymond, A.; Everaert, A.; Dowd, C.S.; Maes, L.; et al. Synthesis and Bioactivity of β-Substituted Fosmidomycin Analogues Targeting 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase. J. Med. Chem. 2015, 58, 2988–3001. [Google Scholar] [CrossRef] [Green Version]
- Sooriyaarachchi, S.; Chofor, R.; Risseeuw, M.D.P.; Bergfors, T.; Pouyez, J.; Dowd, C.S.; Maes, L.; Wouters, J.; Jones, T.A.; Van Calenbergh, S.; et al. Targeting an Aromatic Hotspot in Plasmodium falciparum 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase with β-Arylpropyl Analogues of Fosmidomycin. ChemMedChem 2016, 11, 2024–2036. [Google Scholar] [CrossRef] [Green Version]
- Munier, M.; Tritsch, D.; Krebs, F.; Esque, J.; Hemmerlin, A.; Rohmer, M.; Stote, R.H.; Grosdemange-Billiard, C. Synthesis and Biological Evaluation of Phosphate Isosters of Fosmidomycin and Analogs as Inhibitors of Escherichia coli and Mycobacterium smegmatis 1-Deoxyxylulose 5-Phosphate Reductoisomerases. Bioorg. Med. Chem. 2017, 25, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Meyer, O.; Grosdemange-Billiard, C.; Tritsch, D.; Rohmer, M. Isoprenoid Biosynthesis via the MEP Pathway. Synthesis of (3R,4S)-3,4-Dihydroxy-5-Oxohexylphosphonic Acid, an Isosteric Analogue of 1-Deoxy-D-Xylulose 5-Phosphate, the Substrate of the 1-Deoxy-D-Xylulose 5-Phosphate Reducto-isomerase. Org. Biomol. Chem. 2003, 1, 4367–4372. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.; Ross, K.C. 3 Prodrug Design for Phosphates and Phosphonates. In Progress in Medicinal Chemistry; Ellis, G.P., Luscombe, D.K., Eds.; Elsevier: Amsterdam, The Netherlands, 1997; Volume 34, pp. 111–147. [Google Scholar]
- Krise, J.P.; Stella, V.J. Prodrugs of Phosphates, Phosphonates, and Phosphinates. Adv. Drug Deliv. Rev. 1996, 19, 287–310. [Google Scholar] [CrossRef]
- Kurz, T.; Geffken, D.; Wackendorff, C. Carboxylic Acid Analogues of Fosmidomycin. Z. Für Nat. 2003, 58, 457–461. [Google Scholar] [CrossRef]
- Nguyen-Trung, A.T.; Tritsch, D.; Grosdemange-Billiard, C.; Rohmer, M. Synthesis of Tetrazole Analogues of Phosphonohydroxamic Acids: An Attempt to Improve the Inhibitory Activity Against the DXR. Bioorg. Med. Chem. Lett. 2013, 23, 1643–1647. [Google Scholar] [CrossRef]
- Macchiarulo, A.; Pellicciari, R. Exploring the Other Side of Biologically Relevant Chemical Space: Insights into Carboxylic, Sulfonic and Phosphonic Acid Bioisosteric Relationships. J. Mol. Graph. Model. 2007, 26, 728–739. [Google Scholar] [CrossRef]
- Gadakh, B.; Pouyez, J.; Wouters, J.; Venkatesham, A.; Cos, P.; Van Aerschot, A. N-Acylated Sulfonamide Congeners of Fosmidomycin Lack Any Inhibitory Activity Against DXR. Bioorg. Med. Chem. Lett. 2015, 25, 1577–1579. [Google Scholar] [CrossRef]
- Island, M.D.; Wei, B.Y.; Kadner, R.J. Structure and Function of the uhp Genes for the Sugar Phosphate Transport System in Escherichia coli and Salmonella typhimurium. J. Bacteriol. 1992, 174, 2754–2762. [Google Scholar] [CrossRef] [Green Version]
- Lemieux, M.J.; Huang, Y.; Wang, D.-N. Glycerol-3-Phosphate Transporter of Escherichia coli: Structure, Function and Regulation. Res. Microbiol. 2004, 155, 623–629. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Furukawa, S.; Ogihara, H.; Yamasaki, M. Fosmidomycin Resistance in Adenylate Cyclase Deficient (cya) Mutants of Escherichia coli. Biosci. Biotechnol. Biochem. 2003, 67, 2030–2033. [Google Scholar] [CrossRef] [Green Version]
- Hemmerlin, A.; Tritsch, D.; Hammann, P.; Rohmer, M.; Bach, T.J. Profiling of Defense Responses in Escherichia coli Treated with Fosmidomycin. Biochimie 2014, 99, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Halpern, L. The Transfer of Inorganic Phosphorous Across the Red Cell Membrane. J. Biol. Chem. 1936, 114, 747–770. [Google Scholar] [CrossRef]
- Leibman, K.C.; Heidelberger, C. The Metabolism of P32-Labelled Ribonucleotides in Tissue Slices and Cell. J. Biol. Chem. 1955, 216, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Wiemer, A.J. Metabolic Efficacy of Phosphate Prodrugs and the Remdesivir Paradigm. ACS Pharmacol. Transl. Sci. 2020, 3, 613–626. [Google Scholar] [CrossRef]
- De Clercq, E. Tenofovir Alafenamide (TAF) as the Successor of Tenofovir Disoproxil Fumarate (TDF). Biochem. Pharmacol. 2016, 119, 1–7. [Google Scholar] [CrossRef]
- Hostetler, K.Y. Alkoxyalkyl Prodrugs of Acyclic Nucleoside Phosphonates Enhance Oral Antiviral Activity and Reduce Toxicity: Current State of the Art. Antivir. Res. 2009, 82, A84–A98. [Google Scholar] [CrossRef] [Green Version]
- Spinner, C.D.; Gottlieb, R.L.; Criner, G.J.; Arribas López, J.R.; Cattelan, A.M.; Soriano Viladomiu, A.; Ogbuagu, O.; Malhotra, P.; Mullane, K.M.; Castagna, A.; et al. Effect of Remdesivir vs Standard Care on Clinical Status at 11 Days in Patients With Moderate COVID-19: A Randomized Clinical Trial. JAMA 2020, 324, 1048–1057. [Google Scholar] [CrossRef]
- Goldman, J.D.; Lye, D.C.B.; Hui, D.S.; Marks, K.M.; Bruno, R.; Montejano, R.; Spinner, C.D.; Galli, M.; Ahn, M.Y.; Nahass, R.G.; et al. Remdesivir for 5 or 10 Days in Patients with Severe COVID-19. N. Engl. J. Med. 2020, 383, 1827–1837. [Google Scholar] [CrossRef]
- Smolders, E.J.; Jansen, A.M.E.; Ter Horst, P.G.J.; Rockstroh, J.; Back, D.J.; Burger, D.M. Viral Hepatitis C Therapy: Pharmacokinetic and Pharmacodynamic Considerations: A 2019 Update. Clin. Pharmacokinet. 2019, 58, 1237–1263. [Google Scholar] [CrossRef] [Green Version]
- Kirby, B.J.; Symonds, W.T.; Kearney, B.P.; Mathias, A.A. Pharmacokinetic, Pharmacodynamic, and Drug-Interaction Profile of the Hepatitis C Virus NS5B Polymerase Inhibitor Sofosbuvir. Clin. Pharmacokinet. 2015, 54, 677–690. [Google Scholar] [CrossRef]
- Scott, L.J.; Chan, H.L.Y. Tenofovir Alafenamide: A Review in Chronic Hepatitis B. Drugs 2017, 77, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, K.; Fung, S.K.; Nguyen, T.T.; Cheng, W.; Sicard, E.; Ryder, S.D.; Flaherty, J.F.; Lawson, E.; Zhao, S.; Subramanian, G.M.; et al. Twenty-Eight Day Safety, Antiviral Activity, and Pharmacokinetics of Tenofovir Alafenamide for Treatment of Chronic Hepatitis B Infection. J. Hepatol. 2015, 62, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Mackman, R.L. Anti-HIV Nucleoside Phosphonate GS-9148 and Its Prodrug GS-9131: Scale Up of a 2′-F Modified Cyclic Nucleoside Phosphonate and Synthesis of Selected Amidate Prodrugs. Curr. Protoc. Nucleic Acid Chem. 2014, 56, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Reichenberg, A.; Wiesner, J.; Weidemeyer, C.; Dreiseidler, E.; Sanderbrand, S.; Altincicek, B.; Beck, E.; Schlitzer, M.; Jomaa, H. Diaryl Ester Prodrugs of FR900098 with Improved in Vivo Antimalarial Activity. Bioorg. Med. Chem. Lett. 2001, 11, 833–835. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, J.; Ortmann, R.; Jomaa, H.; Schlitzer, M. Double Ester Prodrugs of FR900098 Display Enhanced in-Vitro Antimalarial Activity. Arch. Der Pharm. 2007, 340, 667–669. [Google Scholar] [CrossRef]
- Ortmann, R.; Wiesner, J.; Reichenberg, A.; Henschker, D.; Beck, E.; Jomaa, H.; Schlitzer, M. Acyloxyalkyl Ester Prodrugs of FR900098 With Improved in Vivo Anti-Malarial Activity. Bioorg. Med. Chem. Lett. 2003, 13, 2163–2166. [Google Scholar] [CrossRef]
- Ortmann, R.; Wiesner, J.; Reichenberg, A.; Henschker, D.; Beck, E.; Jomaa, H.; Schlitzer, M. Alkoxycarbonyloxyethyl Ester Prodrugs of FR900098 With Improved in vivo Antimalarial Activity. Arch. Der Pharm. 2005, 338, 305–314. [Google Scholar] [CrossRef]
- Courtens, C.; Risseeuw, M.; Caljon, G.; Cos, P.; Van Calenbergh, S. Acyloxybenzyl and Alkoxyalkyl Prodrugs of a Fosmidomycin Surrogate as Antimalarial and Antitubercular Agents. ACS Med. Chem. Lett. 2018, 9, 986–989. [Google Scholar] [CrossRef]
- Jackson, E.R.; Dowd, C.S. Inhibition of 1-Deoxy-D-Xylulose-5-phosphate Reductoisomerase (Dxr): A Review of the Synthesis and Biological Evaluation of Recent Inhibitors. Curr. Top. Med. Chem. 2012, 12, 706–728. [Google Scholar] [CrossRef]
- Ponaire, S.; Zinglé, C.; Tritsch, D.; Grosdemange-Billiard, C.; Rohmer, M. Growth Inhibition of Mycobacterium smegmatis by Prodrugs of Deoxyxylulose Phosphate Reducto-Isomerase Inhibitors, Promising Anti-Mycobacterial Agents. Eur. J. Med. Chem. 2012, 51, 277–285. [Google Scholar] [CrossRef]
- Liu, J.; Barry, C.E., III; Besra, G.S.; Nikaido, H. Mycolic Acid Structure Determines the Fluidity of the Mycobacterial Cell Wall. J. Biol. Chem. 1996, 271, 29545–29551. [Google Scholar] [CrossRef] [Green Version]
- Edwards, R.L.; Brothers, R.C.; Wang, X.; Maron, M.I.; Ziniel, P.D.; Tsang, P.S.; Kraft, T.E.; Hruz, P.W.; Williamson, K.C.; Dowd, C.S.; et al. MEPicides: Potent Antimalarial Prodrugs Targeting Isoprenoid Biosynthesis. Sci. Rep. 2017, 7, 8400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faísca Phillips, A.M.; Nogueira, F.; Murtinheira, F.; Barros, M.T. Synthesis and Antimalarial Evaluation of Prodrugs of Novel Fosmidomycin Analogues. Bioorg. Med. Chem. Lett. 2015, 25, 2112–2116. [Google Scholar] [CrossRef] [PubMed]
- Courtens, C.; Risseeuw, M.; Caljon, G.; Maes, L.; Cos, P.; Martin, A.; Van Calenbergh, S. Double Prodrugs of a Fosmidomycin Surrogate as Antimalarial and Antitubercular Agents. Bioorg. Med. Chem. Lett. 2019, 29, 1232–1235. [Google Scholar] [CrossRef] [PubMed]
- Courtens, C.; Risseeuw, M.; Caljon, G.; Maes, L.; Martin, A.; Van Calenbergh, S. Amino Acid Based Prodrugs of a Fosmidomycin Surrogate as Antimalarial and Antitubercular Agents. Bioorg. Med. Chem. 2019, 27, 729–747. [Google Scholar] [CrossRef] [PubMed]
- Courtens, C.; Risseeuw, M.; Caljon, G.; Cos, P.; Martin, A.; Van Calenbergh, S. Phosphonodiamidate Prodrugs of N-Alkoxy Analogs of a Fosmidomycin Surrogate as Antimalarial and Antitubercular Agents. Bioorg. Med. Chem. Lett. 2019, 29, 1051–1053. [Google Scholar] [CrossRef] [PubMed]
- Munier, M.; Tritsch, D.; Lièvremont, D.; Rohmer, M.; Grosdemange-Billiard, C. Synthesis and Biological Evaluation of Aryl Phosphoramidate Prodrugs of Fosfoxacin and its Derivatives. Bioorg. Chem. 2019, 89, 103012. [Google Scholar] [CrossRef]
- Langel, Ü. Cell-Penetrating Peptides: Processes and Applications; CRC Press: Boca Raton, FL, USA, 2002; p. 424. [Google Scholar]
- Pujals, S.; Fernández-Carneado, J.; López-Iglesias, C.; Kogan, M.J.; Giralt, E. Mechanistic Aspects of CPP-Mediated Intracellular Drug Delivery: Relevance of CPP Self-Assembly. Biochim. Biophys. Acta Biomembr. 2006, 1758, 264–279. [Google Scholar] [CrossRef] [Green Version]
- Kamena, F.; Monnanda, B.; Makou, D.; Capone, S.; Patora-Komisarska, K.; Seebach, D. On the Mechanism of Eukaryotic Cell Penetration by α- and β-Oligoarginines—Targeting Infected Erythro cytes. Chem. Biodivers. 2011, 8, 1–12. [Google Scholar] [CrossRef]
- Samuel, B.U.; Hearn, B.; Mack, D.; Wender, P.; Rothbard, J.; Kirisits, M.J.; Mui, E.; Wernimont, S.; Roberts, C.W.; Muench, S.P.; et al. Delivery of Antimicrobials into Parasites. Proc. Natl. Acad. Sci. USA 2003, 100, 14281–14286. [Google Scholar] [CrossRef] [Green Version]
- Chadwick, J.; Jones, M.; Mercer, A.E.; Stocks, P.A.; Ward, S.A.; Park, B.K.; O’Neill, P.M. Design, Synthesis and Antimalarial/Anticancer Evaluation of Spermidine Linked Artemisinin Conjugates Designed to Exploit Polyamine Transporters in Plasmodium falciparum and HL-60 Cancer Cell Lines. Bioorg.Med. Chem. 2010, 18, 2586–2597. [Google Scholar] [CrossRef] [PubMed]
- Yajima, S.; Hara, K.; Sanders, J.M.; Yin, F.; Ohsawa, K.; Wiesner, J.; Jomaa, H.; Oldfield, E. Crystallographic Structures of Two Bisphosphonate:1-Deoxyxylulose-5-Phosphate Reductoisomerase Complexes. J. Am. Chem. Soc. 2004, 126, 10824–10825. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Endo, K.; Kato, M.; Cheng, G.; Yajima, S.; Song, Y. Structures of 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase/Lipophilic Phosphonate Complexes. ACS Med. Chem. Lett. 2011, 2, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Zinglé, C.; Tritsch, D.; Grosdemange-Billiard, C.; Rohmer, M. Catechol-Rhodanine Derivatives: Specific and Promiscuous Inhibitors of Escherichia coli Deoxyxylulose Phosphate Reductoisomerase (DXR). Bioorg. Med. Chem. 2014, 22, 3713–3719. [Google Scholar] [CrossRef]
- Adeyemi, C.M.; Hoppe, H.C.; Isaacs, M.; Mnkandhla, D.; Lobb, K.A.; Klein, R.; Kaye, P.T. Synthesis and Anti-Parasitic Activity of N-Benzylated Phosphoramidate Mg2+-Chelating Ligands. Bioorg. Chem. 2020, 105, 104280. [Google Scholar] [CrossRef]
- Hui, X.; Yue, Q.; Zhang, D.D.; Li, H.; Yang, S.Q.; Gao, W.Y. Antimicrobial Mechanism of Theaflavins: They Target 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase, the Key Enzyme of the MEP Terpenoid Biosynthetic Pathway. Sci. Rep. 2016, 6, 38945. [Google Scholar] [CrossRef] [Green Version]
- Haymond, A.; Dowdy, T.; Johny, C.; Johnson, C.; Ball, H.; Dailey, A.; Schweibenz, B.; Villarroel, K.; Young, R.; Mantooth, C.J.; et al. A High-Throughput Screening Campaign to Identify Inhibitors of DXP Reductoisomerase (IspC) and MEP Cytidylyltransferase (IspD). Anal. Biochem. 2018, 542, 63–75. [Google Scholar] [CrossRef]
















































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knak, T.; Abdullaziz, M.A.; Höfmann, S.; Alves Avelar, L.A.; Klein, S.; Martin, M.; Fischer, M.; Tanaka, N.; Kurz, T. Over 40 Years of Fosmidomycin Drug Research: A Comprehensive Review and Future Opportunities. Pharmaceuticals 2022, 15, 1553. https://doi.org/10.3390/ph15121553
Knak T, Abdullaziz MA, Höfmann S, Alves Avelar LA, Klein S, Martin M, Fischer M, Tanaka N, Kurz T. Over 40 Years of Fosmidomycin Drug Research: A Comprehensive Review and Future Opportunities. Pharmaceuticals. 2022; 15(12):1553. https://doi.org/10.3390/ph15121553
Chicago/Turabian StyleKnak, Talea, Mona A. Abdullaziz, Stefan Höfmann, Leandro A. Alves Avelar, Saskia Klein, Matthew Martin, Markus Fischer, Nobutada Tanaka, and Thomas Kurz. 2022. "Over 40 Years of Fosmidomycin Drug Research: A Comprehensive Review and Future Opportunities" Pharmaceuticals 15, no. 12: 1553. https://doi.org/10.3390/ph15121553
APA StyleKnak, T., Abdullaziz, M. A., Höfmann, S., Alves Avelar, L. A., Klein, S., Martin, M., Fischer, M., Tanaka, N., & Kurz, T. (2022). Over 40 Years of Fosmidomycin Drug Research: A Comprehensive Review and Future Opportunities. Pharmaceuticals, 15(12), 1553. https://doi.org/10.3390/ph15121553